

Abstract
T-cell protein tyrosine phosphatase (TC-PTP) has a critical role in the development of the immune system and has been identified as a negative regulator of inflammation. Single-nucleotide polymorphisms in the TC-PTP locus have been associated with increased susceptibility to inflammatory bowel diseases (IBDs) in patients. To further understand how TC-PTP is related to IBDs, we investigated the role of TC-PTP in maintaining the intestinal epithelial barrier using an in vivo genetic approach. Intestinal epithelial cell (IEC)-specific deletion of TC-PTP was achieved in a mouse model at steady state and in the context of dextran sulphate sodium (DSS)-induced colitis. Knockout (KO) of TC-PTP in IECs did not result in an altered intestinal barrier. However, upon DSS treatment, IEC-specific TC-PTP KO mice displayed a more severe colitis phenotype with a corresponding increase in the immune response and inflammatory cytokine profile. The absence of TC-PTP caused an altered turnover of IECs, which is further explained by the role of the tyrosine phosphatase in colonic stem cell (CoSC) proliferation. Our results suggest a novel role for TC-PTP in regulating the homeostasis of CoSC proliferation. This supports the protective function of TC-PTP against IBDs, independently of its previously demonstrated role in intestinal immunity.
Keywords: Colonic stem cells, DSS-induced colitis, intestinal epithelial barrier, TC-PTP
Introduction
The intestine is the host of a strong innate and adaptive immune cell population. Within the intestine, a physical and biochemical barrier maintains tissue homeostasis and provides protection against the diverse pool of microorganisms that resides in the intestinal lumen.1 This barrier is composed of a single-cell layer of intestinal epithelial cells (IECs) that is maintained by the proliferation and differentiation of pluripotent intestinal stem cells at the bottom of the crypts. The stem cells proliferate and differentiate into the various IECs, which migrate up along the crypt–villus axis. These IECs include enterocytes, which ensure the absorption of nutrients, and secretory cells such as goblet cells, enteroendocrine cells and Paneth cells, which are responsible for the secretion of mucins, hormone regulators and antimicrobial proteins, respectively. With the functions of these specialized epithelial cells, the intestinal barrier can perform its biochemical function.2
The role of the intestinal epithelium is not restricted to the gut lumen; it also has an immunoregulatory function in the underlying tissue.3 Epithelial cells signal to local immune cells using various cytokines to modulate inflammatory reactions. Given the important functions of the intestinal barrier, defective intestinal permeability is one of the major hallmarks in the development of inflammatory bowel diseases (IBDs). Such a phenomenon leads to the invasion of pathogens into the subepithelial space, which triggers an intense inflammatory response. Continual breaches in the barrier and constant exposure to pathogens causes a sustained inflammation and contributes to the further development of IBDs. Maintaining the intestinal epithelial integrity is thus crucial in preventing the progression of these diseases.
The anti-inflammatory function of TC-PTP was first demonstrated using a TC-PTP knockout (KO) mouse model. TC-PTP-deficient mice suffer from severe generalized inflammation characterized by elevated serum levels of interferon gamma (IFNγ) and monocytic infiltration.4 Moreover, colony-stimulating factor 1 receptor (CSF-1R), an important factor in the regulation of the immune response, was identified as a substrate of TC-PTP. Considering that CSF-1R promotes the differentiation and survival of macrophages, this helps explain the hyper-activation of macrophages observed in TC-PTP KO mice.5 In addition, mice hemi-deficient for the phosphatase are more susceptible to dextran sulphate sodium (DSS)-induced colitis than their wild-type (WT) counterparts. Their gut epithelium is more severely damaged and local expression of inflammatory cytokines, such as interleukin (IL)-6, is increased.6 Finally, several genomic studies linked a single nuclear polymorphism (SNP) located downstream of PTPN2 (the gene encoding TC-PTP) to an increased susceptibility to several autoimmune diseases, including Crohn’s disease and ulcerative colitis.7,8 The role of TC-PTP in immune cells has been largely studied.9 Therefore, its implication in preventing intestinal inflammation through monitoring immune cell lineages is not surprising. Until recently, its biological function in maintaining the integrity of the intestinal epithelium had never been studied in vivo. Kasper et al. 10 performed an initial characterization of this function. We decided to further evaluate this potential role of TC-PTP using the well-characterized villin-Cre mouse model to generate a conditional KO model in which TC-PTP is eliminated specifically in IECs.11
Materials and methods
Mice
The targeting construct used to generate the mice was amplified by PCR using R1 ES cell (129/SvJ) genomic DNA. The construct includes two LoxP sites flanking exon 6, an IRES-dependent Cerulean cassette and a neomycin resistance cassette, both of which are flanked by FRT sites (Figure 1a). R1 ES cells were electroporated and correctly targeted clones were injected in C57BL/6 blastocysts. Chimeric mice were mated with C57BL/6 mice and subsequently mated with β-actin/FLPe C57BL/6 mice (McGill Transgenic Core, Montreal, QC, Canada) over three generations to induce germline deletion of the Cerulean and neomycin cassettes. Genotyping of WT and mutant alleles was assessed by PCR (primers: 5′--3′, 5′--3′; Figure 1b). Experiments were performed with mice backcrossed for eight generations to the C57BL/6 background and were used at 6–8 weeks of age unless otherwise noted. Villin-Cre mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Colitis was induced in mice by adding 5% dextran sulfate sodium (DSS; BP1585-100, lot144831 and lot111812, Fisher Bioreagents, Ottawa, ON, Canada) in their drinking water for 10 days, or otherwise specified, as previously done in Hassan et al. 6 Water was changed every 3 days. Mice were monitored and weighed daily during treatment. Animal protocols were approved by the McGill Facility Animal Care Committee and experiments were performed accordingly.
