

Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is a devastating inherited skin blistering disease caused by mutations in the COL7A1 gene that encodes type VII collagen (C7), a major structural component of anchoring fibrils at the dermal‐epidermal junction (DEJ). We recently demonstrated that human cord blood‐derived unrestricted somatic stem cells promote wound healing and ameliorate the blistering phenotype in a RDEB (col7a1 −/−) mouse model. Here, we demonstrate significant therapeutic effect of a further novel stem cell product in RDEB, that is, human placental‐derived stem cells (HPDSCs), currently being used as human leukocyte antigen‐independent donor cells with allogeneic umbilical cord blood stem cell transplantation in patients with malignant and nonmalignant diseases. HPDSCs are isolated from full‐term placentas following saline perfusion, red blood cell depletion, and volume reduction. HPDSCs contain significantly higher level of both hematopoietic and nonhematopoietic stem and progenitor cells than cord blood and are low in T cell content. A single intrahepatic administration of HPDSCs significantly elongated the median life span of the col7a1 −/− mice from 2 to 7 days and an additional intrahepatic administration significantly extended the median life span to 18 days. We further demonstrated that after intrahepatic administration, HPDSCs engrafted short‐term in the organs affected by RDEB, that is, skin and gastrointestinal tract of col7a1 −/− mice, increased adhesion at the DEJ and deposited C7 even at 4 months after administration of HPDSCs, without inducing anti‐C7 antibodies. This study warrants future clinical investigation to determine the safety and efficacy of HPDSCs in patients with severe RDEB. stem cells translational medicine 2018;7:530–542
Keywords: Stem cell transplantation, Placenta, Cellular therapy, Clinical translation, Transgenic mouse, Stem/progenitor cell
Significance Statement.
A major challenge for the treatment of recessive dystrophic epidermolysis bullosa (RDEB) is a generalized blistering in the skin as well as the gastrointestinal (GI) tract. A systemic approach that is safe to patients and alleviates both the outside and inside manifestations is in great demand for patients with RDEB. This article demonstrates significant therapeutic benefits of human placental‐derived stem cells (HPDSCs) in a mouse model of RDEB, in the absence of any conditioning regimen. HPDSCs effectively migrated to the organs affected by RDEB, that is, skin and GI tract, deposited the missing protein (C7), and significantly improved the adherence of the epidermis to the dermis of the skin without inducing anti‐C7 antibodies. As HPDSCs have already been demonstrated to be safe in humans with malignant and nonmalignant diseases, the results from this study will greatly facilitate a future clinical investigation using HPDSCs as human leukocyte antigen‐independent donor cells in patients with RDEB.
Introduction
Recessive dystrophic epidermolysis bullosa (RDEB) is an inherited mucocutaneous blistering disease caused by mutations in COL7A1 1. This gene encodes type VII collagen (C7), a major component of anchoring fibrils at the dermal/epidermal basement membrane zone (BMZ). Patients with RDEB suffer from recurrent erosions in the skin, oral mucosa, and gastrointestinal (GI) and genito‐urinary tracts 2. Moreover, patients with RDEB are at high risk of developing aggressive cutaneous squamous cell carcinomas, which is associated with a very poor prognosis 3, 4, 5, 6, 7. Currently, there is no cure for RDEB, other than supportive and palliative care. Nevertheless, significant progress has been made toward more effective treatment of patients with RDEB using a variety of approaches focusing on repair of genetic defects, protein replacement, and cell therapies 6, 8, 9, 10, 11, 12, 13, 14, 15, 16.
Preclinical studies have demonstrated that transplantation of wild‐type (WT) murine hematopoietic progenitor enriched cells improves survival of col7a1 −/− mice 17. Subsequent clinical studies in patients with RDEB indicated that allogeneic hematopoietic progenitor cell transplantation (AlloHPCT) following myeloablative chemotherapy results in improvement in wound healing and increased C7 protein deposition at the dermal‐epidermal junction (DEJ) 10. Complications from myeloablative conditioning, however, have been a major limitation in this therapeutic approach. Furthermore, mortality from AlloHPCT is frequently due to delay in neutrophil engraftment, which increases the risk of the patients contracting opportunistic infections and developing severe graft versus host disease (GvHD). Our recent experience, as well as that of others, demonstrates better tolerance and less toxicity in patients with RDEB following reduced toxicity conditioning and AlloHPCT 18, 19.
Human placental‐derived stem cells (HPDSCs) are a source of stem cells obtained by Celgene Cellular Therapeutics (CCT)'s proprietary process involving perfusion of donated full‐term placentas and depletion of red blood cells, nonviable cells, and tissue debris 20. Although both HPDSCs and human umbilical cord blood (UCB) originate from placentas, the processing, that is, perfusion of placentas versus gravity collection of blood, has resulted in phenotypic and functional differences between these two cell products. As HPDSCs are largely negative for major histocompatibility complex (MHC) class II molecules, we initiated a pilot study of investigating the safety and efficacy of adding HPDSCs without human leukocyte antigen (HLA) matching (as universal donor cells), in allogeneic UCB transplantation in children and adults with selected malignant and nonmalignant diseases 20. To date, there have been no adverse events associated with HPDSC infusions in the 35 patients enrolled to date. However, compared to the mean of 97% whole donor UCB chimerism at day 30, the average percentage of whole blood HPDSC chimerism was less than 1%, suggesting a different functional property of HPDSC than UCB in hematopoietic reconstitution 21. Nevertheless, HPDSC infusion appeared to facilitate UCB immune reconstitution and reduce the probability of grade II–IV acute GvHD (aGVHD), compared with historical data 21. Furthermore, as HPDSCs are rich in stem cells, we hypothesize that HPDSCs per se may have direct regenerative properties, which would grant their use as a product by itself for the treatment of RDEB, without any toxic conditioning regimen.
In this study, as a first step toward bringing HPDSCs to the clinic in patients with RDEB, we examined the role of HPDSCs on overall survival and different functional outcomes in a therapeutic animal model of RDEB. We demonstrate that administration of HPDSCs, without preconditioning or immunosuppression, has therapeutic benefits in col7a1 −/− mice.
Materials and Methods
Mice
The Col7a1 − / − RDEB mice were generated by breeding C57BL6/J Col7a1 +/− mice with the genotype determined by polymerase chain reaction (PCR) 22. C57BL6/J Col7a1 +/− mice, kindly provided by Dr. Jouni Uitto at Thomas Jefferson University, were developed by targeted ablation of the Col7a1 gene through out‐of‐frame deletion of exons 14–18 22. All animal studies were conducted using protocols approved by New York Medical College Institutional Animal Care & Use Committee (IACUC).
Preparation of HPDSCs
HPDSCs were prepared and cryopreserved at CCT in a Good Manufacturing Practices‐compliant facility, as previously reported 23. Briefly, postpartum placentas were procured following full‐informed consent of donors with donor eligibility documentation and quality control. HPDSC isolation and recovery were achieved by cannulation of the umbilical vessels (two arteries and one vein) under sterile conditions with polyethylene catheters connected to a flow‐controlled fluid circuit allowing perfusion of the placenta. A total of 750 ml of perfusion solution (0.9% NaCl injection solution USP Grade) (VWR, Radnor, PA) was collected from each placenta. After red blood cell depletion using Hetastarch and volume reduction, the cells were cryopreserved in a solution containing 5% human albumin and 10% dimethyl sulfoxide with a controlled rate freezer prior to final storage in the gas phase of a liquid nitrogen tank. Viability of the HPDSCs was determined using 7‐aminoactinomycin D (BD Bioscience, San Jose, CA) by flow cytometry.
