

Abstract
Cystic Fibrosis (CF) is a genetic disorder that arises due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator gene, which encodes for a protein responsible for ion transport out of epithelial cells. This leads to a disruption in transepithelial Cl-, Na + and HCO3− ion transport and the subsequent dehydration of the airway epithelium, resulting in infection, inflammation and development of fibrotic tissue. Unlike in CF, fibrosis in other lung diseases including asthma, chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis has been well characterised. One of the driving forces behind fibrosis is Epithelial Mesenchymal Transition (EMT), a process where epithelial cells lose epithelial proteins including E-Cadherin, which is responsible for tight junctions. The cell moves to a more mesenchymal phenotype as it gains mesenchymal markers such as N-Cadherin (providing the cells with migration potential), Vimentin and Fibronectin (proteins excreted to help form the extracellular matrix), and the fibroblast proliferation transcription factors Snail, Slug and Twist. This review paper explores the EMT process in a range of lung diseases, details the common links that these have to cystic fibrosis, and explores how understanding EMT in cystic fibrosis may open up novel methods of treating patients with cystic fibrosis.
Keywords: Epithelial mesenchymal transition, Cystic fibrosis, Lung, Fibrosis, E-cadherin
Background
Cystic Fibrosis (CF) is a genetic disorder that arises due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which produces a protein responsible for epithelial ion transport. This leads to a disruption in transepithelial Cl-, Na + and HCO3− ion transport and the subsequent dehydration of the epithelium within a range of organs including the respiratory system, pancreas, reproductive system, and sweat glands [1, 2]. Foremost is the disruption to the respiratory tract, which becomes enveloped with thickened mucus due to an osmotically-driven reduction in airway surface liquid volume, thus reducing mucociliary clearance [2, 3], and facilitating colonisation by pathogenic organisms. This leads to a cycle of inflammation and infection as pathogens such as bacteria, viruses, and fungi proliferate in the thickened mucus of the conducting airways. The cycle of infection and inflammation leads to fibrosis of the airways, pulmonary insufficiency and bronchiectasis which together ultimately leads to respiratory failure. Importantly, while there is a failure to clear thickened mucus there also appears to be a lack of, or dysfunction in, an auto-feedback mechanism preventing goblet cells from continually over-producing mucins, leading to mucus plugging and mucus plaques [4].
Fibrosis in other lung diseases including asthma, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) has been well characterised. One of the driving forces behind fibrosis is Epithelial Mesenchymal Transition (EMT), a mechanism first identified in the 1980s [5]. EMT is the process of epithelial cells losing epithelial proteins including E-Cadherin, which is responsible for tight junctions [6, 7], and the miRNA200 family which helps maintain an epithelial phenotype [8]. The cell moves to a more mesenchymal phenotype as it gains mesenchymal markers such as N-Cadherin (providing the cells with migration potential) [9], Vimentin and Fibronectin (proteins excreted to help form the extracellular matrix) [7, 10], and the fibroblast proliferation transcription factors Snail, Slug and Twist [7, 11]. Furthermore, upon the loss of the tight junctions and pseudo-stratified phenotype, epithelial cells become flattened and take on a stratified squamous epithelium appearance, allowing them to migrate throughout the tissue [12, 13].
When tissue is damaged/wounded or invaded by foreign antigens such as viruses and bacteria, a series of signaling cascades activate the immune system, resulting in inflammatory responses that lead to EMT [14, 15]. Macrophages, neutrophils, eosinophils and other immune cells are recruited to the damaged tissue and release an array of cytokines and growth factors including transforming growth factor β1 (TGF-β1) that signal the tissue to repair itself [16–19]. A study looking at kidney fibrosis estimated that during the repair process, 35% of fibroblasts that populate the tissue under repair are from epithelial/endothelial mesenchymal transition, 12% are derived from bone marrow (BM) via cell migration through the CXCL12/CXCR4 axis, and 30% are resident cells [15]. However, when tissue is persistently damaged, this leads to chronic inflammation, increased and prolonged EMT, and increased fibroblast proliferation resulting in hyperplasia [20]. Fibroblastic cells become activated to form myofibroblasts that excrete products that create a disorganised extracellular matrix. The accumulation of this matrix leads to permanently damaged fibrotic tissue with an aberrant architecture that is unable to function correctly [21, 22].
Over the last decade, the origin of proliferating fibroblasts during tissue repair has become a focus of a large body of research designed to better understand and prevent tissue fibrosis. The build-up of fibrotic tissue can lead to hepatic cirrhosis, nephrogenic systemic fibrosis and pulmonary fibrosis resulting in ongoing pathology and scarring of the affected organs, leading to early death unless the affected organ is removed and replaced through transplantation [23–25].
EMT is associated with many processes, including embryonic development, wound healing and tissue repair, and cell migration. These processes are subdivided into three distinct EMT categories:
Type 1: During embryonic implantation onto the uterine epithelium, primitive epithelial cells in the trophoectoderm undergo EMT and migrate within the inner cell mass and undergo mesenchymal to epithelial transition (MET) to form cells that will go on to form the various organs [15, 26, 27].
Type 2: Tissue damage occurs over a prolonged period and leads to fibrotic tissue. Despite being termed epithelial to mesenchymal transition; this process is not confined to epithelial cells. Endothelial cells and pericytes have also been observed to undergo a very similar process, indicating that this process is important for more than just epithelial tissue repair [28–30].
Type 3: The largest body of research has been on type 3 EMT which is involved in malignant cell growth leading to metastasis [26, 27]. With the removal of tight junctions through down regulation of E-Cadherin, transdifferentiated epithelial cells are free to migrate to areas of damage through the tissue and even the bloodstream if required. However, this process can be utilised by cancerous cells which, after undergoing EMT, have the potential to metastasize and form secondary tumor growths within distant organs [31]. Once metastases have occurred, prognosis is diminished because the cells can evade treatments such as chemotherapy, making many treatments redundant and potentially more dangerous than beneficial [32, 33]. The remainder of this review will focus on Type 2 EMT, and for convenience will be referred to simply as EMT.
EMT signaling: A pathway to fibrotic tissue
EMT is a complex process that involves a large interactome including protein to protein and genetic interactions that are initiated and controlled as a response to extracellular cues. At the forefront of these interactions is TGF-β1 which, on addition to epithelial cultures, causes the cells to undergo EMT (Fig. 1) [7, 10]. TGF-β1 is involved in several cellular functions including cell proliferation, cell differentiation and apoptosis [34–36]. TGF-β1 performs these various actions by directly activating a range of signaling pathways including Smad proteins, ERK/MAP kinases and micro RNAs [37–41], indicating that the progression of EMT is caused by a complex cascade of multiple signal transductions. TGF-β1 binds to target molecules on the cellular membrane known as TGF-β type I and type II receptors (TBR-I and II) creating a cascade of signals [37]. Activation of pathway-restricted Smads form heterodimers with Smad4 and then translocate to the nucleus. Within the nucleus they can promote the transcription of Snail and Twist, which together help break down E-cadherin and subsequently the tight junctions and adherens junctions, resulting in a leaky epithelium [42]. Apical polarity of the cells is then lost and cells are able to detach from the basal lamina, allowing them to break free of the tissue and become mobile [43–45]. A range of EMT inducers act directly or indirectly with TGF-β1, or via alternative pathways including reactive oxygen species (ROS) that are generated through hypoxic conditions, Fibroblast Growth Factor-2 (FGF-2), Epidermal Growth Factor (EGF), Connective Tissue Growth Factor (CTGF) and Transglutaminase 2 (TG2) which can activate matrix bound TGF-β1 [46–49].
