

Type I IFNs are important cytokines with antiviral and regulatory functions. The antiviral properties of IFN-α are well established and successfully used in the treatment of infectious diseases and cancer in humans. By contrast, the detrimental effects of chronic exposure to type I IFNs in chronic infections have been widely reported in animal models and HIV/SIV infection. Therefore, the blockade of this pathway has been explored as a potential therapeutic target.1,2
Differentiation and the acquisition of effector functions in CD8 T cells is the result of the integration of signals provided by the TCR and the inflammatory environment during infection. This process is orchestrated by key specific transcription factors.3 However, little is known about the mechanisms of post-transcriptional regulation of CD8 T-cell differentiation. The emerging role of miRNAs in the regulation of CD8 T cells is the tip of an iceberg of these mechanisms in fine-tuning the integration of signals during CD8 T cell effector differentiation.4,5 CD8 T cells are critical players in immunity against viruses; however, in pathological conditions with the failure of viral control/elimination, CD8 T cells become ‘exhausted’. The post-transcriptional regulation mechanisms that lead to T-cell exhaustion during chronic infection are mostly unknown. A recent manuscript, Moffet et al.6 showed that miRNA-31 tunes the response to type I IFNs signaling in CD8 T cells. In the chronic viral infection setting, deletion of Mir31 in CD8 T cells altered the path to T-cell exhaustion and infection outcome.6
MicroRNAs (miRNAs) are small non-coding RNAs that negatively regulate gene expression by binding to the 3′ untranslated region of regulated mRNA (3′-UTR). miRNAs are transcribed as long transcripts of primary miRNA (pri-miRNA) that undergo a maturation process. The DGCR8/DROSHA (DiGeorge syndrome critical region 8) protein complex is part of this process in generating precursor miRNA (pre-miRNA). These precursors are transported to the cytoplasm and further processed into the mature form by the Dicer-TRBP (TAR RNA-binding protein 2) complex. Post-transcriptional regulation by miRNAs is a mechanism that has been shown to be involved in T-cell development, differentiation and function.5 Several miRNAs have been identified to play important functions in CD8 T cell effector and memory differentiation, including miRNA-146a, miRNA-155, miRNA17-92, miRNA-139 and miRNA-1-150.4,5 In this growing list of miRNAs, Moffett et al. identified miRNA-31. They found that miRNA-31 was highly upregulated on CD8 T cells upon TCR stimulation. The ex vivo analysis of CD8 T cell subsets showed that miRNA-31 expression was restricted to more differentiated memory/effector phenotypes, suggesting its regulation during differentiation.
To identify the target genes of miRNA-31, they used expression profiling in CD8 T cells (expressing transgenic OVA-specific TCR, OT1-CD8 T cells). OT1-CD8 T cells were transduced with a lentiviral vector expressing mRNA-31. The mRNAs downregulated by miRNA-31 expression showed an enrichment for the predicted miRNA-31 binding sites. Eight new miRNA-31 target genes were identified (Psd4, Sh2d1a, Ilf3, Coro7, Rab1b, Stra13, Cdkn1a and Ifi30) in addition to the previously reported targets (Ppp6c, Lats2, Oxsr1 and ELavl1). An ingenuity-pathway analysis of OT-CD8 T cell expressing miRNA-31 showed mRNA transcripts associated with type I IFNs, including Ifna2, Irf3 and Irf7, suggesting a potential role for mRNA-31 in this pathway.
To study the in vivo role of miR-31, the researchers deleted Mir31 in the germline of C56BL/6 mice by crossing mice with loxP-flanked Mir31 alleles (Mir31fl/fl) and mice that express Cre recombinase under the control of the promoter of the gene encoding the glycoprotein, Zp3, to generate Mir31fl/fl Zp3-Cre mice. Additionally, conditionally deleted Mir31 in T cells was generated by crossing Mir31fl/fl with mice in which Cre is expressed under the control of the T-cell-specific Cd4 promoter (Cd4Cre, Mir31fl/flCd4Cre). Analysis of the global transcriptional changes of CD8 T cells after in vitro stimulation revealed an enrichment of mRNA transcripts of effector molecules, such as perforin (Pfr), and several granzymes, including Gzmd, gzmc, Gzme, Gzmg and Gzmb, as well as osteopontin (Spp1) in the miRNA-31-deficient CD8 T cells compared with wild-type cells. This enrichment was more noticeable when CD8 T cells were stimulated in the presence of IFN-β, suggesting that miRNA-31 may play a role in modulating the sensitivity of CD8 T cells to type I IFNs during activation. More importantly, several mRNA transcripts associated with T cell dysfunction during chronic infection with lymphocytic choriomeningitis virus (LCMV) had reduced expression in miRNA-31 deficient CD8 T cells, including the prostaglandin E2 receptor (Ptger2), the proto oncogene c-Maf (Maf), and other transcripts described in mouse tumor models, such as metallothioneins (Mt1 and Mt2).