Colony Forming Cell (CFU) Assay
CD34+ cells were selected from HPDSCs with a human CD34 positive selection kit and isolated using automated cell separator RoboSep (StemCell Technologies, Inc., Vancouver, Canada). The CFU assay was performed using MethoCult, following the manufacturer's protocol (StemCell Technologies, Inc.). Briefly, CD34+ cells were mixed with complete MethoCult medium supplemented with stem cell factor, granulocyte colony‐stimulating factor, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), interleukin 3, interleukin 6, and erythropoietin (Epo) and plated in triplicate at a density of 100, 300, and 1,000 cells per 35 mm plate, respectively. After 2–3 weeks, the culture was evaluated for colony formation and scoring using an inverted microscope and a scoring grid.
Flow Cytometry Analysis
Flow cytometry analysis was performed to compare the immunophenotypes of HPDSCs from six placentas with donor‐matched UCB. Post‐thawed HPDSCs and UCB were resuspended in phosphate buffered solution (PBS) with 2% fetal bovine serum at a density of 1 × 106/ml, incubated with conjugated antibodies (Table 1) according to a standard protocol, and analyzed using BD LSRFortessa (BD Biosciences). To investigate the in vivo trafficking of HPDSCs, peripheral blood and organs including lung, spleen, bone marrow, GI, and skin were isolated from the recipient RDEB mice on different days after HPDSC administration. Following lysis of the red blood cells from the peripheral blood and mechanical dissociation of the organs, single cell suspension was immunostained with anti‐HLA‐A, B, C antibody (Biolegend, San Diego, CA) and analyzed using MACSQuant Analyzer (Miltenyi Biotech, Inc., Auburn, CA). The level of human cell persistence was presented as an average percentage of HLA‐A,B,C positive cells of the total single cell suspension from peripheral blood or organs of biological repeats.
Table 1.
List of the antibodies used in this study.
| Antibodies | Conjugation | Cat # | Vendor |
|---|---|---|---|
| CD34 | FITC | 555821 | BD Pharmingen |
| CD38 | PE | 347687 | BD Pharmingen |
| CD4 | PerCP | 550631 | BD Pharmingen |
| CD117 | PE‐Cy7 | 339195 | BD Pharmingen |
| CD45 | APC‐Cy7 | 557833 | BD Pharmingen |
| CD56 | PE‐Cy5 | 555517 | BD Pharmingen |
| CD3 | APC‐Cy7 | 557832 | BD Pharmingen |
| CD105 | FITC | 561443 | BD Pharmingen |
| CD44 | PE | 550989 | BD Pharmingen |
| CD200 | PerCP‐Cy5.5 | 562124 | BD Pharmingen |
| CCR10 | PerCP‐Cy5.5 | 564772 | BD Pharmingen |
| CD73 | PE | 550257 | BD Pharmingen |
| CD197 (CCR7) | PE‐Cy7 | 560922 | BD Pharmingen |
| CD184 (CXCR4) | PE‐Cy5 | 560937 | BD Pharmingen |
| CD191 (CCR1) | APC | 362908 | Biolegend |
| CD193 (CCR3) | PerCP/Cy5.5 | 310718 | Biolegend |
| CD195 (CCR5) | APC/Cy7 | 359110 | Biolegend |
| CD117 | PE‐Cy7 | 313212 | Biolegend |
| HLA‐A,B,C | APC/Cy7 | 311425 | Biolegend |
| CD10 | APC‐cy7 | 312212 | Biolegend |
| CD133/1 | APC | 130‐098‐829 | Miltenyi |
| HLA‐ABC | PE/Cy5 | 311408 | Biolegend |
| Anti‐human nuclei | MAB 1281 | Millipore | |
| Cell viability solution | 7‐AAD solution | 555816 | BD Via‐Probe |
| Col7A1 | 234192 | Calbiochem |
Abbreviations: APC, allophycocyanin; Cy, cyanine; FITC, fluorescein; HLA, human leukocyte antigen; PE, phycoerythrin; PerCP, peridinin‐chlorophyll.
Reverse Transcription Polymerase Chain Reaction (RT‐PCR) Analysis for type VII collagen Gene Expression in HPDSCs
RNA was extracted with an RNeasy kit (Qiagen, Valencia, CA) and cDNA was synthesized using M‐MLV Reverse Transcriptase (Promega, Madison, WI). PCR analysis was performed in standard PCR conditions (initial denaturation at 94°C for 3 minutes, 35 cycles of 94°C 30 seconds, 60°C 30 seconds and 72°C 30 seconds, and followed by a final extension of 72°C for 3 minutes) with Choice‐Taq Blue DNA Polymerase (Denville Scientific, Inc., Holliston, MA). The following two sets of COL7A1 primers were used for PCR amplifications: F1, TGACCCACGGACAGAGTTCG, R1, GATCAGGATGCAGACCTTGG; F2, GGCTTCTGGGCTTAATGTG, R2, GGGCTGAGTAGTGAAGGAT, as previously reported 24.
HPDSC Administration in col7a1 − / − Mice
HPDSCs were thawed in the buffer composed of 1:1 mixture of 10% Dextran 40 in 0.9% NaCl and 5% human serum albumin and resuspended in the same buffer at a concentration of approximately 70,000 cells/μl. Next, 15 μl of the HPDSC suspension was injected into the liver of newborn RDEB mice (D0) using a 31G needle that was attached to MINJ‐PD microINJECTOR (Tritech Research, Los Angeles, CA) at an injection speed of 0.5 s/µl. The dose per body weight is about 7 × 105 cells/g in mice. In the group with repeated HPDSC administration, a second dose (1 × 106) HPDSCs was injected in D5–8 RDEB mice in the liver. The same volume of the thawing buffer was injected in parallel as a vehicle control. All the survivors included for the following assays were genotyped to confirm the genetic knockout of the mice.
Quantitation of Anti‐C7 Antibodies in the Sera of Peripheral Blood
The presence of circulating anti‐C7 antibodies in the RDEB mice following stem cell injection(s) was evaluated by NC1‐based enzyme‐linked immunosorbent assay (ELISA), as published before with modifications 25. Briefly, 96‐well microtiter plates (MaxiSorp; Nunc, Rochester, NY) were coated with purified recombinant NC1 domain of C7, kindly provided by Mei Chen, Ph.D., University of Southern California, at a concentration of 1.5 μg/ml (0.15 μg/well) in 100 mM carbonate buffer, pH 9.3 for 1 hour at room temperature. Following overnight incubation with blocking buffer (PBS with 1% bovine serum albumin [BSA]) at 4°C, the plates were washed three times with PBS with 0.05%Tween‐20 (PBST). NC1‐coated wells were subsequently incubated for an hour with 1:100 diluted sera obtained from WT, untreated RDEB, and RDEB mice on different postnatal days after receiving HPDSCs or unrestricted somatic stem cells (USSCs) injections, respectively. The serum from the human C7 injected WT mice (an active EB acquisita (EBA) model 26), also kindly provided by Mei Chen, was used as a positive control. After washing with PBST three times, goat anti‐mouse horseradish peroxidase‐conjugated antibody (Santa Cruz Biotechnology, Dallas, TX) diluted in PBST with 1% BSA (1: 50,000) was added to the well and incubated for 30 minutes. After washing with PBST, SureBlue substrate (KPL, Inc., Gaithersburg, MD) was added to each well and incubated for 1 hour at RT. The reaction was then stopped by hydrogen chloride based solution (KPL). Optical density (OD) was measured at 450 nm on a microplate reader (Spectramax 320; Molecular Devices, Sunnyvale, CA). Samples were run in triplicate for each time point.
