

Similar to conventional chemokine receptors, atypical chemokine receptors (ACKRs) are cell-surface receptors that possess a seven transmembrane domain structure and regulate immune responses by interacting with chemokines. However, ACKRs differ from conventional chemokine receptors because of their inability to couple to G proteins and therefore to transduce intracellular signals mediating leukocyte migration upon chemokine binding. Instead, ACKRs internalize, transport or scavenge chemokines, regulating their extracellular availability, and thus impacting homeostasis, inflammation and immune response.1,2,3
ACKR1, also known as Duffy antigen receptor for chemokines, is expressed in endothelial cells (ECs), erythrocytes and a subset of neurons.4 On ECs, ACKR1 regulates inflammatory responses by transporting chemokines to the luminal surface of vessels, presenting them to rolling leukocytes.5 In the brain, ACKR1 regulates neurotransmission and the activity of Purkinje neurons, influencing motor function and behavior.6 In erythrocytes, ACKR1 is believed to function as a blood-based chemokine buffer by taking up, accumulating and degrading chemokines.7 Moreover, ACKR1 expressed on erythrocytes provides binding sites for Plasmodium vivax and Plasmodium knowlesi, enhancing susceptibility to malaria.8 Interestingly, most individuals in Sub-Saharan Africa bear a single-nucleotide polymorphism in the ACKR1 promoter that disrupts the binding site for the transcription factor GATA-1, and thus impairs the expression of ACKR1 in erythrocytes but not in ECs.9,10 Due to the associated resistance to malaria, this ‘Duffy-negative phenotype’ has conferred an evolutionary advantage and is widespread in African diaspora. These individuals have low neutrophil cell counts, or neutropenia, which mechanism so far has remained unresolved.11
In a recent issue of Nature Immunology, Rot and colleagues published a study in which ACKR1 was identified as an essential regulator of hematopoiesis by promoting direct interactions between bone marrow (BM) nuclear erythroid cells (NECs) and hematopoietic stem cells (HSCs).12 The authors also found that the absence of ACKR1 on bone marrow erythroid cells induced the development of neutrophils with altered phenotype; these neutrophils ultimately accumulated in the spleen, leading to neutropenia and recapitulating the phenotype found in Duffy-negative individuals (Figure 1). These results provided fundamental insights into the regulation of normal hematopoiesis and unveiled an unexplored mechanism that has sustained an evolutionary advantage, resulting in the fixation of the ACKR1 rs2814778(G) polymorphism in Africa.
Figure 1.
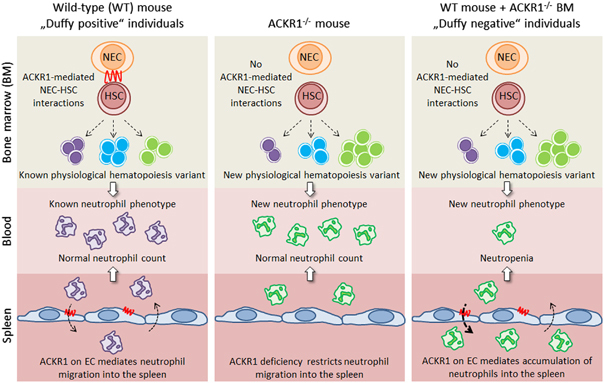
Schematic representation of how atypical chemokine receptor 1 (ACKR1) regulates hematopoiesis, the neutrophil phenotype and migration. In wild-type mice and ‘Duffy positive’ individuals, ACKR1 expressed on nucleated erythroid cells (NECs) in the bone marrow (BM) mediates their interaction with hematopoietic stem cells (HSC) and enables a known physiological hematopoiesis variant, resulting in neutrophils with a known phenotype, which migrate into the spleen as facilitated by ACKR1 expression on endothelial cells (ECs). In contrast, in Ackr1-deficient mice (and possibly in rare patients with a premature stop codon in the ACKR1 open reading frame, causing ACKR1 silencing in all tissues13), a lack of ACKR1 on NECs prevents their interaction with HSCs, affecting hematopoiesis and resulting in neutrophils with a new phenotype (overexpressing CD45, CD16/CD32 and CCR2). However, the neutrophil number in blood remains unchanged because ACKR1 deficiency on ECs prevents neutrophil entry into the spleen. In wild-type mice reconstituted with BM from Ackr1-deficeint mice (WT mice+Ackr1 −/− BM), as well as individuals of African ancestry homozygous for the rs2814778(G) allele of ACKR1 (‘Duffy-negative’ individuals), hematopoiesis is affected by a lack of ACKR1 on NECs in the same way as in Ackr1 −/− mice. However, as ACKR1 expression is preserved on ECs, neutrophils with the new phenotype accumulate in the spleen, resulting in neutropenia.