Fig. 1.
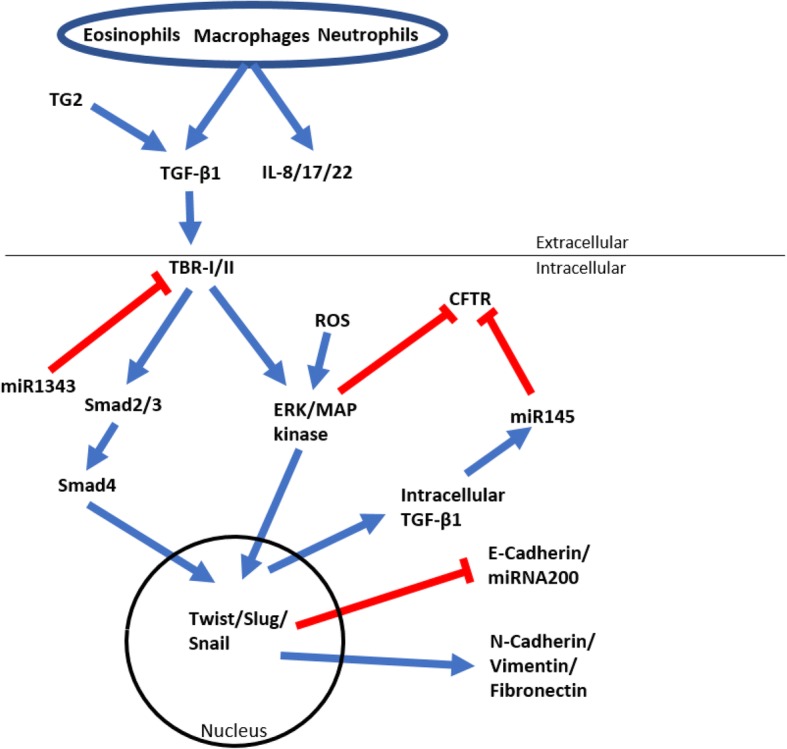
A schematic diagram of the TGF-β1 signaling cascade leading to EMT (blue arrows indicating active signal pathway, red lines indicating inhibition)
EMT, an underlying role in fibrotic lung diseases
Kim et al. reported that terminally-differentiated airway epithelial cells (AECs) could transdifferentiate into fibroblasts and myofibroblasts and develop fibrotic tissue [50]. Using a mouse model expressing β-galactosidase exclusively in lung epithelial cells, they tracked the epithelial cells under pulmonary fibrotic conditions and found β-galactosidase positive cells also expressing the mesenchymal markers α-smooth muscle actin (α-SMA) and Vimentin, showing that lung epithelial cells can transdifferentiate into other cell types. Subsequently it was shown that EMT in various lung diseases is the underlying cause of fibroblast, goblet cell and pneumocyte hyperplasia, which leads to lung fibrosis [51, 52]. The following sections outline and demonstrate the role of EMT in asthma, IPF, viral infections, COPD, and why it is important to gain more insight into its role in cystic fibrosis.
Asthma
Asthma is an inflammation and swelling of the airways that results in airway narrowing, goblet cell hyperplasia and airway hyper-responsiveness that make it difficult to breathe [51]. Severity can range from mild to severe. Human air liquid interface cultures (ALI) cultures developed from the epithelial cells of normal and asthmatic patients have been examined for their response to TGF-β1 exposure. While E-cadherin expression was initially lower in the asthmatic ALIs, treatment with TGF-β resulted in significantly decreased E-cadherin expression, whereas expression in normal ALIs did not decrease significantly [10].
Fibronectin, which was absent without TGF-β1 exposure in both asthmatic and normal ALIs, was then expressed after TGF-β1 addition, but at a much greater level in asthma ALIs [10]. Furthermore, histological analysis of the ALI cultures revealed that markers of EMT were far more extensive throughout the epithelial layers in asthma ALIs compared to normal, and in the latter those markers were primarily localised to the basal epithelial layer. Since tight junctions within the asthmatic epithelial airways are often disturbed, TGF-β1 released from immune cells might be able to access and affect cell layers deeper into the epithelium [42, 53]. The varying severity of asthma also appears to alter epithelial cell responses to TGF-β1, with Johnson et al., [9] showing that TGF-β1 had a greater effect on cells obtained from severe asthmatic subjects [9]. A range of markers including EFNB2, FGFR1, FGFR2, INSR, IRS2, NOTCH2, TLE1, NTRK2 and ADAM33 are all dysregulated in asthma patients [54, 55], and IL-22, a pro-inflammatory cytokine produced by immune cells, is significantly elevated in severe asthmatic subjects compared to mild asthmatic subjects [9]. Together these studies show that EMT is associated with asthma severity, but whether modifying the EMT response has therapeutic applications remains to be seen.
Idiopathic pulmonary fibrosis
Idiopathic Pulmonary Fibrosis (IPF) is a distinct variety of progressive fibrosing interstitial pneumonia associated with declining lung function, and is caused by increasing amounts of fibrotic tissue that cannot be correctly repaired by the lung [56]. To date, IPF is irreversible and has a 5-year survival of 43% [57, 58]. Interleukin-17 (IL-17) can induce TGF-β1 gene and protein expression in IPF animal models and IPF patients through the Smad2/3 and ERK1/2 pathways [59], and blocking TGF-β1 in rat models has been shown to slow disease progression [60]. Immunohistological analysis of human IPF patients has shown increased cell proliferation to repair the tissue through increased cytokeratin 14 (CK14) expression, a marker for airway basal progenitor cells. E-Cadherin expression extends into the basal cells as well as deeper into the underlying tissue and appears to be colocalised with the N-Cadherin expression associated with mesenchymal cells, indicating that cells throughout the epithelium are in the process of transition from epithelial cells to mesenchymal cells [13]. Recently, tannic acid and triptolide has been identified as a potential drug to slow IPF, through binding to the active site of TGF-β1 [61, 62]. In vitro results have shown that following tannic acid treatment, TGF-β1 induced Smad2 and Smad3 phosphorylation is diminished, reversing morphogenic and genetic changes in epithelial cell cultures [62]. Other drugs have also recently been examined, including thalidomide (inhibiting the Smad independent pathway), Pirfenidone (inhibits myofibroblast differentiation through mitophagy induction leading to reduced ROS and PDGFR-PI3K and Akt activation) and Tubastatin (Inhibits HDAC6 activated TGF-β1-PI3K-AKT signaling leading to decreased collagen type 1 expression) [63–65]. These results suggest that despite the extensive distribution of EMT-induced fibrosis throughout IPF lungs, retarding fibrosis may be a possibility with the use of drugs that inhibit EMT.