Histological and Immunocytochemical Analyses
Both front and hind paws as well as GI tract (small intestine) were excised from the selected mice, embedded in Tissue‐Tec OCT Compound (Sakura Finetek, Torrance, CA) and stored at −80°C. Six micrometer serial sections were cut for each specimen. H&E staining was processed following standard procedures, at the Core Histology Lab of New York Medical College. The percentage of separation at DEJ was calculated as average ratio of total detached/overall DEJ per finger digit, for all the paws in the section, quantitated with ImageJ by tracing the DEJ along the periphery of biopsies. To determine human cell engraftment in the recipient skin, the sections were fixed in 4% paraformaldehyde and blocked with Mouse on Mouse (M.O.M.) blocking reagent (Vector Laboratories, Burlingame, CA) in the presence of 0.1% Triton (Sigma, St. Louis, MO). The slides were then incubated with 1:100 diluted mouse anti‐human nuclei antibody (EMD Millipore, Billerica, MA) at 4°C overnight, followed by incubation with Alexa Fluor 488 goat anti‐mouse immunoglobulin G (IgG) (Invitrogen, Carlsbad, CA). Nuclei were counterstained with Vectashield Mounting Medium with DAPI (Vector Laboratories). The immunocytochemistry staining for C7 was performed following the manufacturer's recommendation: the cryostat sections were incubated with 0.5% BSA/PBS for 10 minutes at 42°C followed by hyaluronidase (2 mg/ml) digestion for 30 minutes. The slides were then incubated with 1:60 diluted rabbit anti‐COL7A1 (EMD Chemicals, Fort Wentworth, GA) antibody overnight at 4°C, followed by incubation with Alexa Fluor 488 goat anti‐rabbit IgG (Invitrogen, NY) and mounting in the Vectashield Mounting Medium with 4',6‐Diamidino‐2‐Phenylindole (DAPI) (Vector Laboratories). Images were acquired by Nikon 90i Eclipse microscope using the same setting for all the experimental samples (Nikon Instrument, Inc., Melville, NY). For the preparation of organotypic paw slice, paws were excised from newborn RDEB mice, embedded in 3% low‐melt agarose gel and cut into 250 μm slice using a vibratome (Leica Biosystems Wetzlar, Germany). The paw slices were immediately imaged under Nikon Eclipse TE300 microscope.
Ultrastructural Skin Examination
The preparation of skin biopsies for transmission electron microscopy (EM) was as previously described 27. Skin biopsies were fixed in 2.5% glutaraldehyde and 4% paraformaldehyde overnight at 4°C. The skin was then washed in 0.1 M cacodylate buffer, followed by incubation with 1% osmium tetroxide solution at room temperature for 1.5 hours. After washing in distilled water, samples were incubated in 2% uranyl acetate and dehydrated in ethanol and propylene oxide and embedded in epoxide resin. Ultrathin sections were stained with uranyl acetate and lead citrate and examined in a Hitachi HT7700 transmission electron microscope. All chemicals and supplies used for EM were purchased from Electron Microscopy Sciences, Inc. (Hatfield, PA).
Statistical Analysis
Kaplan‐Meier analysis was applied to determine the median life span and log‐rank (Mantel‐Cox) test was used to compare survival between different experimental groups (GraphPad Prism 6). An unpaired Student's t test was used to determine the difference in the percentage of subset populations between HPDSCs and UCB as well as the separation at DEJ at the basement membrane zone the WT, untreated RDEB, and HPDSC treated RDEB mice. A p value < .05 was considered significant.
Results
HPDSCs Are Rich in Both Hematopoietic and Nonhematopoietic Stem Cells
The overall cell types as determined by flow cytometry analysis are similar between HPDSCs and UCB. In both cell sources, greater than 80% of the cells are lymphocytes, monocytes, or granulocytes. Among the remaining cells, several different cell types are identified, including hematopoietic stem cells, mesenchymal stem cells (MSCs), megakaryocytic precursors, and endothelial progenitors. HPDSCs contain a significantly greater amount of CD34+ hematopoietic stem/progenitor cells compared with donor‐matched UCB (Fig. 1A). Specifically, a subpopulation of cells with a phenotype of CD34+/CD45− was observed in a significantly higher concentration in HPDSCs than UCB (1.9% vs. 0.1%, p < .05), suggesting that the hematopoietic precursors in HPDSCs are of a particularly immature phenotype. In addition, cells that are positive for MSC markers, CD105, CD73, and CD200 were also at a significantly higher concentration in HPDSCs. Moreover, 39%, 78%, 62%, 55%, and 40% of the cells within this MSC‐like population were positive for the chemokine receptors CCR1, CCR3, CCR5, CCR7, and CXCR4, respectively, implying chemoattractant properties in wound repair scenarios (Fig. 1B). In contrast, only 5% of the cells within MSC‐like population were positive for platelet‐derived growth factor receptor alpha (PDGFRα), which has been shown to mediate migration of bone marrow MSCs to wounding via high‐mobility group box 1‐PDGFRα axis 28, 29. In addition, immunophenotyping revealed that HPDSCs have a lower percentage of T cells than UCB, suggesting a lower risk for GVHD (Fig. 1C).
Figure 1.
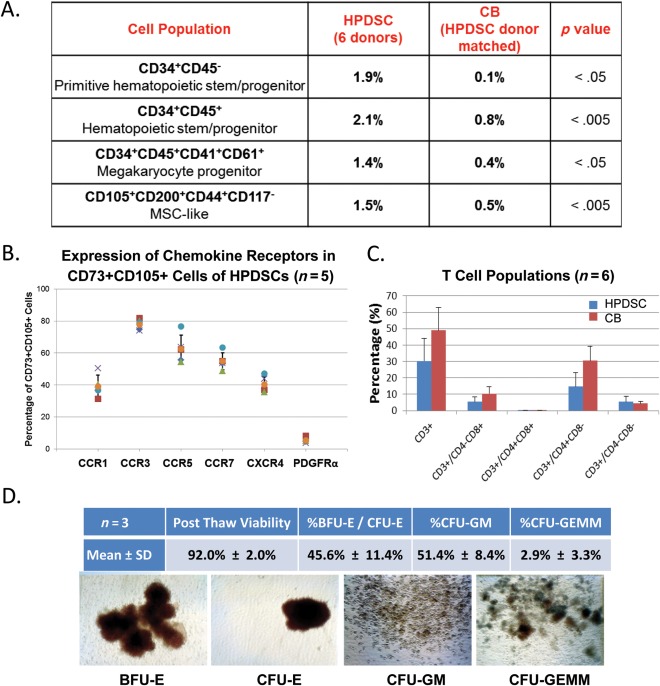
Characterization of HPDSCs. (A): Comparison of the percentage of subset cell populations between donor matched HPDSCs and CB (n = 6 donor units). (B): Percentage of the cells that express the cytokine receptors in CD73+CD105+ cells (n = 5 HPDSC units). (C): Percentage of T cell subsets between HPDSCs and CB (n = 6 donor units). (D): Post‐thaw viability of HPDSC and CFU analyses (n = 3 HPDSC units). Abbreviations: BFU‐E, burst‐forming unit‐erythroid; CB, cord blood; CFU, colony‐forming unit; CFU‐E, CFU‐erythroid; CFU‐GM, CFU‐granulocyte‐macrophages; CFU‐GEMM, CFU‐granulocyte, erythroid, macrophage, megakaryocyte; HPDSCs, human placental‐derived stem cells; MSC, mesenchymal stem cell.
We also determined that cryopreserved HPDSC CD34+ cells have a high post‐thaw viability (91.3%) and cell recovery. In vitro CFU assays further demonstrated the proliferation and differentiation functions of the progenitors including CFU‐erythroid and burst‐forming unit‐erythroid, CFU‐granulocyte‐macrophages and CFU‐granulocyte, erythroid, macrophage, megakaryocyte, from post‐thawed HPDSCs (Fig. 1D).