Using immunohistochemistry and flow cytometry, the authors first confirmed the expression of ACKR1 in BMECs and erythroid cells, and not in any other hematopoietic cell subtype. They also found high levels of ACKR1 expression in early developmental stages of NECs and lower levels upon NEC maturation, suggesting that the expression of ACKR1 may be physiologically relevant during hematopoiesis. Interestingly, immunophenotypic analyses of the hematopoietic development comparing wild-type and Ackr1-deficient mice revealed normal erythropoiesis with no differences in number or relative proportion of erythroid cell subpopulations. However, the absence of ACKR1 induced shifts in the composition of the hematopoietic stem and progenitor cell (HSPC) compartment. For instance, the frequency and proliferative capacity of lineage-negative (Lin−) Sca-1+c-kit+ (LSK) cells were reduced, whereas the level of CD34 expression was higher in the BM of Ackr1 −/− mice. A more pronounced reduction was found in the CD48− subpopulation of LSKs due to an increased relative proportion of HSCs. Reductions were also observed in the absolute cell numbers of the multipotent progenitor (MPP) 1a and MPP1b subpopulations. Among myeloid lineage progenitors, the absence of ACKR1 resulted in an overall slight reduction of myeloid progenitor cells (MPCs) due to decreased absolute cell numbers of granulocytic–monocytic progenitors and megakaryocytic–erythroid progenitors.
The investigators further used a gene expression microarray to evaluate whether the deficiency of ACKR1 affected the transcriptomic profile of the HSPC populations. They compared the transcriptomes in LSKs and granulocytic–monocytic progenitors cell populations from wild-type and Ackr1-deficient mice, and found that in the BM of Ackr1 −/− mice, genes primarily assigned to the neutrophil and myeloid lineages were upregulated. Moreover, genes encoding for neutrophil effector molecules (such as cathelicidin, neutrophil granule protein or resistin-like molecule gamma) were clearly upregulated in Ackr1-deficient cells compared with their wild-type counterparts. Together with the abnormal HSPC composition found in Ackr1-deficient mice, these data identified ACKR1 as an essential regulator of BM homeostasis.
Reciprocal bone marrow chimeras were used to assess the contributions of ACKR1 from NECs (hematopoietic compartment) and ECs (stromal compartment) to normal hematopoiesis. When ACKR1 was absent in the hematopoietic compartment, the HSPC subpopulations resembled those of the Ackr1-deficient mouse, regardless of the background of the recipient. Conversely, the reconstitution of irradiated Ackr1 −/− recipients with ACKR1-expressing BM cells restored hematopoietic parameters similar to those of wild-type mice. Additionally, to rule out the possibility that ACKR1 expressed on circulating mature erythrocytes could affect BM hematopoiesis, the authors generated parabiotic WT and Ackr1-deficient mice that shared the same circulatory system. The presence of wild-type mature erythrocytes in blood circulation did not restore the normal HSPC populations in Ackr1-deficient parabionts. Overall, these data clearly suggest a key role of ACKR1 expression on NECs on BM hematopoiesis.
To prove that NECs formed direct cell contacts with HSCs, Rot and colleagues employed 2-photon laser scanning microscopy of the whole-mounted bone marrow and found that the HSCs formed immediate contacts with the NECs in the BM from wild-type mice. In contrast, in the BM of Ackr1-deficient mice, only a third of all HSCs was in close proximity with NECs. The authors pinpointed the requirement for ACKR1 in interactions between NEC and MMP1 or LRP, but not MMP2 cells. These results further expanded our knowledge on the complexity of the HSC niche in BM.
Despite numerical changes of hematopoietic progenitors in BM, Ackr1-deficient mice showed normal differential peripheral blood leukocyte counts. However, the neutrophils of Ackr1 −/− mice overexpressed the FCγ receptors CD16/CD32 (important for antimicrobial defense), CD45 (a molecule that amplifies FCγ receptor function) and CCR2 (an inflammatory chemokine receptor expressed on monocytes but not on neutrophils in wild-type mice). The same set of membrane molecules was also overexpressed on the neutrophils of Duffy-negative individuals, suggesting that the lack of ACKR1 exclusively on erythroid cells could change BM hematopoiesis in humans in the same way observed in Ackr1 −/− mice. However, this seems contradictory: seemingly healthy Duffy-negative individuals have blood neutropenia, whereas Ackr1-deficeint mice have normal blood neutrophil counts. Because ACKR1 expression on ECs mediates neutrophil exit into peripheral tissue,5 the authors examined blood neutrophils in reciprocal wild-type and Ackr1 −/− BM chimeric mice. Indeed, they found that the blood of wild-type mice reconstituted with BM from Ackr1-deficient mice had reduced numbers of neutrophils, which accumulated in the spleen due to ACKR1 expression on venular ECs. However, the positioning of the neutrophils (in white or red pulp) and their fate (cell death or contribution to the splenic host defenses) remain to be answered by future studies.
Overall, this study described an important role of ACKR1 expressed on the cells of erythroid lineage on bone marrow cell output and thereby, broadening our understanding of the HSC niche in the BM and the chemokine system role in hematopoiesis.14 Furthermore, this study carries clinical implications, providing a mechanistic insight for peripheral blood neutropenia in healthy individuals of African ancestry that carry the allelic variant rs2814778(G) of ACKR1.