One of the target genes of miRN-31 is Ppp6c (protein phosphatase-6 catalytic subunit), a phosphatase that regulates the activity of Map3k7 (TAK1), which inhibits type I IFN signaling. Transduction of miRNA-31 or silencing its target, Ppp6c, with shRNA increased the sensitivity of CD8 T cells to IFN-β, suggesting a role for miR-31 in fine-tuning the responsiveness to type I IFNs during CD8 T-cell activation (Figure 1). While type I IFNs are critical for antiviral immunity, in the setting of chronic infections, persistent exposure to type I IFNs leads to impaired T cell responses. To address whether miR-31 might be involved in T-cell sensitivity to type I IFNs in the context of chronic infections, a mouse model of chronic infection with LCMV clone 13 was used. In this model, CD8 T cells show an exhausted phenotype, which was characterized by the expression of inhibitory receptors, such as PD1, LAG3, 2B4, CD160 and TIM3 and diminished effector T-cell function.7 Both miR-31-deficient and wild-type mice had similar signs of disease in the initial acute phase (days 9–11) post infection. By contrast, in the chronic phase, miR-31-deficient mice recovered and showed no signs of disease, with viral titers and tissue associated viral RNA significantly lower than the wild-type animals. Viral control in the miR-31-deficient mice was mainly associated with increased LCMV-specific CD8 T cells with an effector and effector memory phenotype. The virus-specific cells had lower expression of the exhaustion marker PD1 and they showed polyfunctionality by their ability to secrete cytokines, ultimately promoting viral control (Figure 1). These data indicated that miR-31 plays a critical role in promoting CD8 T cell dysfunction during LCMV chronic infection by modulating sensitivity to type I IFNs.
Figure 1.
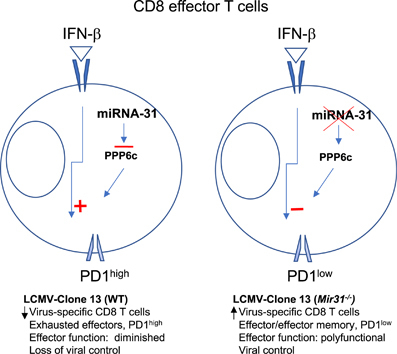
miRNA-31 modulates the type I IFN response in CD8 T cells. The in vitro miRNA-31 target gene, Ppp6c, regulates the sensitivity of the type I IFN response. miR-31-deficient CD8 T cells were infected with a lentiviral vector expressing miRNA-31 or shRNA targeting Ppp6c to assess the in vitro response to IFN-β. Activation was measured by the detection of CD69 upregulation using flow cytometry. Expression of miRNA-31 or silencing Ppp6c enhanced CD69 upregulation (relative to control lentivirus) after in vitro stimulation with IFN-β. miR-31 regulates T-cell exhaustion during chronic infection. During the chronic phase of an infection with LCMV clone 13, miR-31-deficient mice recovered and showed no signs of disease compared with wild-type mice. Viral control in the miR-31-deficient mice was associated with increased polyfunctional virus-specific CD8 T cells with effector and effector memory phenotypes and low PD1 expression.