Intrahepatic Administration(s) of HPDSCs Significantly Improved the Survival of RDEB Mice
Col7a1 − / − mice are born with hemorrhagic erosions and without treatment have a median life span of 2 days 27. Due to poor accessibility to the veins of neonatal mice, we injected HPDSCs or vehicle in parallel in the liver of newborn RDEB mice, an approach that is comparable to intravenous administration 27, 30. We demonstrated that injection of vehicle composed of dextran and human serum albumin extended the survival of some recipients; however, it was not statistically significant compared with the historical survival of untreated RDEB mice (Fig. 2A). We postulate that the vehicle improved the life span of some recipients by adjusting fluid balance, similar to the Ringer's lactate/dextrose solution as reported before 22. Significantly, after a single intrahepatic HPDSC administration on the day of birth (D0), the median life span of the RDEB mice was elongated to 7 days (n = 32; p ≤ .001 HPDSC treated vs. vehicle) and about 10% of the recipient mice lived to adulthood (over 3 weeks) (Fig. 2A). Of note, one recipient survived for 16 weeks before sacrifice. The HPDSC‐treated RDEB mice showed normal eating and grooming behaviors. However, they were smaller in size and weight, compared to healthy littermates. In patients with RDEB, the cumulative risk of pseudosyndactyly reaches a plateau level of 98% by the age of 20 and has been suggested to be triggered by pro‐inflammatory and pro‐fibrotic phenotype of dermal fibroblasts 31. In the RDEB mice treated by HPDSCs, mitten deformity started to appear at about 8 weeks of age and the digits were almost completely fused by 12 weeks, particularly in the front paws (Fig. 2B), similar to our previous report regarding RDEB mice treated with human cord blood‐derived USSCs 27. We also demonstrated that a second dose of HPDSC on day 5–7 significantly extended the median life span to 18 days (n = 24; p ≤ .001 single vs. repeated HPDSC administration(s)). Almost 40% and 16% of the recipients survived to adulthood and past 15 weeks of age, respectively (Fig. 2A). The repeated HPDSC administrations also appeared to slow down the progression of pseudosyndactyly, although a larger group size is required for a quantitative analysis. As demonstrated in Figure 1B, by 12 weeks of age, there were three distinct digits remaining in the RDEB mice that received repeated (2×) HPDSC administrations.
Figure 2.
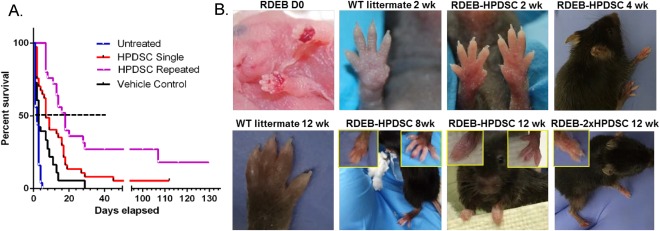
HPDSC administration(s) significantly extended the median life span of RDEB mice and improved the blistering skin phenotype. (A): Kaplan‐Meier survival curves of the RDEB mice that had not received any intervention (untreated, n = 18, blue line, historical data), vehicle (n = 21, black line), a single intrahepatic HPDSC at birth (n = 34, red line) and repeated HPDSCs (n = 24, purple line). p ≤ .001 HPDSC treated versus vehicle or untreated; p ≤ .001 single versus repeated HPDSC administration; p > .05 vehicle versus phosphate buffered solution. (B): Phenotypes of the paws in the WT, newborn RDEB and RDEB mice on different days post HPDSC administration(s). The left and right inset figures illustrate the representative front and hind paws, respectively. Abbreviations: HPDSC, human placental‐derived stem cell; RDEB, recessive dystrophic epidermolysis bullosa; WT, wild‐type.
HPDSC Administration Resulted in C7 Expression in RDEB Mice Without Eliciting Anti‐C7 Antibodies
We first tested whether HPDSCs themselves express type VII collagen and found by RT‐PCR and quantitative PCR analyses that HPDSC are not a source for C7 expression (Fig. 3A and data not shown). Surprisingly, in contrast to a complete absence of C7 in the newborn untreated RDEB skin, a continuous C7 staining appeared at the DEJ of the paw skin of 1‐ and 2‐week‐old HPDSC‐treated RDEB mice (Fig. 3B). In the paw skin of 7‐week‐old HPDSC‐treated RDEB mouse, C7 was mostly identified in patches, particularly at or close to the region with dermal‐epidermal separation. The C7 staining was much less intense in the HPDSC‐treated RDEB mice that survived over three months (12, 14, 15, and 16 weeks, respectively), but it was still detectable particularly close to the dermal‐epidermal separation (Fig. 3B and data not shown).
Figure 3.
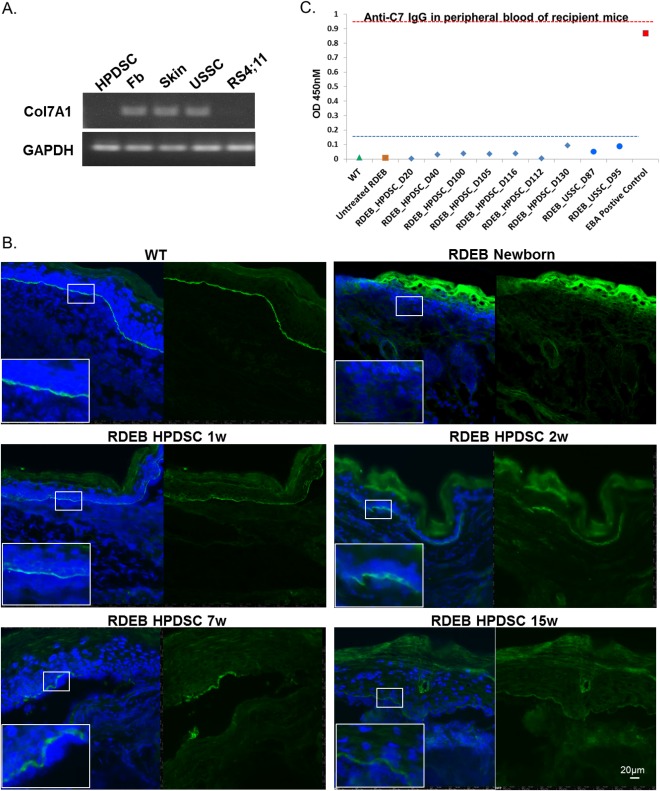
HPDSC administration resulted in C7 deposition in the RDEB skin without inducing anti‐C7 antibodies in the recipient RDEB mice. (A): Representative RT‐PCR analysis for the expression of type VII collagen in HPDSCs, human fibroblasts, human skin, USSCs, and RS4;11 (a leukemia cell line as a negative control). (B): Immunocytochemical staining for C7 in the paw skin of WT, newborn untreated RDEB and 1, 2, 7, and 15‐week‐old RDEB mice post HPDSC administration. C7 is stained in green and nuclei are counterstained with DAPI (blue). (C): Quantitation of the anti‐C7 IgG in the sera of the RDEB mice from 20 to 130 days post HPDSC administration (blue diamonds, n = 7). Sera from the USSC‐treated RDEB mice (blue circles, n = 2) were also included for comparison. The sera of WT (green triangle) and untreated RDEB mice (orange square) were used as negative controls and serum from the human C7 injected wild‐type mice (an active EB Acquisita model, red square) was used as a positive control. Each sample was assayed in triplicate and the experiments were repeated twice. Abbreviations: DAPI, 4'6‐diamidino‐2‐phenylindole; EB, epidermolysis bullosa; HPDSC, human placental‐derived stem cell; RDEB, recessive dystrophic epidermolysis bullosa; RT‐PCR, reverse transcription polymerase chain reaction; USSCs, unrestricted somatic stem cells; WT, wild‐type.