Viral infections
Viral infections of the airways elicit immune responses leading to tissue repair, and they have also been found to induce EMT in vitro. Specific cell surface binding proteins such as the Epstein-Barr virus (EBV) encoded latent membrane protein 1 (LMP1) in kidneys have been found to directly initiate EMT [66], as do secondary infections such as enterotoxigenic Escherichia coli in intestinal epithelial cells [67]. Human cytomegalovirus (HCMV), human papillomavirus (HPV), hepatitis C virus (HCV) and respiratory syncytial virus (RSV) have been shown to induce morphological changes, switch from epithelial to mesenchymal markers, and increase proliferation and migration of non-invasive cancer cell lines [68–71]. Repeated rhinovirus infections as a child can also increase the likelihood of developing asthma [72, 73]. Minor, et al. showed in vitro that the addition of rhinovirus (RV) was sufficient to induce EMT, although the effect was significantly enhanced with the addition of TGF-β1 [74]. This effect may be due to TGF-β1 mediated silencing of the protective mucosal interferon (IFN)-I and III production through the down-regulation of inducible interferon regulatory factor 1 (IRF1) expression in mesenchymal cells, which has been shown to increase both RV and RSV replication [75]. Interestingly, the measles virus, which can infect polarised epithelial cells, is unable to infect epithelial cells after they have undergone EMT, likely because the measles virus cellular receptor (which is still unknown) is down-regulated during EMT [76]. Other viruses that have been shown to induce EMT include the human immunodeficiency virus (HIV) through the hedgehog pathway [77], and Transmissible gastroenteritis virus (TGEV) through the TGF-β/PI3K/ERK pathways [67].
Whether EMT results from a viral infection, or EMT provides a suitable environment for viral infection are both important notions to consider as they can each have ramifications for diseases that result in organ fibrosis such as cystic fibrosis. Identifying which of these two paths is the primary cause will allow us to understand the fibrotic process more deeply and provide ways of controlling it, as well as preventing secondary infections.
Chronic obstructive pulmonary disease
Chronic Obstructive Pulmonary Disease (COPD) results from chronic inflammation, pulmonary remodeling, permanent airflow obstruction, and air trapping that leads to difficulties breathing [78, 79]. EMT is present in COPD, and in patients with COPD who are still smoking, the leaky epithelium, goblet cell hyperplasia, and poorly formed architecture of the airways results in more prevalent EMT [80–83]. Milara et al., (2013) showed in vitro and in vivo that E-cadherin was almost absent in both smokers and COPD patients, while collagen type 1 and Vimentin expression was far more prevalent compared to non-smokers [84]. From airway biopsies of COPD patients, expression of TGF-β1 and its downstream signaling partners Smad2/3 were greatly increased in COPD patients compared to normal, with the most prominent expression around blood vessels [38]. Mahmood et al., [85] found that there was a distinct difference between the small and large airways, with Type 2 EMT primarily found within the small airways leading to fibrotic tissue, and Type 3 EMT more closely associated with large airways leading to COPD-related cancers [85].
The canonical Wnt signaling pathway in COPD/EMT has also been shown to be up-regulated, as indicated by cellular-compartment expression of β-catenin in epithelial cells that is positively correlated with the EMT markers Twist and Snail [80]. Amongst current smokers with and without COPD, there was a shift from cytoplasmic to nuclear staining for β-catenin, Twist and Snail in basal cells, reticular basement membrane cells and lamina propria cells.
ALI cultures using COPD cell lines that are simply maintained and not exposed to cigarette smoke show that mesenchymal markers present early in the cultures are lost over time, indicating that EMT-related fibrosis can be halted in vitro, provided that the relevant EMT-causing stimuli (i.e. cigarette smoking) are removed [83]. Elevated heparin-binding epidermal growth factor (HB-EGF) has recently been linked to COPD disease severity by increasing EMT and collagen deposition [86]. However, the use of inhaled corticosteroids can decrease EMT activity in COPD patients through a reduction in epidermal growth factor receptor (EGFR) expression, suggesting a potential method for slowing or halting the development of COPD [87].
Unlike the lung diseases discussed earlier, it seems that EMT related to COPD is a direct result of cigarette smoke, and the elimination of this stimulus can slow down the progression of EMT induced fibrosis.
Cystic fibrosis
Research examining the involvement of EMT in CF has been limited, focusing only on CFTR involvement in cancer and other fibrotic diseases [88–90]. If new respiratory therapeutics extend the life expectancy of CF patients by 20+ years then the CFTR deficiency in other organs may have more severe effects on life and functioning, with CF patients already at a 17× higher risk of developing gut cancers [91]. The mechanisms of this interaction with cancers remain unknown, and what effects these processes will have should be elucidated well before they become a problem.
Linking CFTR with EMT
Pseudomonas aeruginosa infection (a key and common infection in CF patients that results in production of an excess of largely ineffective neutrophils) can induce TGF-β1 driven EMT by activating monocytes [92]. TGF-β1 is a known CF modifier gene that can influence the severity of respiratory CF disease based on TGF-β1 polymorphisms as well as environmental factors such as smoking which exacerbate or reduce respiratory severity by modulating TGF-β1 signaling [93–95].
Recently, an increase in UDP-glucose levels (an extracellular nucleotide that helps regulate mucociliary clearance) in CF lung secretions was shown to recruit neutrophils through the upregulation of interleukins [96]. Neutrophils have been shown to excrete neutrophil-derived elastase which can cleave E-Cadherin [97]. The epithelial hyperplasia present in the airways of CF mice, where a 5-fold increase in basal epithelial cells with clonogenic/proliferative potential has been reported [98], indicates that CF lungs undergo increased tissue remodeling and repair, consistent with an EMT process.
A portion of airway basal cells are progenitor cells that can self-renew and differentiate through two basal cell sub-types; basal stem cells and basal luminal progenitors. After epithelial injury the basal luminal progenitor cells become either ciliated cells or mucin secretory cells [99, 100]. How these cells divide and expand throughout the injured epithelium to repair the wound is still largely unknown, but it is possible that basal cells in these circumstances undergo at least a partial EMT process. This clonal expansion may not just be an inflammatory response, but potentially driven by the CFTR deficiency itself. Recently, TG2 (an EMT inducer that works through TGF-β1) was found to be elevated in vitro in CF epithelial cell cultures leading to increased TGF-β1 and EMT induction. Inhibition of TG2 could reverse the EMT process, lower TGF-β1 gene expression, reduce the amount of extracellular matrix bound TGF-β1 and stabilise CFTR [49].