Figure 1.
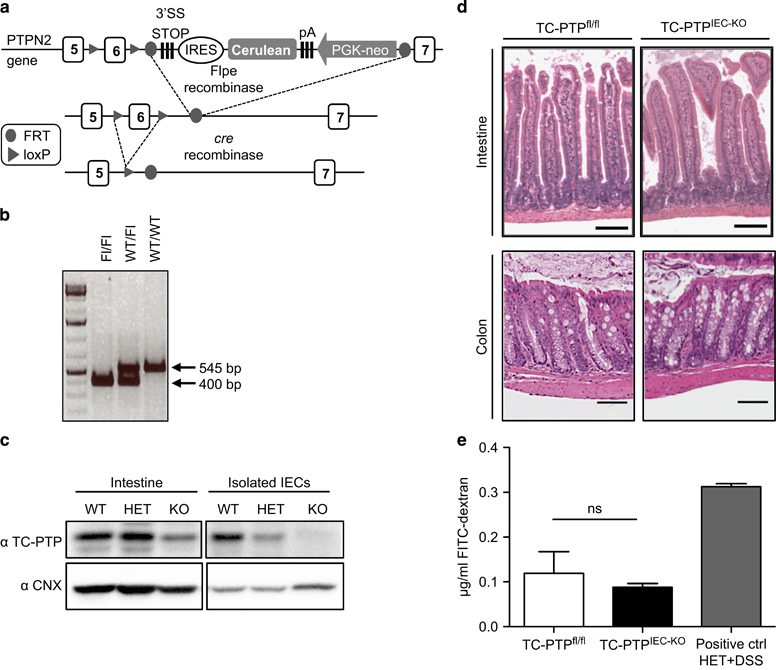
Loss of TC-PTP in mouse IECs does not affect intestinal homeostasis. (a) A schematic of TC-PTP WT allele with the exon 6 flanked by two LoxP sites (arrows) followed by an IRES-Cerulean and a PGK-neo construct. Cerulean cassette is removed by Flpe-recombinase and the exon 6 is deleted by the cre-recombinase to generate a truncated and untranslated TC-PTP transcript. (b) Scheme of PCR used to genotype the generated mice. Amplification of the floxed allele (Fl) results in a 400 bp fragment, whereas the WT allele generates a fragment of 545 bp. (c) Whole intestine and isolated IECs samples from WT (TC-PTPfl/fl), HET (Vil-Cre/TC-PTPWT/fl) and KO (Vil-Cre/TC-PTPfl/fl, later on referred as TC-PTPIEC−KO) mice. Lysates were analyzed by western blotting with antibodies against TC-PTP and calnexin (CNX) as loading control. (d) H&E staining of paraffin-embedded intestine and colon sections from control and TC-PTPIEC−KO mice. Histological analysis (as described in Materials and methods) did not show any differences. Representative sections of n=3. (e) Detection of FITC–dextran in serum (μg/ml) of TC-PTPfl/fl, TC-PTPIEC−KO and positive control mice measured 4 h after oral administration of FITC–dextran. Data shown represent mean values of two mice per group. Results representative of three independent experiments. FITC, fluorescein isothiocyanate; IEC, intestinal epithelial cell; TC-PTP, T-cell protein tyrosine phosphatase; H&E, hematoxylin and eosin; KO, knockout; WT, wild type.
In vivo intestinal permeability
Mice were administered 600 mg of fluorescein isothiocyanate (FITC)–dextran (#46944-100MG, Sigma-Aldrich, St Louis, MO, USA) per kilogram of body weight by gavage. Blood was collected 4 h later by cardiac puncture. The serum concentration of FITC–dextran was measured using the Varioskan plate reader (ThermoFisher Scientific, Waltham, MA, USA) with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. Serially diluted FITC–dextran was used to generate a standard curve.
Histochemistry
Histopathological analysis was performed on intestines flushed with phosphate-buffered saline (PBS) and then rolled into a Swiss-roll, fixed in 10% formalin for 12 h and embedded in paraffin. Four-micrometer sections were cut and stained with hematoxylin and eosin (H&E). Colitis scoring was performed by a veterinary pathologist (J-M Lapointe) at the McGill Comparative Medicine and Animal Centre according to the following criteria: inflammatory score (0–12 points, based on cell infiltration, cell type and extent of inflammation), inflammatory cell location (0–3 points), surface epithelium loss (0–3 points), gland loss (0–3 points), ratio of goblet cell to enterocyte (0–3 points), goblet cell distension (0–3 points) and mucosal architecture (0–3 points) for a maximum of 30 total ranking points. Immunohistochemistry was done using primary antibodies directed against Ki67 and cleaved caspase-3, both performed by the GCRC Histology Core. The number of Ki67+ cells per crypt was manually quantified in 10 crypts per mouse in a total of n=5 mice. Quantification was performed by two separate individuals.
Isolation of intestinal cells
IECs and lamina propria cells were isolated as follows. The gut was dissected and Peyer’s patches were excised on an illuminated surface. The gut was further cut in 2 cm pieces and opened lengthwise. Pieces were washed in tissue lysis buffer: Hank’s balanced salt solution (#14175, Gibco, Waltham, MA, USA) with 2% FBS (#12483–020, Gibco) and 15 mM Hepes (#15630, Gibco). Samples were then incubated in dissociation buffer: 550 ml tissue lysis buffer supplemented with 1 g EDTA (#6381–92–6, Bioshop, Burlington, ON, Canada) with heavy shaking (250 r.p.m.) for 30 min at room temperature and vortexing at half-time. Supernatant was filtered with a 70 μM strainer (#352350, Falcon, Ottawa, ON, Canada) and IECs were collected by centrifugation at 670 g for 5 min. Further samples were then washed again in tissue lysis buffer before digestion in Roswell Park Memorial Institute medium (#R0883, Sigma-Aldrich) with 10% FBS and 15 mM Hepes (#15630, Gibco) to which collagenase (0.2 mg in 25 ml of buffer, #C5138, Sigma-Aldrich) and DNase (1 mg in 25 ml of buffer, #10–104159–001, Roche, Laval, QC, Canada) were freshly added. Digestion was done for 1 h with heavy shaking (250 r.p.m.) at 37 °C and vortexing at half-time. The cell suspension was filtered using a 70 μM strainer and centrifuged at 670 g for 5 min to collect cells from the lamina propria.