A safety concern for the RDEB therapeutic strategy that exposes the recipients to foreign C7 protein is the risk of unwanted immune responses, that is, generating anti‐C7 antibody that subsequently binds to BMZ and initiates an immune attack in the skin 32. Therefore, we investigated the sera from HPDSC administered mice at various time points (n = 7), from 20 to 130 days, and subjected them to the ELISA analysis and comparison with those from negative (WT and untreated RDEB mice) and positive (EBA mice) controls. The positive and negative controls showed an OD of 0.87 ± 0.01 and 0.011± 0.004, respectively, with a 1:100 dilution (Fig. 3C). The level of anti‐C7 reactivity in sera of HPDSC treated mice fluctuated from 0.003 on D20 to 0.09 on D130 with an average OD of 0.04 ± 0.029. It was previously reported that positive reactivity of EBA patients with recombinant C7 domain ranged between 0.3 and 2, and an OD value <0.2 was considered as negative for the production of autoantibody 26. Therefore, we can conclude that C7 originated from HPDSC administration was well‐tolerated in the recipients and did not elicit significant adverse immune response, at least within our time window of the experiments. In line with our previous work 27, USSC administration did not result in significant anti‐C7 IgG production in these recipient mice.
HPDSC Administration Increased Adhesion at DEJ but Did Not Rescue the Defective Anchoring Fibrils
As demonstrated in Figure 4A, a majority portion of epidermis exhibited detachment from the dermis in the paw skin of RDEB mice at newborn stage and 1 and 4 weeks after vehicle administration (53%–76%; p > .05). The adhesion at DEJ was significantly improved following administration of HPDSCs (Fig. 4A). Particularly, only about 11% of the paw skin exhibited separation at DEJ, at 1 and 2 weeks after a single HPDSC treatment (p < .001 compared to newborn and vehicle controls). In addition, although the adhesion deteriorated at DEJ in the HPDSC‐treated mice at later days (7 and 15 weeks), probably as results of increased abrasion in the paws and loss of HPDSC cells in vivo (as described below), the percentage of DEJ separation was still significantly lower in these mice compared with the controls (p < .05).
Figure 4.
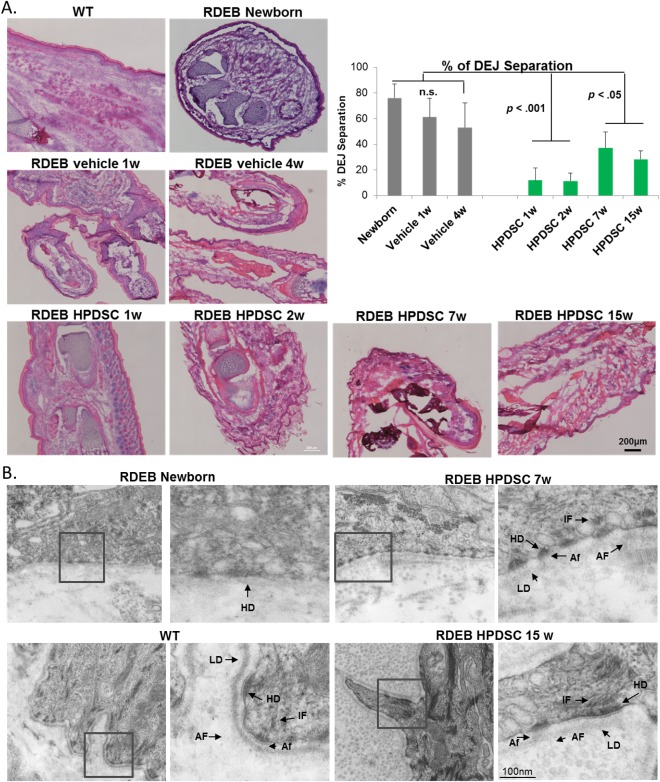
HPDSC administration improved adhesion at DEJ but did not rescue defective anchoring fibrils. (A): H&E analysis and quantitation on the percentage of DEJ separation in the skin of the WT, untreated newborn RDEB and RDEB mice on different days post vehicle or single HPDSC administration. (B): Transmission electron microscopy analysis of skin biopsies of wild type, untreated newborn RDEB and RDEB mice 7 week and 15 week post HPDSC administration, respectively. Abbreviations: AF, anchoring fibrils; Af, anchoring filaments; DEJ, dermal‐epidermal junction; HD, hemidesmosome; HPDSC, human placental‐derived stem cell; IF, intermediate filaments; LD, lamina densa; RDEB, recessive dystrophic epidermolysis bullosa; WT, wild‐type.
We also examined the ultrastructure of RDEB skin after HPDSC treatment. In the skin of 7‐ and 15‐week‐RDEB mice treated with HPDSCs, numerous hemidesmosomes with defined inner and outer plaques, similar to the WT, were present and were associated with bundles of inserting keratin intermediate filaments. Hemidesmosomes were also connected with the well‐formed anchoring filaments abutting the lamina densa (Fig. 4B). However, in contrast to the anchoring fibrils in WT skin, there were only wisp structures emanating from the lamina densa in the skin of HPDSC treated RDEB mice. Although there were more of these wisp‐like structures following HPDSCs compared to untreated RDEB mice, HPDSC administration did not lead to the formation of any anchoring fibrils with normal morphology.
Circulation of HPDSCs after Intrahepatic Injection and Engraftment in the Skin and GI
To investigate the trafficking of HPDSCs after intrahepatic administration, we prepared single cell suspensions from the peripheral blood, lung, spleen, skin, GI (small intestines) and bone marrow of recipient RDEB mice and performed flow cytometry analysis based on the cell surface expression of class I HLA‐A,B,C in HPDSC cells (Fig. 5A). One week after intrahepatic administration, HLA‐A, B, C positive cells were present in the lung and peripheral blood of col7a1 −/− mice (Fig. 5B). Importantly, 0.52% ± 0.44% and 0.11% ± 0.06% of the skin and GI cell suspension from the 1‐week‐old recipients (n = 2) were positive for human specific antigen, respectively (Fig. 5B). The results indicate that HPDSCs entered the circulation after intrahepatic injection and migrated to the organs affected by RDEB. Furthermore, among the HLA‐A,B,C positive cells in the skin suspensions, 22% expressed CD45, indicating that both hematopoietic and nonhematopoietic cells migrated to the skin (Fig. 5C). In addition, 18% of the human cells expressed CXCR4, suggestive of an enrichment of the HPDSCs that responded to the chemoattractant protein SDF1 in the skin. The human cells in the GI tract also exhibited similar percentage of cell surface marker expression (data not shown). Consistent with the low level of PDGFRα expression in the HPDSCs before injection, PDGFRα was not identified in the human HPDSCs that migrated to skin or GI tract (data not shown). We further demonstrated that the percentage of human cells in skin and GI was increased to 3.47% ± 0.87% and 0.79% ± 0.12%, respectively, in the 2‐week‐old recipients (n = 3) (Fig. 5B). After 3 weeks, while most of the human cells had disappeared from the circulation (lung and peripheral blood), there was still a significant number of human cells in the skin (0.67% ± 0.27%) and GI tract (0.23% ± 0.08%) (n = 2).
Figure 5.