The close association of EMT (Particularly type-3 EMT which leads to metastasis) with cancer led to the identification that CFTR is often down-regulated in metastatic cancer cells [88, 101]. Maloney et al., (2016) showed higher levels of circulating TGF-β1 in CF patients [102]. TGF-β1 decreases CFTR expression through the p38 MAPK pathway and interestingly this was shown to occur prior the classical EMT E-Cadherin to N-Cadherin shift with low TGF-β1 concentrations [103]. E-Cadherin/N-Cadherin co-localisation along with CFTR downregulation throughout ALI cultures treated with TGF-β1 has been reported and suggested that this is evidence that EMT is not occurring [104]. However, this phenomenon was also shown by Jonsdottir et al. who suggest that this may just be an intermediate phase in the EMT process [13].
The recent insights into the roles of micro RNAs have shown that they are key factors in both CFTR regulation as well as EMT with miR1343 binding to the 3’UTR of TGFRβ 1 and 2, resulting in unstable mRNA transcripts thus reducing the level of TGF-β1 signaling while miR145 which is upregulated due to TGF-β1 binds to the 3’UTR of CFTR causing reduced CFTR expression [41, 105].
The addition of TGF-β1 to non-invasive breast cancer cells caused the cells to undergo type-3 EMT as seen by the decrease in E-cadherin, but interestingly CFTR was also down-regulated [88]. To determine whether CFTR down-regulation was connected with E-cadherin down-regulation and EMT, rather than just a side effect of TGF-β1 addition, the non-invasive cells were treated with a CFTR inhibitor, resulting in a decrease in E-cadherin expression [101]. When a metastatic cancer cell line was made to over-express CFTR, upon subcutaneous injection into mice a reduced number of metastatic lung growths resulted compared to the same cell line without CFTR over-expression [88].
The direct implication of CFTR in cancer progression is still somewhat controversial since CFTR has not yet been connected in any direct signaling pathways, however, CFTR may act by regulating intracellular Cl− concentrations [106], and so influencing the intracellular environment.
C-Src (a tyrosine kinase) has also been linked to EMT [107, 108], and also found to be regulated by CFTR. Although CFTR normally suppresses the oncogene c-Src, when CFTR is impaired c-Src is up-regulated [109]. c-Src is highly expressed in 60% of cancers and is involved in cell proliferation, cell survival, angiogenesis and invasion pathways [110]. The transcription factor NFkB is activated by c-Src which in turn up-regulates genes such as MUC1, a glycoprotein normally present in lung mucus and is required for mucociliary clearance, but is also highly secreted in CF causing increased mucus to build up and creating an environment for bacterial infection [111]. As a result, in a paracrine fashion c-Src could affect cells that don’t normally express CFTR. c-Src levels in cells from CFTR knockout animal cell lines can be returned to normal with the addition of an IL-1β inhibitor [112].
CFTR is down regulated in COPD patients [113, 114], and cigarette smoke has been identified as a possible initial cause of this down regulation through a rise in cytoplasmic Ca2+ which potentially prevents normal sorting/degradation of CFTR, and results in the rerouting of the CFTR protein from cellular membrane to aggresomes. Chelation of Ca2+ prevented this rerouting and maintained normal CFTR activity on the cellular membrane [90].
Whether CFTR/c-Src/MUC1 interaction is directly involved in the development of hyperplasia and the increased number of stem cells in CF lungs is not known, but significant therapeutic possibilities warrant investigating the role of both type-2 and type-3 EMT in CF lung disease. Ultimately, if EMT is linked to CFTR dysregulation, then using methods to block EMT, such as small molecule drugs like Kaempferol and TGF-β1 receptor kinase inhibitors may assist in reducing both hyperplasia and lung fibrosis [115, 116].
EMT increases cell plasticity
EMT is a complex physiological response process that occurs when tissues are damaged. There is increasing evidence that the traditional concept that once cells had terminally differentiated they would carry out their function before dying and being replaced by a progenitor cell that differentiates into the required cell type, is outdated. It now appears that ‘terminally differentiated’ cells may in fact be a source of these progenitor cells, and indeed Cre lineage tracing experiments in mice show that Club cells can act as a source of progenitor cells for the ciliated cells of the lung [117]. The inhibition of TGF-β1 after EMT induction does result in a transition of the mesenchymal cells back into epithelial cells [10], however the stem cell potential of these de-differentiated epithelial cells may be far wider than the original epithelial cell type. Battula et al., reported that once epithelial cells had undergone EMT in vitro, they were then capable of differentiating down the osteogenic, adipogenic and chondrogenic lineages. These de-differentiated epithelial cells also expressed markers that are associated with mesenchymal stem cells (MSC), while maintaining some markers of epithelial cells [118]. The residence of MSCs within organs is unknown, raising the question; are MSCs a niche population of cells within tissues, or are they various cell types that can transdifferentiate into other cell types. In bone marrow MSCs, various populations of cells have markers that are associated with specific cell lineages, resulting in them preferentially differentiating down those cell lineages [119, 120]. The capability for preferential differentiation is well established amongst MSC isolates from different tissues such as dental pulp, which preferentially differentiate down the odontogenic and neurogenic pathway [121]. A micro-array of human mammary epithelial cells that have been induced to undergo EMT were compared to bone marrow isolated MSCs showed 70% similarity in expressed genes, with 15% of the differentially regulated genes being epithelial markers left from prior to EMT induction [118].
Therefore, if MSCs are indeed de-differentiated cells that were once thought to be terminally-differentiated, this change in viewpoint may allow us to approach controlling the EMT process, to manipulate and direct tissue regeneration, particularly in CF patients where diagnosis is typically neonatal and well before fibrosis of tissue begins to occur.
Conclusion
In conclusion, EMT is a universal and normal process involved in tissue repair, but EMT dysregulation can lead to fibrosis. These processes have already been well studied in a range of lung diseases. While EMT is less understood in the pathology of CF, studies do show the involvement of CFTR in the EMT process, particularly in Type 3 EMT, where there has been compelling evidence of its involvement in cancer progression in lung, gut, liver and breast cancer. Type 2 EMT involvement in CF should be explored further to understand the process in the airways of CF patients as the results may provide novel insights into the causes and effects of the dysregulated cellular pathways, potentially providing a future means for preventing or limiting CF related airway disease. As treatments for CF lung disease improve, an improved understanding of EMT in CF lung disease may also benefit our understanding of CF disease in other organs. While there are several strategies being developed to slow or halt CF lung disease [122], where that disease already exists there remains a need to restore tissue architecture through resolution of the fibrotic response. A fuller understanding of the mechanisms behind EMT and lung fibrosis may allow us to prevent, halt or even reverse the fibrosis process.