Flow cytometry
Isolated lamina propria cells and IECs were resuspended in fluorescence-activated cell sorting (FACS) buffer (PBS, #311–010-CL, Wisent Inc., Saint-Jean-Baptiste, QC, Canada; with 2% FBS). Fc receptors were blocked with anti-CD16/32 (93, #101302, BioLegend, San Diego, CA, USA) for 30 min and cells were stained for 1 h with the following FACS antibodies: anti-CD3 (145–2C11, #553064, BD Pharmingen, San Jose, CA, USA), anti-CD11c (N418, #117305, BioLegend), anti-CD11b (M1/70, #557657 BD Pharmingen), anti-CD19 (6D5, #115519, BioLegend), anti-F4/80 (BM8, #12–4801–82, eBiosciences, Waltham, CA, USA) and anti-Ly6G (RB6–8C5, #108416, BioLegend) for lamina propria cells, and anti-EpCam (G8–8, #118203, BioLegend), anti-CD45 (30-F11, #103113, BioLegend), anti-CD44 (IM7, #103012, BioLegend), anti-CD24 (M1/69, #101805, BioLegend), anti-CD166 (eBioALC48, #1201661–81, eBiosciences) and anti-CD117 (2B8, #47–1171–80, eBiosciences) for IECs. Live cells were gated using the Zombie Aqua fixable viability kit (#423101, BioLegend) as per manufacturer’s protocol. Live populations were analyzed as follows: dendritic cells (DCs; CD11c+), T cells (CD3+), polymorphonuclear neutrophils (PMNs; CD11b+Ly6ChiLy6G+), B cells (CD19+) and colonic stem cells (CoSCs; EpCam+CD45−CD44hiCD24loCD166+cKit[CD177]−). Samples were acquired with the LSRII (BD, Mississauga, ON, Canada) and analyzed using FlowJo (Tree Star, FlowJo LLC, Ashland, OR, USA).
Cytokine quantification
RNA was collected from colons with Trizol as per manufacturer’s protocol (#15596018, Life technologies, Burlington, ON, Canada). Generation of complementary DNA was performed using Superscript III Reverse Transcriptase (P/N56575, Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was performed using the Light Cycler 480 (Roche) with gene-specific primers and SYBR Green I Master mix (#04–707–516–001, Roche). Primers used for each cytokine: IL-6 (F-, R-, IL-1β (F-, R-, IFNγ (F-, R- and IL-10 (F-, R-. GAPDH was used as a reference gene (F-, R-.
Western blotting
Cells were lysed in mRIPA buffer (150 mM NaCl, 50 mM Tris HCl, 0.5% NP-40, 0.25% Na-deoxycholate) with pH 7.4 supplemented with complete EDTA-free protease inhibitor (#04693132001, Roche) and sodium orthovanadate (S6508, Sigma-Aldrich). Protein samples were resolved on 10% SDS-polyacrylamide gel electrophoresis and subjected to immunoblotting with the following antibodies: anti-p-STAT1/3/5 (9167s/9131s/9351s, Cell Signaling, Danvers, MA, USA), anti-STAT1/3/5 (9172S/4904S/9363S, Cell Signaling), anti-p-Jak1 (pY1022/pY1023; 3331S, Cell Signaling), anti-Jak1 (3332, Cell Signaling), anti-cleaved caspase-3 (9691, Cell Signaling), anti-calnexin (kind gift from Dr John J. M. Bergeron, McGill University), anti-actin (#A2066, Sigma-Aldrich), and anti-TC-PTP (14B9, N′, homemade). Primary antibodies were followed by the secondary antibody horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse (Jackson Laboratory).
Statistical analysis
Data were analyzed using Prism (GraphPad Software Inc., La Jolla, CA, USA) software. Mean±s.e.m. is reported. Statistical significance was calculated using a two-tailed Student’s t-test or a one-way analysis of variance test followed by a post hoc analysis (Tukey’s honestly significant difference). Significance was assumed for P-values <0.05.
Results
Loss of TC-PTP in mouse IECs does not affect the homeostatic function of the epithelium
To assess the IEC intrinsic function of TC-PTP in IECs, a mouse in which exon 6 of PTPN2 is framed by LoxP sites was generated. These sites were introduced by homologous recombination and do not affect the expression of the protein as verified by western blotting. As exon 6 contains the catalytic domain of the phosphatase, its deletion via cre-recombinase leads to the complete inactivation of the protein (Figure 1a). Genotyping of the mouse was done by PCR, generating WT and floxed fragments of 545 and 400 bp, respectively (Figure 1b). These TC-PTPfl/fl mice were crossed with villin-Cre mice, which express Cre-recombinase solely in IECs.11 The tissue specificity of the model was confirmed by immunoblotting analysis on IECs isolated from WT (TC-PTPfl/fl), heterozygous (vil-Cre/TC-PTPWT/fl) and KO (vil-Cre/TC-PTPfl/fl, named TC-PTPIEC−KO throughout this report) mice (Figure 1c). No significant differences were observed between heterozygous and WT mice (data not shown). Therefore, this study focuses on the complete KO of TC-PTP in IECs. H&E staining was performed on the intestine and the colon of TC-PTPfl/fl and TC-PTPIEC−KO animals. At steady state, no immune cell infiltration or lesions were observed in the animals, as shown in representative sections (Figure 1d). Tight junctions between IECs have an important role in maintaining selective paracellular permeability in the gut epithelium.12 To test the role of TC-PTP in membrane permeability, TC-PTPfl/fl and TC-PTPIEC−KO mice were administered with FITC-labeled dextran by gavage. Mice with a whole-body hemideficiency of TC-PTP were treated for 4 days with DSS to induce colitis and subsequently used as a positive control. As previously demonstrated,6 these mice displayed epithelial damage and the short-term addition of DSS enhanced the severity of the phenotype, resulting in increased FITC-dextran in the serum upon gavage. There was no difference in the paracellular flux of FITC–dextran between TC-PTPfl/fl and TC-PTPIEC−KO mice (Figure 1e). These results indicate that the loss of TC-PTP in the intestinal epithelium does not affect its overall structure and permeability.