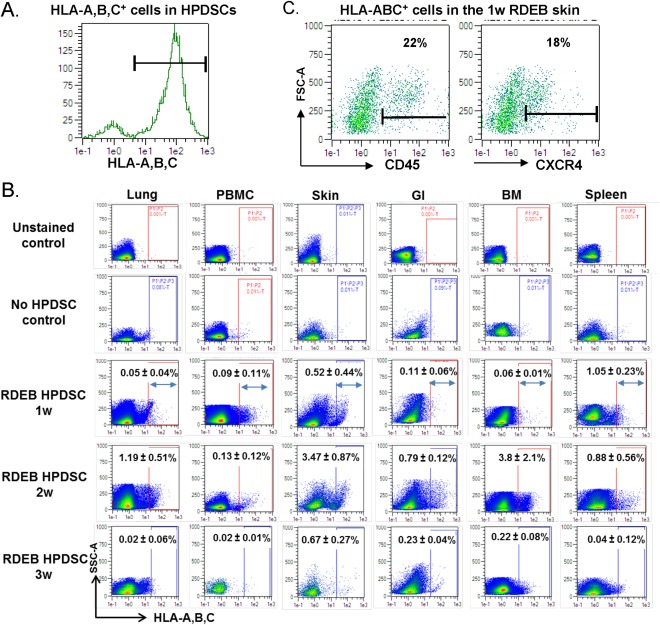
HPDSCs enter the circulation after intrahepatic administration in RDEB mice. (A): Expression of HLA‐A,B,C in HPDSCs. (B): Representative graphics of flow cytometry assay for the HLA‐A,B,C staining and quantitation of the percentage of HLA‐A,B,C positive cells in the organs of RDEB mice 1 week (1w, n = 2), 2 weeks (2w, n = 3), and 3 weeks (3w, n = 2) after HPDSC intrahepatic administration. Single cell suspension from HPDSC treated RDEB mice without HLA‐A,B,C antibody (unstained control) and single cell suspension from untreated WT mice with HLA‐A,B,C antibody (no HPDSC control) were analyzed in parallel as negative controls. (C): Expression of CD45 and CXCR4, respectively, in the HLA‐A,B,C positive cells in the RDEB skin 1 week after HPDSC administration. Abbreviations: BM, bone marrow; GI, gastrointestinal; HLA, human leukocyte antigen; HPDSCs, human placental‐derived stem cells; PBMC, peripheral blood mononuclear cells; RDEB, recessive dystrophic epidermolysis bullosa; WT, wild‐type.
Flow cytometry analysis also demonstrated migration of HPDSCs to the spleen of recipient mice (Fig. 5B). This is not surprising, as previous studies suggest spleen can accumulate most of the intravenously injected MSCs, secondary to the lung 33. However, whether migration of HPDSCs to the spleen could be correlated with anti‐inflammatory effects on splenic function, as previously suggested by the effects of MSCs in a chronic stroke rat model 34, requires further investigation. Another intriguing finding in this analysis is the migration of HPDSCs to the bone marrow of col7a1 −/− mice in the absence of any preconditioning. Similar to the dynamics in skin and GI tract, the highest level of HPDSC engraftment (3.8% ± 2.1%) occurred in bone marrow of the recipients 2 weeks after HPDSC administration (Fig. 5B). The identity of the cells and duration of persistence in bone marrow, skin, and GI tract of the recipients are currently under investigation.
Cutaneous wounds and esophageal strictures are the most apparent manifestations in patients with RDEB. What is rarely reported in patients, but demonstrated in the col7a1 −/− mice, is extensive hemorraghic erosion in the intestines, in addition to the paws (Fig. 6A). To investigate the niche of HPDSC in the skin and GI tract, we performed immunocytochemical staining using human specific nuclei antibody on the sections of skin and GI tract. Consistent with the detection of human cells by flow cytometry, human specific staining was observed in the skin of RDEB mice 1, 2, and 2 weeks after HPDSC administration (Fig. 6B). However, only dermal engraftment was identified and there was no evidence for their conversion into epidermal or endothelial cells (data not shown). Human cells were also identified in the tongue and intestine of the 2‐week‐old HPDSC treated RDEB mice (Fig. 6C). Importantly, human cells were not observed in the intestinal villus, but were mainly in the outmost layer of intestine, that is, serosa, as well as the mesentery, which is continuous with the serosa. However, human cells were no longer revealed by immunostaining in the biopsies of 6 week or older RDEB mice treated with HPDSCs.
Figure 6.
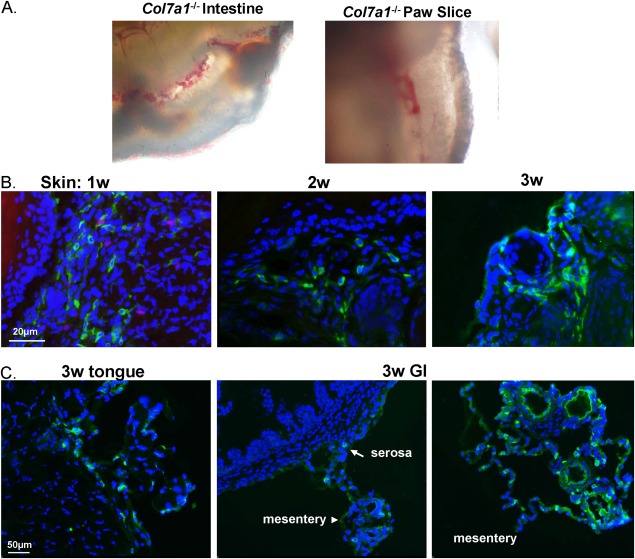
Hemorraghic erosions in intestines and skin of recessive dystrophic epidermolysis bullosa (RDEB) mice and identification of human placental‐derived stem cells (HPDSCs) by immunocytochemical staining. (A): Hemorraghic erosions in freshly isolated small intestines and 250 μm organotypic paw slice of newborn RDEB mice. (B): Immunocytochemical staining of the RDEB skin 1, 2, and 3 weeks after HPDSC administration using anti‐human specific nuclei antibody (hNuc, green). Nuclei were counterstained with DAPI (blue). (C): Identification of human cells in tongue and intestine of RDEB mice 3 weeks after HPDSC administration. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GI, gastrointestinal.
Discussion
This study demonstrated that HPDSCs contain a significantly higher percentage of hematopoietic and nonhematopoietic progenitor cells than cord blood and have important regenerative functions of HPDSCs in a col7a1 −/− mouse model. The human placenta is not only a crucial organ system for the survival and growth of the fetus, but also a hematopoietic organ during fetal development 35, 36, 37. Hematopoietic stem and progenitor cells persist throughout gestation from placental blood, vessel perfusate and cells from digested placenta tissue. The stromal cells generated from placenta also possess pericyte characteristics and may provide a supportive microenvironment for hematopoiesis 35. Here, HPDSCs are perfusate from full‐term placenta with minimal manipulation. Ongoing clinical studies have demonstrated that adding HPDSCs as an adjuvant with UCBT in patients with malignant and nonmalignant diseases has had no adverse effects and may reduce the incidence of aGVHD 20.
Regardless of the potential role in supporting hematopoiesis, we demonstrated in this study that HPDSCs per se had regenerative activity in a col7a1 −/− mouse model of RDEB. The median life span of the RDEB mice was significantly elongated after HPDSC administration, similar to what we previously reported after USSC administration 27. In contrast to USSCs, which are a relatively pure population of nonhematopoietic stem cells, HPDSCs consist of various cellular subsets and do not express significant amount of type VII collagen. Yet, surprisingly, administration of HPDSCs in RDEB mice led to deposition of C7 at the basement membrane zone. As RDEB mice completely lack C7, the detected C7 protein could only come from the administered HPDSCs. However, the identity of the HPDSCs that engrafted in the dermis and cellular origin of the C7 remain to be determined. It is possible that the nonhematopoietic MSC‐like cells in HPDSCs engrafted in the skin and secreted C7, as they express significant levels of various chemokine receptors and flow cytometry analysis revealed an enrichment of CXCR4 positive cells in the RDEB skin a week after HPDSC administration. On the other hand, some subset of stem cells in HPDSCs of an immature phenotype may have the ability to undergo differentiation into a dermal cell fate and contribute to skin regeneration.