Type I IFNs have antiviral properties and enhance CD8 T-cell effector differentiation early during infection. By contrast, in the setting of chronic infection, type I IFNs and the associated inflammatory environment contribute to the pathology of the disease and promote T-cell exhaustion and dysfunction.8 miRNA-31 was upregulated early after activation; however, it does not appear to play a role during the acute phase of LCMV clone 13 and acute influenza infection. The authors showed that miRNA-31 targets the phosphatase, Ppp6c. Ppp6c has been shown to regulate Map3k7 (TAK1) activity, a kinase involved in tonic type I IFN signaling during T cell development; however, its role in the context of infection requires further investigation.9
During infection, failure to eliminate a virus leads to chronic disease, resulting in a sustained inflammatory environment that is rich in type I IFNs and accompanied by the accumulation of T cells with an activated ‘exhausted’ phenotype and diminished in vitro effector function. This is also observed in chronic human infections, including HIV, HBV and HCV.7 In particular, HIV infection is characterized by chronic T-cell immune activation that is reflected by the increased expression of a variety of immunomodulatory receptors, including PD1, CTLA-4, LAG3, CD244/2B4, CD160 and others.10 The role of the type I IFNs in the pathogenesis of HIV/SIV infection has been suggested by observations in human and non-human primate models of HIV infection.11,12 The chronic type I IFN gene signature is a distinguishable feature observed in pathogenic versus non-pathogenic SIV infection models.2 The detrimental effects of chronic exposure to type I IFNs have been assessed in mouse models and more recently in a humanized mouse model infected with HIV.1,13,14,15 This model mimics the disturbances of the immune system observed in patients with HIV infection. Type I IFN signaling blockade during the chronic phase led to reduced immune activation, expression of PD1 and TIM3, and improved cytokine secretion by virus-specific T cells, resulting in viral replication control. More importantly, in this model, type I IFN blockade reduced the viral reservoir.1,14,15
Targeting the type I IFN pathway has been considered a potential therapeutic strategy in chronic infectious diseases, such as HIV. However, blockade of its receptor may compromise immunity against HIV and other viruses. The manuscript by Moffett et al. suggests an alternative strategy to regulate type I IFN signaling during the chronic phase of an infection. The importance of miRNAs in the post-transcriptional regulation of protein expression and their association with human diseases, including viral infections, suggests that miRNAs may be the new frontier for modulating the immune system in the context of chronic infection.
Conflict of interest
The author declares no conflict of interest.
References
- 1.Deeks SG, Odorizzi PM, Sekaly RP. The interferon paradox: can inhibiting an antiviral mechanism advance an HIV cure? J Clin Invest. 2017;127:103–105. doi: 10.1172/JCI91916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who's interfering with whom? Nat Rev Microbiol. 2015;13:403–413. doi: 10.1038/nrmicro3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gracias DT, Katsikis PD. MicroRNAs: key components of immune regulation. Adv Exp Med Biol. 2011;780:15–26. doi: 10.1007/978-1-4419-5632-3_2. [DOI] [PubMed] [Google Scholar]
- 5.Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev. 2016;16:279–294. doi: 10.1038/nri.2016.40. [DOI] [PubMed] [Google Scholar]
- 6.Moffett HF, Cartwright ANR, Kim HJ, Godec J, Pyrdol J, Aijo T, et al. The microRNA miR-31 inhibits CD8+ T cell function in chronic viral infection. Nat Immunol. 2017;18:791–799. doi: 10.1038/ni.3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38:542–557. doi: 10.1016/j.it.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing Y, Wang X, Jameson SC, Hogquist KA. Late stages of T cell maturation in the thymus involve NF-kappaB and tonic type I interferon signaling. Nat Immunol. 2016;17:565–573. doi: 10.1038/ni.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuchroo VK, Anderson AC, Petrovas C. Coinhibitory receptors and CD8 T cell exhaustion in chronic infections. Curr Opin HIV AIDS. 2014;9:439–445. doi: 10.1097/COH.0000000000000088. [DOI] [PubMed] [Google Scholar]
- 11.Catalfamo M, Le Saout C, Lane HC. The role of cytokines in the pathogenesis and treatment of HIV infection. Cytokine Growth Factor Rev. 2012;23:207–214. doi: 10.1016/j.cytogfr.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev. 2013;254:326–342. doi: 10.1111/imr.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Saout C, Hasley RB, Imamichi H, Tcheung L, Hu Z, Luckey MA, et al. Chronic exposure to type-I IFN under lymphopenic conditions alters CD4 T cell homeostasis. PLoS Pathog. 2014;10:e1003976. doi: 10.1371/journal.ppat.1003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest. 2017;127:260–268. doi: 10.1172/JCI89488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng L, Ma J, Li J, Li D, Li G, Li F, et al. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest. 2017;127:269–279. doi: 10.1172/JCI90745. [DOI] [PMC free article] [PubMed] [Google Scholar]