Acknowledgements
Research supported by NHMRC Project Grant APP1098127, the USA CF Foundation (PARSON15GO), and philanthropic donors via the Cure 4 Cystic Fibrosis Foundation (www.cure4cf.org).
Abbreviations
- ALI
Air Liquid Interface
- BM
Bone Marrow
- CF
Cystic Fibrosis
- CFTR
Cystic Fibrosis Transmembrane Conductance Regulator
- COPD
Chronic Obstructive Pulmonary Disease
- EMT
Epithelial Mesenchymal Transition
- IPF
Idiopathic Pulmonary Fibrosis
- MET
Mesenchymal Epithelial Transition
- TGF-β1
Transforming Growth Factor-β1
- α-SMA
α-Smooth Muscle Actin
Authors’ contributions
NR was the major contributor to writing the manuscript, NF, DP and MD all aided in writing parts of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 2.Quinton PM. Too much salt, too little soda: cystic fibrosis. Sheng Li Xue Bao. 2007;59:397–415. [PubMed] [Google Scholar]
- 3.Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell. 2001;8:149–158. doi: 10.1016/S1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- 4.Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev. 2002;54:1359–1371. doi: 10.1016/S0169-409X(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 5.Greenburg G, Hay ED. Cytodifferentiation and tissue phenotype change during transformation of embryonic lens epithelium to mesenchyme-like cells in vitro. Dev Biol. 1986;115:363–379. doi: 10.1016/0012-1606(86)90256-3. [DOI] [PubMed] [Google Scholar]
- 6.Karicheva O, Rodriguez-Vargas JM, Wadier N, Martin-Hernandez K, Vauchelles R, Magroun N, Tissier A, Schreiber V, Dantzer F. PARP3 controls TGFbeta and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget. 2016; [DOI] [PMC free article] [PubMed]
- 7.Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng XY, Xu L, Qu ZH. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the Snail pathway and promotes airway remodeling in asthma. Mol Med Rep. 2013;8:1663–1668. doi: 10.3892/mmr.2013.1728. [DOI] [PubMed] [Google Scholar]
- 8.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JR, Nishioka M, Chakir J, Risse PA, Almaghlouth I, Bazarbashi AN, Plante S, Martin JG, Eidelman D, Hamid Q. IL-22 contributes to TGF-beta1-mediated epithelial-mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res. 2013;14:118. doi: 10.1186/1465-9921-14-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–133. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 11.Baulida J. Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol Oncol. 2017;11:847–859. doi: 10.1002/1878-0261.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawson GW, Van Winkle LS, Toskala E, Senior RM, Parks WC, Plopper CG. Mouse strain modulates the role of the ciliated cell in acute tracheobronchial airway injury-distal airways. Am J Pathol. 2002;160:315–327. doi: 10.1016/S0002-9440(10)64375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ, Gudmundsson G, Gudjonsson T, Magnusson MK. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Investig. 2015;95:1418–1428. doi: 10.1038/labinvest.2015.114. [DOI] [PubMed] [Google Scholar]
- 14.Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation - a two-way street. Mol Med. 2017;23 [DOI] [PMC free article] [PubMed]
- 15.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Xie H, He D, Li L. Infiltrating macrophages increase RCC epithelial mesenchymal transition (EMT) and stem cell-like populations via AKT and mTOR signaling. Oncotarget. 2016; [DOI] [PMC free article] [PubMed]
- 17.Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489–495. doi: 10.1378/chest.07-1631. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Jia M, Yan X, Cao L, Barnes PJ, Adcock IM, Huang M, Yao X. Increased neutrophil gelatinase-associated lipocalin (NGAL) promotes airway remodelling in chronic obstructive pulmonary disease. Clin Sci (Lond) 2017;131:1147–1159. doi: 10.1042/CS20170096. [DOI] [PubMed] [Google Scholar]
- 19.Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, Matsumoto T, Boveda-Ruiz D, Gil-Bernabe P, Nagao M, Sugimoto M, et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One. 2013;8:e64281. doi: 10.1371/journal.pone.0064281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao YL, Zhu RT, Sun YL. Epithelial-mesenchymal transition in liver fibrosis. Biomed Rep. 2016;4:269–274. doi: 10.3892/br.2016.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganesan S, Sajjan US. Repair and remodeling of airway epithelium after injury in chronic obstructive pulmonary disease. Curr Respir Care Rep. 2013;2 [DOI] [PMC free article] [PubMed]
- 22.Shaykhiev R, Zuo WL, Chao I, Fukui T, Witover B, Brekman A, Crystal RG. EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype. Proc Natl Acad Sci U S A. 2013;110:12102–12107. doi: 10.1073/pnas.1303058110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Li X, Li M, Chen M, Fan Q, Wei W. Antiinflammation and antioxidant effects of thalidomide on pulmonary fibrosis in mice and human lung fibroblasts. Inflammation. 2017; [DOI] [PubMed]
- 24.Chen L, Cui X, Li P, Feng C, Wang L, Wang H, Zhou X, Yang B, Lv F, Li T. Suppression of MicroRNA-219-5p activates keratinocyte growth factor to mitigate severity of experimental cirrhosis. Cell Physiol Biochem. 2016;40:253–262. doi: 10.1159/000452542. [DOI] [PubMed] [Google Scholar]
- 25.Parsons AC, Yosipovitch G, Sheehan DJ, Sangueza OP, Greenberg CS, Sane DC. Transglutaminases: the missing link in nephrogenic systemic fibrosis. Am J Dermatopathol. 2007;29:433–436. doi: 10.1097/DAD.0b013e318156e43f. [DOI] [PubMed] [Google Scholar]
- 26.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10:2865–2873. doi: 10.4161/cc.10.17.17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, Hartman J, Religa P, Morikawa H, Ishii Y, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U S A. 2016;113:E5618–E5627. doi: 10.1073/pnas.1608384113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agarwal S, Loder S, Cholok D, Peterson J, Li J, Fireman D, Breuler C, Hsieh HS, Ranganathan K, Hwang C, et al. Local and circulating endothelial cells undergo endothelial to mesenchymal transition (EndMT) in response to musculoskeletal injury. Sci Rep. 2016;6:32514. doi: 10.1038/srep32514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho WT, Su CC, Chang JS, Chang SW, Hu FR, Jou TS, Wang IJ. In Vitro and In Vivo Models to Study Corneal Endothelial-mesenchymal Transition. J Vis Exp. 2016; [DOI] [PMC free article] [PubMed]
- 31.Yang X, Han M, Han H, Wang B, Li S, Zhang Z, Zhao W. Silencing Snail suppresses tumor cell proliferation and invasion by reversing epithelial-to-mesenchymal transition and arresting G2/M phase in non-small cell lung cancer. Int J Oncol. 2017;50:1251–1260. doi: 10.3892/ijo.2017.3888. [DOI] [PubMed] [Google Scholar]