It has to be noted that the anchoring fibrils were still far from normal in the skin of HPDSC‐treated RDEB mice. Kühl et al. recently determined the half‐life of C7 in the skin to be approximately 1 month and the disappearance of anchoring fibrils followed the rate of C7 loss 38. In this study, the C7 was more continuous along the DEJ within 2 weeks of HPDSC administration however became less intense and mostly patchy in the skin of 7 weeks and older HPDSC treated RDEB mice. This is consistent with the loss of C7 over time. The other consideration is that it took 18 months for the anchoring fibrils to mature in human skin regeneration from cultured epithelial autografts on full thickness burn wounds 39. It is thus likely that the level of C7 secreted by HPDSCs was insufficient or too short‐term for its correct assembly into anchoring fibrils. Nevertheless, administration of HPDSCs significantly improved the adhesion at DEJ, particularly within first 2 weeks of age. One can also infer from the survival curves of the RDEB mice of different treatment groups that the first week of life was important and once the animals pass this critical age, they have a better chance for the long‐term survival. Indeed, the complex cellular composition in HPDSCs suggests that the mechanism of action for skin regeneration is likely to be multifactorial. The col7a1 −/− mice are born with extensive hemorrhagic breakage in not only the skin, but also the GI tract. In addition to depositing C7 at DEJ, the monocytes, megakaryocytes, or any unidentified subset of HPDSCs may have played a role in alleviating hemorrhagic blistering and promoting wound healing in RDEB. In patients with RDEB, chronic inflammation and fibrosis are the morbid manifestations that further result in pseudosyndactyly and development of aggressive cutaneous squamous cell carcinoma 40. In this study, RDEB mice still developed pseudosyndactyly after HPDSC administration. Nystrom et al. previously demonstrated that administration of losartan significantly reduces fibrosis and prolongs the progression of fibrotic digit fusion via antagonizing transforming growth factor (TGF)‐β signaling in C7‐hypomorphic mice 41. The current study suggests that repeated HPDSC administration may have delayed the progression of pseudosyndactyly. Future study will quantitatively determine whether a more frequent administration of HPDSCs may further delay or slow down the progression of mitten deformities and whether this effect of HPDSCS is mediated through TGFβ signaling. Moreover, continued investigation on the cellular identity of the human cells isolated from the mucocutaneous membranes of RDEB mice using single‐cell transcriptome analysis will potentially lead to the development of more effective therapies and facilitate the clinical translation of HPDSCs in patients with RDEB.
It is important to note that the size of HPDSCs is much smaller (4–12 μm) compared to other cells such as MSCs that are derived and cultured in vitro (30 μm in diameter in suspension 42). Therefore, the risk of HPDSCs to form emboli in the lung vessels, even at a high dose, is much lower than that of MSCs. Lee et al. previously reported that more than 80% of MSCs accumulated in the lung of mouse recipients within a few minutes of injection, with formation of emboli in some recipients 43. The authors also reported an exponential decrease of MSC signal in the overall body followed by a complete disappearance after 4 days 43. In addition, MSCs generally do not home to bone marrow 42. This study demonstrated that in the absence of prior irradiation and concurrent immunosuppression, HPDSCs circulated from lung to the peripheral blood and migrated to skin, GI tract, and bone marrow. Importantly, 3 weeks post HPDSC administration, even though very few human cells persisted in the lung or peripheral blood, their engraftment was still identified in the skin, GI, and bone marrow. Our previous data suggest that the immune system in the col7a1 −/− mice may be compromised thus allowing a longer persistence of allogeneic cells than otherwise in healthy recipients 27. It is unclear whether this would be the case for the patients with RDEB. El‐Darouti et al. recently demonstrated that i.v. MSCs with and without the use of cyclosporine similarly improved wound healing in patients with RDEB, suggesting that concomitant immunosuppression was not necessary for the MSC therapy 44. In this study, the human cells were identified in 3‐week‐old mice but were no longer detected in the 6‐week or older RDEB mice. Therefore, the human cells disappeared from the recipients between 3 and 6 weeks of age. Our future work will evaluate the effects of concurrent immunosuppression on long‐term engraftment of HPDSC and therapeutic outcomes in the RDEB mice.
There were several limitations associated with this study that might impact the translational relevance of the work. Due to early demise of the col7a1 knockout mice consistently reported by us and others 17, 22, 27, 45, improvement on the median life span and long‐term survival were used as important readouts for the therapeutic function of HPDSCs in this animal model. Although multiple mechanisms as discussed above could be inferred for the benefits, prolonging the survival of col7a1 knockout mice is not clinically directly relevant to the human patients with RDEB. Moreover, as a result of poor survival of col7a1 −/− mice, the study was not designed to have a specific number of survivors at prescribed time points and conclusions made for later time points were based on low number of mice. Thus, more animals are required to investigate further therapeutic functions of HPDSCs on fibrosis and mitten deformity, which develop later in life and are more clinically relevant to patients with RDEB. Last, in this proof of principle study, each col7a −/− newborn mouse received about a million HPDSCs. In our clinical studies, the enrolled patients with malignant and nonmalignant diseases each received a full HPDSC donor unit, varying from 50 to 500 million cells 20. Therefore, the dose administered in the neonatal mice in the current study was high considering their small body size. Whether there is a threshold for the dose response in the survival or the level of C7 at DEJ in the col7a1 −/− mice requires further investigation. Nevertheless, based on disappearance of HPDSCs and decrease of C7 with time in the treated mice, we speculate that serial administration, rather than escalating a single dose of HPDSCs would result in more sustained therapeutic benefits in the treatment of RDEB. Although the dose per HPDSC unit is limited without ex vivo expansion, an advantage of using HPDSCs as a cell product is that there is no shortage of donor sources, that is, placentas. Therefore, considering a potential demand for serial HPDSC infusions in patients with RDEB, we will next investigate the feasibility and efficacy of infusing multiunit HPDSCs in col7a1 −/− mice.
Conclusion
HPDSCs are rich in hematopoietic stem cells, megakaryocytic precursors, and nonhematopoietic stem cells, which suggest multifactorial mechanisms of action for RDEB skin regeneration. Systemic administration of HPDSCs in col7a1 −/− mice alleviated hemorrhagic blistering and significantly improved the dermal‐epidermal attachment in the skin. In addition, HPDSCs effectively migrated from lung to the organs affected by RDEB, that is, skin and GI tract and deposited C7 without inducing anti‐C7 antibodies in the recipient mice. This study provides a basis for a translational pilot phase I trial to investigate the safety and feasibility of administration of HPDSC alone or in combination with UCB transplantation in patients with RDEB.
Author Contributions
Y.L.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; L.I. and H.Z.: collection and assembly of data, data analysis and interpretation, manuscript writing; R.S.: collection and assembly of data, data analysis and interpretation; T.P.: collection and assembly of data; X.Z.: conception and design, collection and assembly of data, manuscript writing; A.M.C.: conception and design, manuscript writing; J.M.: data analysis and interpretation; J.P.G.: conception and design, manuscript writing, final approval of manuscript; M.S.C.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
J.P.G. holds a leadership position in Celgene Cellular Therapeutics. R.S. and X.Z. are employed by Celgene Cellular Therapeutics. X.Z. also declared advisory role with Celularity, Human Longevity, Inc. The other authors indicated no potential conflicts of interest.
Acknowledgments
We thank Dr. Jouni Uitto at Jefferson Medical College for the col7a1 +/− mice. We also thank Mei Chen at University of Southern California for providing purified recombinant NC1 domain and EBA serum. We would also thank Erin Morris, RN, Janet Ayello, MS, ASCP, and Miguel Muniz, AAS for the assistance in the preparation of this manuscript. We also appreciate the input from Daria Ivanova on the flow analysis. This work was supported by DEBRA International funding to M.S.C., Pediatric Cancer Research Foundation to M.S.C., and NYMC/Touro SEED funding program to Y.L.