TC-PTPIEC−KO mice are more susceptible to DSS-induced colitis
To test the function of the phosphatase in maintaining the epithelial barrier in a context of inflammation, mice were treated for 10 days with 5% DSS in their drinking water, as previously performed in our laboratory.6 DSS directly compromises the integrity of the IEC barrier and leads to colitis-like inflammation.13 DSS treatment resulted in decreased body weight and a shortening of the colon, indicating successful induction of colitis (Figures 2a and b). Upon IEC-specific KO of TC-PTP, DSS treatment resulted in an average decrease in body weight of ~6.3% compared with the DSS-treated WT mice (Figure 2a). No significant change in body weight was observed between the untreated TC-PTPIEC−KO mice and the untreated WT mice (Figure 2a). On the other hand, DSS-treated TC-PTPIEC−KO mice did not exhibit any significant change in colon length compared with DSS-treated WT mice (Figure 2b). Consistent with the findings of Mähler et al.,14 we observed these phenotypes to be particularly stronger in males. Upon H&E staining of colon sections, DSS-treated TC-PTPIEC−KO were found to have a higher colitis score than DSS-treated WT mice due to the inflammatory cell infiltration and increased epithelial injury (Figures 2c and d). This result correlates with the noted decrease in body weight.
Figure 2.
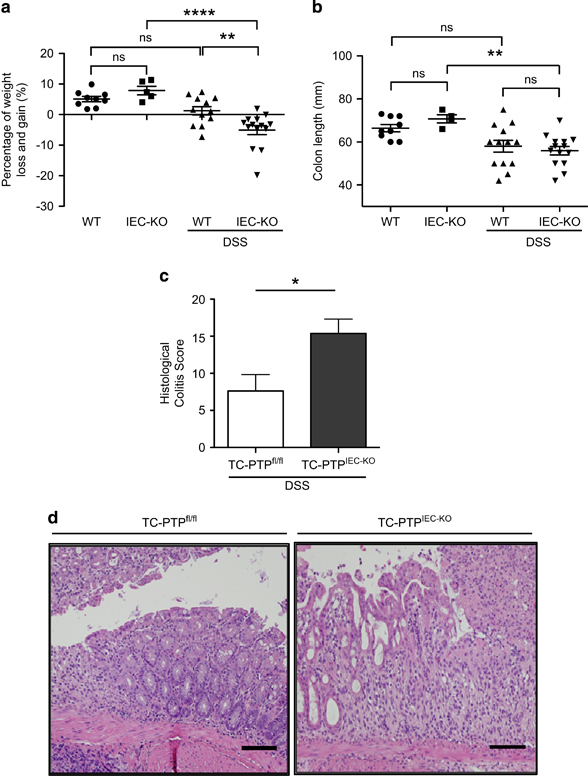
TC-PTPIEC−KO mice are more susceptible to DSS-induced colitis. (a) Percentage of body weight loss and gain of 12 age-matched TC-PTPfl/fl mice (WT, DSS) and TC-PTPIEC−KO mice (IEC-KO, DSS) after 10 days of 5% DSS treatment. In parallel, nine control TC-PTPfl/fl mice (WT) and five TC-PTPIEC−KO mice (IEC-KO) were given regular water. There was a statistically significant difference between the groups as determined by one-way ANOVA (F(3,36)=14.51, P<0.0001). (b) Colon length in millimeters (mm). There was a statistically significant difference between the IEC-KO DSS treated and untreated groups as determined by one-way ANOVA (F(3,36)=6.23, P=0.0016). (c) Histopathological score of controls and TC-PTPIEC−KO mice after DSS treatment based on inflammatory cell infiltration and epithelial injury. Error bars indicate mean±s.e.m. *P<0.05. (d) Representative H&E staining of colonic tissues from control and TC-PTPIEC−KO mice following DSS treatment demonstrating the epithelial damage. n=12. Bar, 50 μM. ANOVA, analysis of variance; DSS, dextran sulphate sodium; TC-PTP, T-cell protein tyrosine phosphatase; WT, wild type.