- 32.Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–10711. doi: 10.18632/oncotarget.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandrakesan P, Panneerselvam J, Qu D, Weygant N, May R, Bronze MS, Houchen CW. Regulatory roles of Dclk1 in epithelial mesenchymal transition and Cancer stem cells. J Carcinog Mutagen. 2016;7 [DOI] [PMC free article] [PubMed]
- 34.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 35.Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2002;4:51–58. doi: 10.1038/ncb731. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Chen Y, Li G, Chen M, Huang W, Liu Y, Li Y. TGF-beta1/FGF-2 signaling mediates the 15-HETE-induced differentiation of adventitial fibroblasts into myofibroblasts. Lipids Health Dis. 2016;15:2. doi: 10.1186/s12944-015-0174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 38.Mahmood MQ, Reid D, Ward C, Muller HK, Knight DA, Sohal SS, Walters EH. Transforming growth factor (TGF) beta1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology. 2016; [DOI] [PubMed]
- 39.Banerjee P, Surendran H, Chowdhury DR, Prabhakar K, Pal R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J Mol Med (Berl). 2016; [DOI] [PubMed]
- 40.Milara J, Peiro T, Serrano A, Guijarro R, Zaragoza C, Tenor H, Cortijo J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28:138–148. doi: 10.1016/j.pupt.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Lutful Kabir F, Ambalavanan N, Liu G, Li P, Solomon GM, Lal CV, Mazur M, Halloran B, Szul T, Gerthoffer WT, et al. MicroRNA-145 antagonism reverses TGF-beta inhibition of F508del CFTR correction in airway epithelia. Am J Respir Crit Care Med. 2018;197:632–643. doi: 10.1164/rccm.201704-0732OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, Haitchi HM, Vernon-Wilson E, Sammut D, Bedke N, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549–556. doi: 10.1016/j.jaci.2011.05.038. [DOI] [PubMed] [Google Scholar]
- 43.Wu RY, Zhang Y, Feng XH, Derynck R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol Cell Biol. 1997;17:2521–2528. doi: 10.1128/MCB.17.5.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Klausen C, Zhu H, Leung PC. Activin a increases human trophoblast invasion by inducing SNAIL-mediated MMP2 up-regulation through ALK4. J Clin Endocrinol Metab. 2015;100:E1415–E1427. doi: 10.1210/jc.2015-2134. [DOI] [PubMed] [Google Scholar]
- 45.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1120–L1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 48.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 49.Nyabam S, Wang Z, Thibault T, Oluseyi A, Basar R, Marshall L, Griffin M. A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochim Biophys Acta. 2016;1863:2234–2244. doi: 10.1016/j.bbamcr.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 50.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu T, Liu Y, Miller M, Cao L, Zhao J, Wu J, Wang J, Liu L, Li S, Zou M, et al: Autophagy plays a role in FSTL1-induced EMT and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol 2017:ajplung 00510 02016. [DOI] [PubMed]
- 52.Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res. 2002;3:3. doi: 10.1186/rr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu L, Fu X, Chen X, Han X, Dong P. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol Int. 2017;41:960–968. doi: 10.1002/cbin.10788. [DOI] [PubMed] [Google Scholar]
- 54.Loffredo LF, Abdala-Valencia H, Anekalla KR, Cuervo-Pardo L, Gottardi CJ, Berdnikovs S. Beyond epithelial-to-mesenchymal transition: common suppression of differentiation programs underlies epithelial barrier dysfunction in mild, moderate, and severe asthma. Allergy. 2017;72:1988–2004. doi: 10.1111/all.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang L, Wu J, Huang T, Zhang P, Xin X, Shi Y. TGF-beta1 stimulates epithelial-mesenchymal transition mediated by ADAM33. Exp Ther Med. 2018;15:985–992. doi: 10.3892/etm.2017.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fujimoto H, Kobayashi T, Azuma A. Idiopathic pulmonary fibrosis: treatment and prognosis. Clin Med Insights Circ Respir Pulm Med. 2015;9:179–185. doi: 10.4137/CCRPM.S23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spagnolo P, Rossi G, Cavazza A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev Clin Immunol. 2014;10:1005–1017. doi: 10.1586/1744666X.2014.917050. [DOI] [PubMed] [Google Scholar]
- 58.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang T, Liu Y, Zou JF, Cheng ZS. Interleukin-17 induces human alveolar epithelial to mesenchymal cell transition via the TGF-beta1 mediated Smad2/3 and ERK1/2 activation. PLoS One. 2017;12:e0183972. doi: 10.1371/journal.pone.0183972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 61.Chen H, Chen Q, Jiang CM, Shi GY, Sui BW, Zhang W, Yang LZ, Li ZY, Liu L, Su YM, et al. Triptolide suppresses paraquat induced idiopathic pulmonary fibrosis by inhibiting TGFB1-dependent epithelial mesenchymal transition. Toxicol Lett. 2018;284:1–9. doi: 10.1016/j.toxlet.2017.11.030. [DOI] [PubMed] [Google Scholar]
- 62.Pattarayan D, Sivanantham A, Krishnaswami V, Loganathan L, Palanichamy R, Natesan S, Muthusamy K, Rajasekaran S. Tannic acid attenuates TGF-beta1-induced epithelial-to-mesenchymal transition by effectively intervening TGF-beta signaling in lung epithelial cells. J Cell Physiol. 2017; [DOI] [PubMed]
- 63.Kurita Y, Araya J, Minagawa S, Hara H, Ichikawa A, Saito N, Kadota T, Tsubouchi K, Sato N, Yoshida M, et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir Res. 2017;18:114. doi: 10.1186/s12931-017-0600-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saito S, Zhuang Y, Shan B, Danchuk S, Luo F, Korfei M, Guenther A, Lasky JA. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFbeta-PI3K-Akt pathway. PLoS One. 2017;12:e0186615. doi: 10.1371/journal.pone.0186615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou XL, Xu P, Chen HH, Zhao Y, Shen J, Jiang C, Jiang S, Ni SZ, Xu B, Li L. Thalidomide inhibits TGF-beta1-induced epithelial to mesenchymal transition in alveolar epithelial cells via Smad-dependent and Smad-independent signaling pathways. Sci Rep. 2017;7:14727. doi: 10.1038/s41598-017-15239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SM, Oh SW, Park SH, Hur DY, Hong SW, Han SY. Epstein-Barr virus-encoded latent membrane protein 1 induces epithelial to mesenchymal transition by inducing V-set Ig domain containing 4 (VSIG4) expression via NF-kB in renal tubular epithelial HK-2 cells. Biochem Biophys Res Commun. 2017;492:316–322. doi: 10.1016/j.bbrc.2017.08.116. [DOI] [PubMed] [Google Scholar]