The findings reported by Duchene and colleagues also open some very intriguing concerns. First, the mechanism behind the ACKR1-mediated interaction between NECs and HSCs remains unclear. ACKR1 could serve as a sink or a reservoir for chemokines that might influence migration, survival and/or the proliferation of HSPCs;14 however, these interactions could be mediated by other molecules such as the tetraspanin family member CD82.15 Of note, ACKR1 does not bind CXCL12,2 a key chemokine required for the maintenance of HSCs,16 suggesting that some other ACKR1-binding chemokines could regulate NEC–HSC interactions (such as ligands for CCR3, which is expressed on HSCs17). Second, it is unclear whether other ACKRs also play a role in hematopoiesis. In contrast with ACKR1, ACKR3 binds to CXCL12.2 Although B cells and granulocytes develop normally in Ackr3-deficient mice,18 it is possible that ACKR3-deficiency induced changes in HSPC subpopulations, leading to phenotypically distinct leukocytes, similar to altered neutrophils in Ackr1 −/− mice. Third, the means by which the altered neutrophils in humans carrying the rs2814778(G) allele provide resistance to Plasmodium parasites, and viral and bacterial infections remain unknown. Interestingly, Ackr1-deficient mice that also show this altered neutrophil phenotype are not more susceptible to bacterial infection compared with wild-type mice, despite altered neutrophil trafficking following exposure to various inflammatory stimuli.2,4 Finally, it will be interesting to see whether this new mouse model (wild-type mice reconstituted with BM from Ackr1-deficient mice) can be used to investigate altered susceptibility to inflammatory and immune conditions in persons with Duffy-null phenotype.2 Future studies providing answers to these and additional questions should contribute to the understanding of the role of ACKR1 and other ACKRs in hematopoiesis, inflammation and immunity.
Acknowledgements
The work of RF is supported by the grants of the Deutsche Forschungsgemeinschaft (SFB900-B1, SFB738-B5, KFO250, FO334/5-1), the European Research Council (ERC Advanced Grant 322645), and the Government of Lower Saxony (N-RENNT, BioFabrication for NIFE).
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Ulvmar MH, Hub E, Rot A. Atypical chemokine receptors. Exp Cell Res. 2011;317:556–568. doi: 10.1016/j.yexcr.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nibbs RJB, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol. 2013;13:815–829. doi: 10.1038/nri3544. [DOI] [PubMed] [Google Scholar]
- 3.Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol. 2014;15:623–630. doi: 10.1038/ni.2889. [DOI] [PubMed] [Google Scholar]
- 4.Novitzky-Basso I, Rot A. Duffy antigen receptor for chemokines and its involvement in patterning and control of inflammatory chemokines. Front Immunol. 2012;3:266. doi: 10.3389/fimmu.2012.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol. 2009;10:101–108. doi: 10.1038/ni.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider EH, Fowler SC, Lionakis MS, Swamydas M, Holmes G, Diaz V, et al. Regulation of motor function and behavior by atypical chemokine receptor 1. Behav Genet. 2014;44:498–515. doi: 10.1007/s10519-014-9665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reutershan J, Harry B, Chang D, Bagby GJ, Ley K. DARC on RBC limits lung injury by balancing compartmental distribution of CXC chemokines. Eur J Immunol. 2009;39:1597–1607. doi: 10.1002/eji.200839089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261:1182–1184. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- 9.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy–negative individuals. Nat Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- 10.Peiper SC, Wang ZX, Neote K, Martin AW, Showell HJ, Conklyn MJ, et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J Exp Med. 1995;181:1311–1317. doi: 10.1084/jem.181.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thobakgale CF, Ndung’u T. Neutrophil counts in persons of African origin. Curr Opin Hematol. 2014;21:50–57. doi: 10.1097/MOH.0000000000000007. [DOI] [PubMed] [Google Scholar]
- 12.Duchene J, Novitzky-Basso I, Thiriot A, Casanova-Acebes M, Bianchini M, Etheridge SL, et al. Atypical chemokine receptor 1 on nucleated erythroid cells regulates hematopoiesis. Nat Immunol. 2017;18:753–761. doi: 10.1038/ni.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rios M, Chaudhuri A, Mallinson G, Sausais L, Gomensoro-Garcia AE, Hannon J, et al. New genotypes in Fy(a-b-) individuals: nonsense mutations (Trp to stop) in the coding sequence of either FY A or FY B. Br J Haematol. 2000;108:448–454. doi: 10.1046/j.1365-2141.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- 14.Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol. 2008;15:49–58. doi: 10.1097/MOH.0b013e3282f29012. [DOI] [PubMed] [Google Scholar]
- 15.Hur J, Choi J-I, Lee H, Nham P, Kim T-W, Chae C-W, et al. CD82/KAI1 maintains the dormancy of long-term hematopoietic stem cells through interaction with DARC-expressing macrophages. Cell Stem Cell. 2016;18:508–521. doi: 10.1016/j.stem.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]