Corresponding immune response and inflammatory profile in TC-PTPIEC−KO mice
To evaluate the immune response and inflammatory profile in the absence of TC-PTP, immune cells were isolated from the lamina propria of TC-PTPIEC−KO and TC-PTPfl/fl mice upon DSS treatment. The presence of the following immune cells was quantified by flow cytometry: B cells (CD19+), T cells (CD3+), DCs (CD11c+) and PMNs (CD11b+ Ly6Chigh Ly6G+; Figures 3a and b). In the context of DSS-induced inflammation, the presence of B cells, T cells and DCs did not differ upon loss of TC-PTP in the epithelium. On the other hand, an enhanced presence of PMNs was observed in TC-PTPIEC−KO mice. The inflammatory profile of TC-PTPIEC−KO mice was further characterized by analyzing the expression of pro-inflammatory (IL-6, IFNγ, IL-1β) and anti-inflammatory (IL-10) cytokines by qRT-PCR (Figure 3c). There were no significant changes in these cytokine levels between WT and TC-PTPIEC−KO mice upon DSS treatment. However, both IL-1β and IL-6 levels were consistently higher upon DSS treatment, specifically IL-6 that was elevated in TC-PTPIEC−KO mice compared with their WT counterparts. Variable amounts of IL-10 and IFNγ were detected in the colon of both WT and TC-PTPIEC−KO mice upon DSS treatment.
Figure 3.
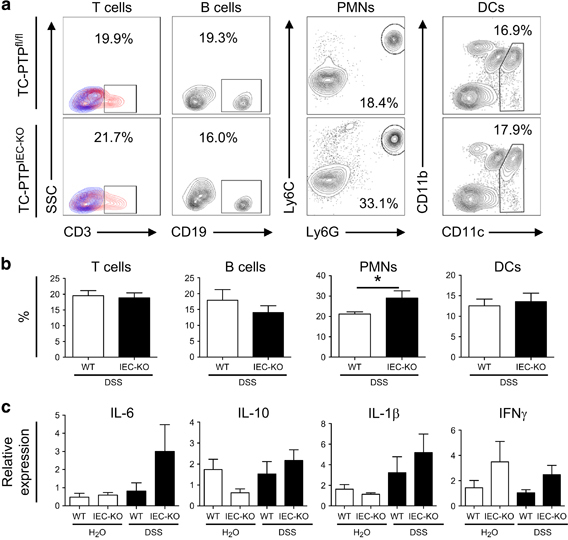
Increased presence of PMNs and expression of inflammatory cytokines in colons of TC-PTPIEC−KO mice upon DSS treatment. (a) Flow cytometry analysis of the cells isolated from the lamina propria from control (WT) and TC-PTPIEC−KO mice. In the first panel (T cells), samples shown in red and fluorescence minus one (FMO) controls shown in blue. (b) Percentage of each cell type identified from 1 × 106 cells stained for flow cytometry. These data are representative of five independent experiments each performed with n=3. Error bars indicate the mean±s.d. *P<0.05. (c) Expression of pro-inflammatory cytokines IL-6, IL-1β, IFNγ and anti-inflammatory cytokine IL-10. RNA isolated from colon of mice treated or not for 10 days with 5% DSS. WT refers to TC-PTPfl/fl mice and IEC-KO to TC-PTPIEC−KO mice. The relative abundance of specific mRNAs, as analyzed by qRT-PCR and normalized to the reference gene GAPDH, is demonstrated. n=15. DSS, dextran sulphate sodium; PMN, polymorphonuclear neutrophil; qRT-PCR, quantitative real-time PCR; TC-PTP, T-cell protein tyrosine phosphatase; WT, wild type; IFNγ, interferon gamma.
TC-PTP regulates the proliferation of CoSCs
To verify whether the absence of TC-PTP in IECs influences their proliferation, colon sections were stained for Ki67, a marker of proliferation. Quantification of Ki67-positive cells in colonic crypts showed increased IEC proliferation in TC-PTPIEC−KO mice (Figures 4a and b). We also evaluated the role of TC-PTP in apoptosis by immunostaining with the cleaved caspase-3 (CC3) marker. Very few IECs were observed to be undergoing apoptosis in the colonic crypts (Supplementary Table 1A). In addition, no difference was observed in the levels of apoptosis between the WT and TC-PTPIEC−KO mice, as assessed by western blotting (Supplementary Table 1B). The phosphorylation profile of TC-PTP targets was assessed in isolated IECs to investigate a potential correlation with the increased number of Ki67-positive cells observed in colonic crypts. The IECs exhibited increased levels of p-STAT1, but not of p-JAK1, p-STAT3 and p-STAT5 (Figure 4c). Using a combination of gene expression patterns and immunohistochemistry, Wang et al. 15 recently developed a flow cytometry gating strategy to identify CoSCs (Figure 4d). Using this technique, we were able to detect a small increase in the CoSCs of TC-PTPIEC−KO mice (Figures 4e and f), which correlates with the previously observed increase in proliferative cells.
Figure 4.