- 67.Xia L, Dai L, Yu Q, Yang Q. Persistent transmissible gastroenteritis virus infection enhances Enterotoxigenic Escherichia coli K88 adhesion by promoting epithelial-mesenchymal transition in intestinal epithelial cells. J Virol. 2017;91 [DOI] [PMC free article] [PubMed]
- 68.Teo WH, Chen HP, Huang JC, Chan YJ. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int J Oncol. 2017;51:1415–1426. doi: 10.3892/ijo.2017.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang W, Wu X, Hu L, Ma Y, Xiu Z, Huang B, Feng Y, Tang X. Overexpression of human papillomavirus type 16 Oncoproteins enhances epithelial-mesenchymal transition via STAT3 signaling pathway in non-small cell lung Cancer cells. Oncol Res. 2017;25:843–852. doi: 10.3727/096504016X14813880882288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu B, Xie S, Hu Y, Chen W, Chen X, Zheng Y, Wu X. Hepatitis C virus NS4B protein induces epithelial-mesenchymal transition by upregulation of Snail. Virol J. 2017;14:83. doi: 10.1186/s12985-017-0737-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiang Z, Liang Z, Yanfeng H, Leitao K. Persistence of RSV promotes proliferation and epithelial-mesenchymal transition of bronchial epithelial cells through nodal signaling. J Med Microbiol. 2017;66:1499–1505. doi: 10.1099/jmm.0.000581. [DOI] [PubMed] [Google Scholar]
- 72.Rubner FJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Gern JE, Lemanske RF, Jr.: Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol 2017, 139:501–507. [DOI] [PMC free article] [PubMed]
- 73.Liu L, Pan Y, Zhu Y, Song Y, Su X, Yang L, Li M. Association between rhinovirus wheezing illness and the development of childhood asthma: a meta-analysis. BMJ Open. 2017;7:e013034. doi: 10.1136/bmjopen-2016-013034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Minor DM, Proud D. Role of human rhinovirus in triggering human airway epithelial-mesenchymal transition. Respir Res. 2017;18:110. doi: 10.1186/s12931-017-0595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, Tian B, Sun H, Garofalo RP, Brasier AR. Epigenetic silencing of IRF1 dysregulates type III interferon responses to respiratory virus infection in epithelial to mesenchymal transition. Nat Microbiol. 2017;2:17086. doi: 10.1038/nmicrobiol.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shirogane Y, Takeda M, Tahara M, Ikegame S, Nakamura T, Yanagi Y. Epithelial-mesenchymal transition abolishes the susceptibility of polarized epithelial cell lines to measles virus. J Biol Chem. 2010;285:20882–20890. doi: 10.1074/jbc.M110.102590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lan X, Wen H, Cheng K, Plagov A, Marashi Shoshtari SS, Malhotra A, Singhal PC. Hedgehog pathway plays a vital role in HIV-induced epithelial-mesenchymal transition of podocyte. Exp Cell Res. 2017;352:193–201. doi: 10.1016/j.yexcr.2017.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ojo O, Lagan AL, Rajendran V, Spanjer A, Chen L, Sohal SS, Heijink I, Jones R, Maarsingh H, Hackett TL. Pathological changes in the COPD lung mesenchyme--novel lessons learned from in vitro and in vivo studies. Pulm Pharmacol Ther. 2014;29:121–128. doi: 10.1016/j.pupt.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 79.Roscioli E, Tran HB, Jersmann H, Nguyen PT, Hopkins E, Lester S, Farrow N, Zalewski P, Reynolds PN, Hodge S. The uncoupling of autophagy and zinc homeostasis in airway epithelial cells as a fundamental contributor to COPD. Am J Physiol Lung Cell Mol Physiol. 2017;313:L453–L465. doi: 10.1152/ajplung.00083.2017. [DOI] [PubMed] [Google Scholar]
- 80.Mahmood MQ, Walters EH, Shukla SD, Weston S, Muller HK, Ward C, Sohal SS. Beta-catenin, twist and Snail: transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci Rep. 2017;7:10832. doi: 10.1038/s41598-017-11375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sohal SS, Reid D, Soltani A, Ward C, Weston S, Muller HK, Wood-Baker R, Walters EH. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15:930–938. doi: 10.1111/j.1440-1843.2010.01808.x. [DOI] [PubMed] [Google Scholar]
- 82.Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, Salit J, Harvey BG, Crystal RG. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gohy ST, Hupin C, Fregimilicka C, Detry BR, Bouzin C, Gaide Chevronay H, Lecocq M, Weynand B, Ladjemi MZ, Pierreux CE, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015;45:1258–1272. doi: 10.1183/09031936.00135814. [DOI] [PubMed] [Google Scholar]
- 84.Milara J, Peiro T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68:410–420. doi: 10.1136/thoraxjnl-2012-201761. [DOI] [PubMed] [Google Scholar]
- 85.Mahmood MQ, Sohal SS, Shukla SD, Ward C, Hardikar A, Noor WD, Muller HK, Knight DA, Walters EH. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction. Int J Chron Obstruct Pulmon Dis. 2015;10:1515–1524. doi: 10.2147/COPD.S81032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lai T, Li Y, Chen M, Pan G, Wen X, Mai Z, Yuan Y, Lv Y, Lv Q, Cen R, et al. Heparin-binding epidermal growth factor contributes to COPD disease severity by modulating airway fibrosis and pulmonary epithelial-mesenchymal transition. Lab Investig. 2018; [DOI] [PubMed]
- 87.Sohal SS, Soltani A, Reid D, Ward C, Wills KE, Muller HK, Walters EH. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study. Int J Chron Obstruct Pulmon Dis. 2014;9:533–542. doi: 10.2147/COPD.S63911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang JT, Jiang XH, Xie C, Cheng H, Da Dong J, Wang Y, Fok KL, Zhang XH, Sun TT, Tsang LL, et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim Biophys Acta. 2013;1833:2961–2969. doi: 10.1016/j.bbamcr.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 89.Hou Y, Guan X, Yang Z, Li C. Emerging role of cystic fibrosis transmembrane conductance regulator - an epithelial chloride channel in gastrointestinal cancers. World J Gastrointest Oncol. 2016;8:282–288. doi: 10.4251/wjgo.v8.i3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rasmussen JE, Sheridan JT, Polk W, Davies CM, Tarran R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J Biol Chem. 2014;289:7671–7681. doi: 10.1074/jbc.M113.545137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst. 2013;105:122–129. doi: 10.1093/jnci/djs481. [DOI] [PubMed] [Google Scholar]
- 92.Borthwick LA, Sunny SS, Oliphant V, Perry J, Brodlie M, Johnson GE, Ward C, Gould K, Corris PA, De Soyza A, Fisher AJ. Pseudomonas aeruginosa accentuates epithelial-to-mesenchymal transition in the airway. Eur Respir J. 2011;37:1237–1247. doi: 10.1183/09031936.00088410. [DOI] [PubMed] [Google Scholar]