Authored by a member of CBA
References
- 1. Hilal L, Rochat A, Duquesnoy P et al. A homozygous insertion‐deletion in the type VII collagen gene (COL7A1) in Hallopeau‐Siemens dystrophic epidermolysis bullosa. Nat Genet 1993;5:287. [DOI] [PubMed] [Google Scholar]
- 2. Fine JD, Bruckner‐Tuderman L, Eady RA et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014;70:1103. [DOI] [PubMed] [Google Scholar]
- 3. Fine JD, Eady RA, Bauer EA et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58:931. [DOI] [PubMed] [Google Scholar]
- 4. Freeman EB, Köglmeier J, Martinez AE et al. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol 2008;158:1308. [DOI] [PubMed] [Google Scholar]
- 5. Fine JD, Johnson LB, Weiner M et al. Cause‐specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr 2008;152:276. [DOI] [PubMed] [Google Scholar]
- 6. Rashidghamat E, McGrath JA. Novel and emerging therapies in the treatment of recessive dystrophic epidermolysis bullosa. Intractable Rare Dis Res 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mellerio JE, Robertson SJ, Bernardis C et al. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: Best clinical practice guidelines. Br J Dermatol 2016;174:56. [DOI] [PubMed] [Google Scholar]
- 8. Woodley DT, Wang X, Amir M et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol 2013;133:1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong T, Gammon L, Liu L et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2008;128:2179. [DOI] [PubMed] [Google Scholar]
- 10. Wagner JE, Ishida‐Yamamoto A., McGrath J. A. et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 2010;363:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiuru M, Itoh M, Cairo MS et al. Bone marrow stem cell therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin 2010;28:371. [DOI] [PubMed] [Google Scholar]
- 12. Itoh M, Kiuru M, Cairo MS et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa‐induced pluripotent stem cells. Proc Natl Acad Sci USA 2011;108:8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tamai K, Uitto J. Stem cell therapy for epidermolysis bullosa‐does it work? J Invest Dermatol 2016;136:2119. [DOI] [PubMed] [Google Scholar]
- 14. Petrof G, Lwin SM, Martinez‐Queipo M et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2015;135:2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venugopal SS, Yan W, Frew JW et al. A phase II randomized vehicle‐controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol 2013;69:898. [DOI] [PubMed] [Google Scholar]
- 16. Petrof G, Martinez‐Queipo M, Mellerio JE et al. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: Results of a randomized, vehicle‐controlled trial. Br J Dermatol 2013;169:1025. [DOI] [PubMed] [Google Scholar]
- 17. Tolar J, Ishida‐Yamamoto A, Riddle M et al. Amelioration of epidermolysis bullosa by transfer of wild‐type bone marrow cells. Blood 2009;113:1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geyer MB, Radhakrishnan K, Giller R et al. Reduced toxicity conditioning and allogeneic hematopoietic progenitor cell transplantation for recessive dystrophic epidermolysis bullosa. J Pediatr 2015;167:765. [DOI] [PubMed] [Google Scholar]
- 19. Tolar J, Wagner JE. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: Repair of the extracellular matrix. Lancet 2013;382:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cairo MS, Tarek N, Lee DA et al. Cellular engineering and therapy in combination with cord blood allografting in pediatric recipients. Bone Marrow Transplant 2016;51:27. [DOI] [PubMed] [Google Scholar]
- 21. Flower A, Abikoff C, Minzer S et al. A pilot trial of unrelated donor human placenta‐derived stem cells (HPDSC) in conjuction with single unrelated cord blood transplantation (UCBT) in children with malignant and non‐malignant disease (IND 14949). Biol Blood Marrow Transplant 2017;23:S121. [Google Scholar]
- 22. Heinonen S, Männikkö M, Klement JF et al. Targeted inactivation of the type VII collagen gene (Col7a1) in mice results in severe blistering phenotype: A model for recessive dystrophic epidermolysis bullosa. J Cell Sci 1999;112:3641. [DOI] [PubMed] [Google Scholar]
- 23. Kang L, Voskinarian‐Berse V, Law E et al. Characterization and ex vivo expansion of human placenta‐derived natural killer cells for cancer immunotherapy. Front Immunol 2013;4:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liao Y, Itoh M, Yang A et al. Human cord blood‐derived unrestricted somatic stem cells promote wound healing and have therapeutic potential for patients with recessive dystrophic epidermolysis bullosa. Cell Transplant 2014;23:303. [DOI] [PubMed] [Google Scholar]
- 25. Remington J, Wang X, Hou Y et al. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther 2009;17:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen M, Chan LS, Cai X et al. Development of an ELISA for rapid detection of anti‐type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol 1997;108:68. [DOI] [PubMed] [Google Scholar]
- 27. Liao Y, Ivanova L, Zhu H et al. Rescue of the mucocutaneous manifestations by human cord blood derived nonhematopoietic stem cells in a mouse model of recessive dystrophic epidermolysis bullosa. Stem Cells 2015;33:1807. [DOI] [PubMed] [Google Scholar]
- 28. Aikawa E, Fujita R, Kikuchi Y et al. Systemic high‐mobility group box 1 administration suppresses skin inflammation by inducing an accumulation of PDGFRalpha(+) mesenchymal cells from bone marrow. Sci Rep 2015;5:11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamai K, Yamazaki T, Chino T et al. PDGFRalpha‐positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci USA 2011;108:6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wulf‐Goldenberg A, Keil M, Fichtner I et al. Intrahepatic transplantation of CD34+ cord blood stem cells into newborn and adult NOD/SCID mice induce differential organ engraftment. Tissue Cell 2012;44:80. [DOI] [PubMed] [Google Scholar]
- 31. Fine JD, Johnson LB, Weiner M et al. Pseudosyndactyly and musculoskeletal contractures in inherited epidermolysis bullosa: Experience of the National Epidermolysis Bullosa Registry, 1986–2002. J Hand Surg 2005;30:14. [DOI] [PubMed] [Google Scholar]
- 32. Hovnanian A. Systemic protein therapy for recessive dystrophic epidermolysis bullosa: How far are we from clinical translation? J Invest Dermatol 2013;133:1719. [DOI] [PubMed] [Google Scholar]
- 33. Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res Ther 2016;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Acosta SA, Tajiri N, Hoover J et al. Intravenous bone marrow stem cell grafts preferentially migrate to spleen and abrogate chronic inflammation in stroke. Stroke 2015;46:2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robin C, Bollerot K, Mendes S et al. Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell 2009;5:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barcena A, Muench MO, Kapidzic M et al. A new role for the human placenta as a hematopoietic site throughout gestation. Reprod Sci 2009;16:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barcena A, Kapidzic M, Muench MO et al. The human placenta is a hematopoietic organ during the embryonic and fetal periods of development. Dev Biol 2009;327:24. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Kühl T, Mezger M, Hausser I et al. Collagen VII half‐life at the dermal‐epidermal junction zone: Implications for mechanisms and therapy of genodermatoses. J Invest Dermatol 2016;136:1116. [DOI] [PubMed] [Google Scholar]
- 39. Compton CC, Gill JM, Bradford DA et al. Skin regenerated from cultured epithelial autografts on full‐thickness burn wounds from 6 days to 5 years after grafting. A light, electron microscopic and immunohistochemical study. Lab Invest 1989;60:600. [PubMed] [Google Scholar]
- 40. Fine JD, Johnson LB, Weiner M et al. Epidermolysis bullosa and the risk of life‐threatening cancers: The National EB Registry experience, 1986–2006. J Am Acad Dermatol 2009;60:203. [DOI] [PubMed] [Google Scholar]
- 41. Nystrom A, Thriene K, Mittapalli V et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med 2015;7:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Furlani D, Ugurlucan M, Ong L et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc Res 2009;77:370. [DOI] [PubMed] [Google Scholar]
- 43. Lee RH, Pulin AA, Seo MJ et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti‐inflammatory protein TSG‐6. Cell Stem Cell 2009;5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El‐Darouti M, Fawzy M, Amin I et al. Treatment of dystrophic epidermolysis bullosa with bone marrow non‐hematopoeitic stem cells: A randomized controlled trial. Dermatol Ther 2016;29:96. [DOI] [PubMed] [Google Scholar]
- 45. Chino T, Tamai K, Yamazaki T et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol 2008;173:803. [DOI] [PMC free article] [PubMed] [Google Scholar]