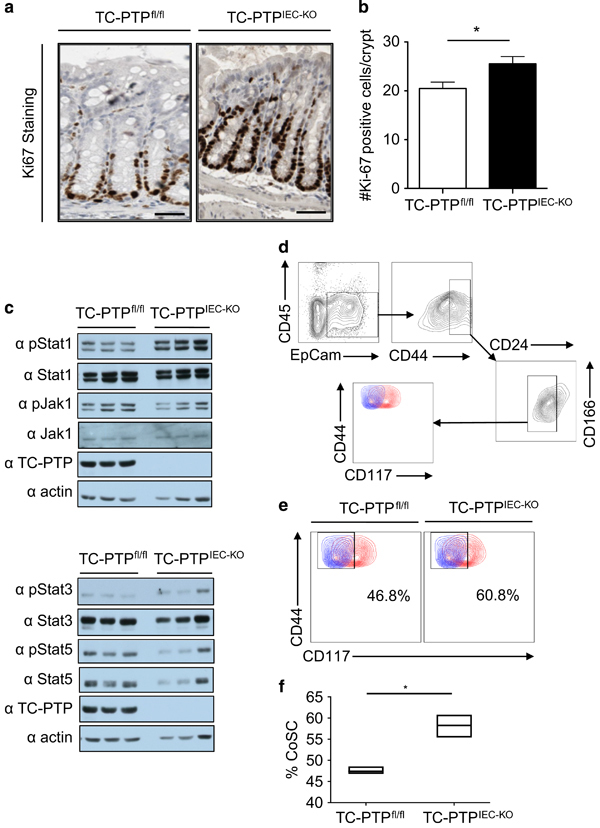
TC-PTP prevents hyper-proliferation of IECs. (a) Immunostaining with the proliferation marker Ki67 using colonic tissues of WT and TC-PTPIEC−KO mice not treated with DSS. Bar, 50 μM. Representative section of n=5. (b) Statistical analysis of Ki67+ cells in 10 clearly defined colonic crypts per mouse. n=5. *P<0.05. (c) Immunoblot of lysates from IECs isolated from three WT and three TC-PTPIEC−KO mice not treated with DSS. Membranes were blotted for p-STAT1, STAT1, p-STAT3, STAT3, p-STAT5, STAT5, p-JAK1, JAK1 and TC-PTP, and actin was used as a loading control. (d, e) Live colonic stem cells (EpCam+CD45−CD44hiCD24loCD166+cKit[CD117]−) defined by flow cytometry from WT and TC-PTPIEC−KO mice not treated with DSS. (d) Gating strategy. (e) Representative plot of three independent experiments performed in triplicates, samples are shown in red and FMO controls shown in blue. (f) Quantification of the percentage of colonic stem cells (% CoSC). Ratio of colonic stem cells identified in a pool of colon epithelial cells (CoSC/colon epithelial cells). Three independent experiments performed with a group of three mice. *P<0.05. DSS, dextran sulphate sodium; FMO, fluorescence minus one; IEC, intestinal epithelial cell; KO, knockout; TC-PTP, T-cell protein tyrosine phosphatase; WT, wild type.
Discussion
IBDs are multifactorial diseases that are not completely understood. Breaching of the intestinal epithelium, however, has been found to be a key contributing factor in the pathogenicity of IBDs. Loss of barrier integrity can lead to triggering of the local mucosa with foreign pathogens, ultimately resulting in excessive immune responses and defective immune regulation. TC-PTP has been extensively studied in immune cells and implicated in local gut inflammation. However, up to recently, its function in maintaining the intestinal epithelium had largely been studied in vitro.16,17,18 Kasper et al. 10 performed an initial in vivo characterization of PTPN2’s function in IECs. To further expand on the role of TC-PTP in maintaining the epithelial barrier of the intestine, we conducted assays using an IEC-specific TC-PTP KO mouse.
We first observed that the elimination of TC-PTP in IECs had no overt effect on the crypt–villus morphology or animal survival. Further investigation showed that the local loss of the phosphatase did not affect intestinal epithelial barrier permeability. However, upon DSS treatment, TC-PTPIEC−KO mice displayed a stronger colitis-like phenotype characterized by increased weight loss, immune cell infiltration and a higher histology colitis score. This confirms the findings of Kasper et al.,10 this time in both male and female mice. In their paper, Kasper et al. do state that PTPN2xVilCre mice tended to suffer more from acute colitis induced by 2.5% DSS, as observed with their colonoscopy and histology results. Our results, using a similar model, indicate that TC-PTP may not have a role in maintaining the epithelium’s permeability, but is essential in maintaining the intestinal barrier response upon induction of damage.
As demonstrated by the evaluation of cells from the lamina propria, the loss of TC-PTP in IECs does not have a direct effect on the innate and adaptive immune cells underlying the epithelium. The increase in PMN infiltration observed in TC-PTPIEC−KO mice seems to be a consequence of an increased sensitivity to DSS-induced colitis. PMNs are part of the innate immune system’s first line of response in the intestine and will accumulate in the lamina propria upon a breach in the epithelial barrier. At the site of injury, PMNs function to secrete growth factors and anti-inflammatory molecules, phagocytose debris, and attract other immune cells to the site of inflammation.19 Although PMNs have been shown to prevent the progression of diseases, they can also contribute to their development. Accumulation of PMNs can cause acute inflammatory responses that can be threatening to the host, as observed in IBD patients.20 TC-PTPIEC−KO mice, which have increased epithelial damage upon DSS treatment, display a higher presence of PMNs to the sites of injury. In the intestine, cytokines are secreted by IECs and other immune cells, which maintains the balance between triggering an immune response and maintaining tolerance.21 The elevated presence of IL-6 correlates with the increased number of PMNs, given that this pro-inflammatory cytokine is well known for its function as a PMN chemoattractant.22 In general, only an increasing trend was observed in the levels of IL-1β and IL-6, which is consistent with Scharl group, who previously reported that PTPN2 has a minor role in IECs during inflammation.10 Collectively, these data indicate that the loss of TC-PTP in the intestinal epithelium leads to a specific phenotype, which might not implicate the immune system as strongly as anticipated. The variation in the local immune system, as well as inflammatory cytokines, appears to be caused by a hypersensitivity to DSS treatment—an indirect effect from the consequences of IEC-specific TC-PTP KO. Any changes resulting from phosphatase loss might not be immediately evident until the barrier is tested, for instance under inflammatory conditions. Therefore, we decided to further examine the role that TC-PTP has in maintaining intestinal structure.