- 93.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 94.Bremer LA, Blackman SM, Vanscoy LL, McDougal KE, Bowers A, Naughton KM, Cutler DJ, Cutting GR. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet. 2008;17:2228–2237. doi: 10.1093/hmg/ddn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Collaco JM, Vanscoy L, Bremer L, McDougal K, Blackman SM, Bowers A, Naughton K, Jennings J, Ellen J, Cutting GR. Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA. 2008;299:417–424. doi: 10.1001/jama.299.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sesma JI, Weitzer CD, Livraghi-Butrico A, Dang H, Donaldson S, Alexis NE, Jacobson KA, Harden TK, Lazarowski ER. UDP-glucose promotes neutrophil recruitment in the lung. Purinergic Signal. 2016; [DOI] [PMC free article] [PubMed]
- 97.Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hansch GM, Gaida MM. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768. doi: 10.1155/2012/720768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Voynow JA, Fischer BM, Roberts BC, Proia AD. Basal-like cells constitute the proliferating cell population in cystic fibrosis airways. Am J Respir Crit Care Med. 2005;172:1013–1018. doi: 10.1164/rccm.200410-1398OC. [DOI] [PubMed] [Google Scholar]
- 99.Pardo-Saganta A, Law BM, Tata PR, Villoria J, Saez B, Mou H, Zhao R, Rajagopal J. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell. 2015;16:184–197. doi: 10.1016/j.stem.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Watson JK, Rulands S, Wilkinson AC, Wuidart A, Ousset M, Van Keymeulen A, Gottgens B, Blanpain C, Simons BD, Rawlins EL. Clonal dynamics reveal two distinct populations of basal cells in slow-turnover airway epithelium. Cell Rep. 2015;12:90–101. doi: 10.1016/j.celrep.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J, Zhang JT, Jiang X, Shi X, Shen J, Feng F, Chen J, Liu G, He P, Jiang J, et al. The cystic fibrosis transmembrane conductance regulator as a biomarker in non-small cell lung cancer. Int J Oncol. 2015;46:2107–2115. doi: 10.3892/ijo.2015.2921. [DOI] [PubMed] [Google Scholar]
- 102.Maloney JP, Narasimhan J, Biller J. Decreased TGF-beta1 and VEGF release in cystic fibrosis platelets: further evidence for platelet defects in cystic fibrosis. Lung. 2016;194:791–798. doi: 10.1007/s00408-016-9925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun H, Harris WT, Kortyka S, Kotha K, Ostmann AJ, Rezayat A, Sridharan A, Sanders Y, Naren AP, Clancy JP. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS One. 2014;9:e106842. doi: 10.1371/journal.pone.0106842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Snodgrass SM, Cihil KM, Cornuet PK, Myerburg MM, Swiatecka-Urban A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS One. 2013;8:e63167. doi: 10.1371/journal.pone.0063167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stolzenburg LR, Wachtel S, Dang H, Harris A. miR-1343 attenuates pathways of fibrosis by targeting the TGF-beta receptors. Biochem J. 2016;473:245–256. doi: 10.1042/BJ20150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Valdivieso AG, Clauzure M, Massip-Copiz M, Santa-Coloma TA. The chloride anion acts as a second messenger in mammalian cells - modifying the expression of specific genes. Cell Physiol Biochem. 2016;38:49–64. doi: 10.1159/000438608. [DOI] [PubMed] [Google Scholar]
- 107.Moon SY, Kim HS, Nho KW, Jang YJ, Lee SK. Endoplasmic reticulum stress induces epithelial-mesenchymal transition through autophagy via activation of c-Src kinase. Nephron Exp Nephrol. 2014;126:127–140. doi: 10.1159/000362457. [DOI] [PubMed] [Google Scholar]
- 108.Liu X, Feng R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim Biophys Sin Shanghai. 2010;42:496–501. doi: 10.1093/abbs/gmq043. [DOI] [PubMed] [Google Scholar]
- 109.Massip Copiz MM, Santa Coloma TA. C- Src and its role in cystic fibrosis. Eur J Cell Biol. 2016;95:401–413. doi: 10.1016/j.ejcb.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 110.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/A:1023772912750. [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez-Guerrico AM, Cafferata EG, Radrizzani M, Marcucci F, Gruenert D, Pivetta OH, Favaloro RR, Laguens R, Perrone SV, Gallo GC, Santa-Coloma TA. Tyrosine kinase c-Src constitutes a bridge between cystic fibrosis transmembrane regulator channel failure and MUC1 overexpression in cystic fibrosis. J Biol Chem. 2002;277:17239–17247. doi: 10.1074/jbc.M112456200. [DOI] [PubMed] [Google Scholar]
- 112.Massip-Copiz MM, Clauzure M, Valdivieso AG, Santa-Coloma TA. CFTR impairment upregulates c-Src activity through IL-1beta autocrine signaling. Arch Biochem Biophys. 2017;616:1–12. doi: 10.1016/j.abb.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 113.Raju SV, Lin VY, Liu L, McNicholas CM, Karki S, Sloane PA, Tang L, Jackson PL, Wang W, Wilson L, et al. The cystic fibrosis transmembrane conductance regulator Potentiator Ivacaftor augments Mucociliary clearance abrogating cystic fibrosis transmembrane conductance regulator inhibition by cigarette smoke. Am J Respir Cell Mol Biol. 2017;56:99–108. doi: 10.1165/rcmb.2016-0226OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sloane PA, Shastry S, Wilhelm A, Courville C, Tang LP, Backer K, Levin E, Raju SV, Li Y, Mazur M, et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One. 2012;7:e39809. doi: 10.1371/journal.pone.0039809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. 2005;7:509–521. doi: 10.1593/neo.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang SQ, Marti TM, Dorn P, Froment L, Hall SR, Berezowska S, Kocher G, Schmid RA, Peng RW. Blocking the epithelial-to-mesenchymal transition pathway abrogates resistance to anti-folate chemotherapy in lung cancer. Cell Death Dis. 2015;6:e1824. doi: 10.1038/cddis.2015.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BL. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 2009;4:525–534. doi: 10.1016/j.stem.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, Mani SA. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28:1435–1445. doi: 10.1002/stem.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Katagiri H, Kushida Y, Nojima M, Kuroda Y, Wakao S, Ishida K, Endo F, Kume K, Takahara T, Nitta H, et al. A distinct subpopulation of bone marrow mesenchymal stem cells, muse cells, directly commit to the replacement of liver components. Am J Transplant. 2016;16:468–483. doi: 10.1111/ajt.13537. [DOI] [PubMed] [Google Scholar]
- 121.Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, DenBesten P, Robey PG, Shi S. Stem cell properties of human dental pulp stem cells. J Dent Res. 2002;81:531–535. doi: 10.1177/154405910208100806. [DOI] [PubMed] [Google Scholar]
- 122.Hudock KM, Clancy JP. An update on new and emerging therapies for cystic fibrosis. Expert Opin Emerg Drugs. 2017;22:331–346. doi: 10.1080/14728214.2017.1418324. [DOI] [PubMed] [Google Scholar]