Given its direct contact with the intestinal lumen, the intestinal epithelium is constantly exposed to physical, chemical and bacterial insults that result in a tightly controlled turnover of IECs to maintain barrier integrity. IECs are known for their high proliferation rate resulting in complete cell turnover of the mouse villi in 2–6 days.23 In Drosophila, it has been shown that the JAK/STAT pathway has an important role in ensuring self-renewal and differentiation of intestinal stem cells. The pathway works in parallel with the Wnt/β-catenin pathway to regulate stem cell proliferation, and its over-activation leads to an increased number of progenitor cells.24,25 Several of the JAK/STAT signaling molecules are direct substrates of TC-PTP26,27,28,29 and the loss of this tyrosine phosphatase enhances their tyrosine-phosphorylation profile. The results obtained from the Ki-67 staining show that TC-PTP affects the proliferation of IECs, suggesting a correlation with the activity of TC-PTP targets. TC-PTP has been found to regulate pro-inflammatory signaling via the IL-6-STAT3 pathway and p38, one of the known targets of TC-PTP, is highly involved in regulating IL-6 secretion.30 STAT3 has been frequently linked to intestinal inflammation both through its function in the epithelium and in local immune cells.31,32 STAT3IEC−KO mice have an increased susceptibility to induced colitis, as well as increased tumor growth when crossed with the Apc Min/+ intestinal cancer mouse model.33 In addition, STAT1 has been shown to have a compensatory role in STAT3-deficient tumors of Apc Min/+ mice by promoting the progression of these tumors.34 In the case of IECs lacking TC-PTP, STAT1 appears to be the major target that is hyper-phosphorylated (Figure 4c).
Colonic crypt stem cells (CoSCs) proliferate at a high rate to maintain the required epithelial turnover. Given their high rate of division, CoSCs have been shown to be more susceptible to induced damage compared with differentiated, specialized cells that are no longer dividing.35 A higher presence of damage-sensitive CoSCs in the colonic crypts of TC-PTPIEC−KO mice might be the reason why these mice have an increased susceptibility to DSS-induced colitis. Our results are consistent with and build upon the findings of the Scharl group10 by quantifying a Ki-67 staining of TC-PTPIEC−KO mice and quantifying CoSCs using flow cytometry. A similar phenotype with increased presence of hematopoietic stem cells (HSCs) was also observed in mice with a full KO of TC-PTP.36 Accordingly, whole-body TC-PTP KO mice exhibit an increased number of hematopoietic progenitor cells in their bone marrow. It was also shown that upon treatment with a TC-PTP inhibitor, this phenotype was reproduced in mouse and human bone marrow, peripheral blood and cord blood. It was further demonstrated that TC-PTP affects HSC activity through cytokine signaling via p-STAT1. We anticipate that through a similar signaling pathway, TC-PTP likely maintains the homeostasis of stem cells in the crypts of the colon.
We have shown that TC-PTP has a role in CoSC proliferation, potentially through a p-STAT1-mediated pathway. The loss of TC-PTP leads to an increase in CoSCs proliferation, which renders the epithelium more susceptible to injuries even if permeability is retained. The consequences of the SNPs located on the PTPN2 locus have not been fully characterized. It would be interesting to determine whether the presence of the SNPs leads to a decrease in the expression of TC-PTP in the intestinal epithelium and a corresponding increase in CoSCs proliferation. This new function of TC-PTP in maintaining the integrity of the intestinal epithelium also opens the door to potential tumor-suppressive roles of the phosphatase in the development of intestinal tumors. Colorectal cancers are largely initiated by mutations that activate the Wnt signaling pathway, leading to increased proliferation of intestinal stem cells as well as inhibition of their differentiation.37,38 Given its role in CoSC proliferation, the absence of TC-PTP could promote the development of tumors, indicating a potential tumor suppressor function for TC-PTP in the colon.
Electronic supplementary material
{kind=link}
Acknowledgements
SBM is a recipient of a Fonds de la Recherche en Santé du Québec (FRSQ) doctoral award. VV is a recipient of the Karassik Family Foundation Oncology PhD Fellowship. MLT is the holder of the Jeanne and Jean-Louis Lévesque Chair in Cancer Research. This work was supported by an operating grant from the Canadian Cancer Society Research Institute (CCS) Grant (# 2011-700922) as well as the Leukemia & Lymphoma Society of Canada. We thank Dr Jörg H. Fritz for scientific expertise and Ailsa Lee Loy for technical support.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2016.72
References
- 1.Goto Y, Kiyono H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev. 2012;245:147–163. doi: 10.1111/j.1600-065X.2011.01078.x. [DOI] [PubMed] [Google Scholar]
- 2.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 3.Panja A, Barone A, Mayer L. Stimulation of lamina propria lymphocytes by intestinal epithelial cells: evidence for recognition of nonclassical restriction elements. J Exp Med. 1994;179:943–950. doi: 10.1084/jem.179.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood. 2004;103:3457–3464. doi: 10.1182/blood-2003-09-3153. [DOI] [PubMed] [Google Scholar]
- 5.Simoncic PD, Bourdeau A, Lee-Loy A, Rohrschneider LR, Tremblay ML, Stanley ER, et al. T-cell protein tyrosine phosphatase (Tcptp) is a negative regulator of colony-stimulating factor 1 signaling and macrophage differentiation. Mol Cell Biol. 2006;26:4149–4160. doi: 10.1128/MCB.01932-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassan S-W, Doody K, Hardy S, Uetani N, Cournoyer D, Tremblay ML. Increased susceptibility to dextran sulfate sodium induced colitis in the T cell protein tyrosine phosphatase heterozygous mouse. PLoS One. 2010;5:e8868. doi: 10.1371/journal.pone.0008868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glas J, Wagner J, Seiderer J, Olszak T, Wetzke M, Beigel F, et al. PTPN2 gene variants are associated with susceptibility to both crohn's disease and ulcerative colitis supporting a common genetic disease background. PLoS ONE. 2012;7:e33682. doi: 10.1371/journal.pone.0033682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doody KM, Bourdeau A, Tremblay ML. T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: lessons from the knockout mouse model and implications in human disease. Immunol Rev. 2009;228:325–341. doi: 10.1111/j.1600-065X.2008.00743.x. [DOI] [PubMed] [Google Scholar]
- 10.Kasper SH, Spalinger MR, Leonardi I, Gerstgrasser A, Raselli T, Gottier C, et al. Deficiency of protein tyrosine phosphatase non-receptor type 2 in intestinal epithelial cells has no appreciable impact on dextran sulphate sodium colitis severity but promotes wound healing. Digestion. 2016;93:249–259. doi: 10.1159/000445289. [DOI] [PubMed] [Google Scholar]
- 11.Pinto D, Robine S, Jaisser F, Marjou FE, Louvard D. Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J Biol Chem. 1999;274:6476–6482. doi: 10.1074/jbc.274.10.6476. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. 2013;70:631–659. doi: 10.1007/s00018-012-1070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose WA, Sakamoto K, Leifer CA. Multifunctional role of dextran sulfate sodium for in vivo modeling of intestinal diseases. BMC Immunol. 2012;13:1–13. doi: 10.1186/1471-2172-13-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mähler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 15.Wang F, Scoville D, He XC, Mahe MM, Box A, Perry JM, et al. Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology. 2013;145:383–95.e21. doi: 10.1053/j.gastro.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scharl M, Rudenko I, McCole DF. Loss of protein tyrosine phosphatase N2 potentiates epidermal growth factor suppression of intestinal epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol. 2010;299:G935–G945. doi: 10.1152/ajpgi.00106.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scharl M, Wojtal KA, Becker HM, Fischbeck A, Frei P, Arikkat J, et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm Bowel Dis. 2012;18:1287–1302. doi: 10.1002/ibd.21891. [DOI] [PubMed] [Google Scholar]
- 18.Scharl M, McCole DF, Weber A, Vavricka SR, Frei P, Kellermeier S, et al. Protein tyrosine phosphatase N2 regulates TNFα-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut. 2011;60:189–197. doi: 10.1136/gut.2010.216606. [DOI] [PubMed] [Google Scholar]
- 19.Williams IR, Parkos CA. Colonic neutrophils in inflammatory bowel disease: double-edged swords of the innate immune system with protective and destructive capacity. Gastroenterology. 2007;133:2049–2052. doi: 10.1053/j.gastro.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Brannigan AE, O'Connell PR, Hurley H, O'Neill A, Brady HR, Fitzpatrick JM, et al. Neutrophil apoptosis is delayed in patients with inflammatory bowel disease. Shock. 2000;13:361–366. doi: 10.1097/00024382-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 22.Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, et al. IL-6 Regulates Neutrophil Trafficking during Acute Inflammation via STAT3. J Immunol. 2008;181:2189–2195. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 23.Williams JM, Duckworth CA, Burkitt MD, Watson AJM, Campbell BJ, Pritchard DM. Epithelial cell shedding and barrier function: a matter of life and death at the small intestinal villus tip. Vet Pathol 2014. [DOI] [PMC free article] [PubMed]
- 24.Lin G, Xu N, Xi R. Paracrine unpaired signaling through the JAK/STAT pathway controls self-renewal and lineage differentiation of drosophila intestinal stem cells. J Mol Cell Biol. 2010;2:37–49. doi: 10.1093/jmcb/mjp028. [DOI] [PubMed] [Google Scholar]
- 25.Cordero JB, Stefanatos RK, Myant K, Vidal M, Sansom OJ. Non-autonomous crosstalk between the Jak/Stat and Egfr pathways mediates Apc1-driven intestinal stem cell hyperplasia in the Drosophila adult midgut. Development. 2012;139:4524–4535. doi: 10.1242/dev.078261. [DOI] [PubMed] [Google Scholar]
- 26.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. 2002;19:446–453. doi: 10.1016/S0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 27.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, et al. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem Biophys Res Commun. 2002;297:811–817. doi: 10.1016/S0006-291X(02)02291-X. [DOI] [PubMed] [Google Scholar]
- 29.Aoki N, Matsuda T. A nuclear protein tyrosine phosphatase TC-PTP is a potential negative regulator of the PRL-mediated signaling pathway: dephosphorylation and deactivation of signal transducer and activator of transcription 5a and 5b by TC-PTP in nucleus. Mol Endocrinol. 2002;16:58–69. doi: 10.1210/mend.16.1.0761. [DOI] [PubMed] [Google Scholar]
- 30.Spalinger MR, McCole DF, Rogler G, Scharl M. Protein tyrosine phosphatase non-receptor type 2 and inflammatory bowel disease. World J Gastroenterol. 2016;22:1034–1044. doi: 10.3748/wjg.v22.i3.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ. New insights into the role of STAT3 in IBD. Inflamm Bowel Dis. 2012;18:1177–1183. doi: 10.1002/ibd.21884. [DOI] [PubMed] [Google Scholar]
- 32.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musteanu M, Blaas L, Mair M, Schlederer M, Bilban M, Tauber S, et al. Stat3 is a negative regulator of intestinal tumor progression in ApcMin mice. Gastroenterology. 2010;138:1003–11.e5. doi: 10.1053/j.gastro.2009.11.049. [DOI] [PubMed] [Google Scholar]
- 35.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 36.Bourdeau A, Trop S, Doody KM, Dumont DJ, Tremblayef ML. Inhibition of T cell protein tyrosine phosphatase enhances interleukin-18-dependent hematopoietic stem cell expansion. Stem Cells. 2013;31:293–304. doi: 10.1002/stem.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.BarbaraLustig JB. The Wnt connection to tumorigenesis. Int J Dev Biol. 2004;48:477–487. doi: 10.1387/ijdb.041815jb. [DOI] [PubMed] [Google Scholar]
- 38.Fevr T, Robine S, Louvard D, Huelsken J. Wnt/β-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27:7551–7559. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.