

Abstract
Background
Positron emission tomography ligands are now available that bind to tau proteins in the brain, providing the exciting opportunity to assess the presence and distribution of tau in vivo in living patients.
Methods
This manuscript performed a systematic review of studies that have performed tau PET imaging in patients with parkinsonian disorders. PubMed was searched through November 13, 2017 and the review included case reports and patient‐control studies.
Results
Most tau‐PET studies have utilized the [18F]AV‐1451 ligand, with a few using the [11C]PBB3 and [18F]THK‐5351 ligands. Elevated cortical tau‐PET uptake has been observed in Parkinson's disease dementia and dementia with Lewy bodies, presumed to be related to Alzheimer's disease‐related pathology. Mild patterns of tau‐PET uptake have been observed in subcortical structures in progressive supranuclear palsy and subcortical structures and motor cortex in corticobasal syndrome, although discrepancy with autoradiographic studies that show lack of binding to 4‐repeat tau and “off‐target” binding observed in subcortical structures limit the interpretation of these findings. Findings in frontotemporal dementia with tau mutations are variable, but elevated signal is most pronounced in mutations with deposition of both 3 and 4‐repeat tau. Elevated tau‐PET uptake has also been observed in multiple system atrophy, a synucleinopathy.
Conclusion
The value of the current generation of tau‐PET ligands varies across parkinsonian syndromes, depending upon underlying variability in tau pathology and “off‐target” binding. More work is needed to understand the biological basis of binding and more specific tau PET ligands are needed to study parkinsonian disorders.
Keywords: AV‐1451, Parkinsonism, PBB3, positron emission tomography, THK‐5351
The recent development of positron emission tomography (PET) ligands that can bind to tau proteins in brain tissue have provided an exciting opportunity to assess the presence and distribution of tau pathology in patients during life. This is particularly valuable for patients with neurodegenerative diseases that are defined by the presence of hyperphosphorylated tau inclusions in the brain (i.e., tauopathies), but also for detecting mixed pathologies in a range of different neurodegenerative diseases. Primary tauopathies associated with parkinsonian syndromes include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and frontotemporal dementia with mutations in the microtubule associated protein tau (MAPT) gene. However, Alzheimer's disease (AD) that is characterized by both tau and beta‐amyloid deposition can also occur in patients with parkinsonian syndromes and hence is also an important target for tau‐PET imaging. Although these diseases are characterized by the presence of abnormal tau proteins, the specific tau isoforms, structure of the tau filaments, morphological characteristics of the tau inclusions, and the distribution of tau pathology differ in each disease (Table 1). The ultimate hope is that tau‐PET imaging could provide an early diagnostic biomarker of the presence of tau pathology and also provide biomarkers of disease progression, which could be used as outcome measures in clinical treatment trials, particularly those assessing therapies that target tau.
Table 1.
Pathological Characteristics of Tauopathies Discussed in this Review
| Neurodegenerative disease | Tau isoforms | Tau filaments | Tau inclusion types | Distribution of tau pathology |
|---|---|---|---|---|
| Alzheimer's disease | 3R + 4R | Predominantly paired helical filaments | Neurofibrillary tangles, neuropil threads and dystrophic neurites | Medial temporal lobe, neocortex (particularly temporal and parietal lobes) |
| Progressive supranuclear palsy33 | 4R | Straight filaments more common than paired helical filaments | Globose neurofibrillary tangles, tufted astrocytes, oligodendroglial coiled bodies and threads | Brainstem, basal ganglia, diencephalon, temporal, motor and premotor cortices |
| Corticobasal degeneration33 | 4R | Twisted ribbon filaments | Ballooned neurons and glial inclusions, astrocytic plaques, threads, coiled bodies | Basal ganglia and neocortex |
| Frontotemporal dementia with MAPT mutations70 | 4R | Twisted ribbon filaments | Neuronal and glial (coiled bodies in oligodendroglia, tufted astrocytes and astrocytic plaques) deposits | Frontal and temporal cortices, medial temporal lobes, basal ganglia, midbrain, pons, dentate nucleus of the cerebellum |
| 3R + 4R | Paired helical and straight filaments | Neurofibrillary tangles or Pick‐like bodies | Frontal and temporal cortices, medial temporal lobes, basal ganglia, pons | |
| 3R | Straight filaments with some twisted filaments | Pick bodies and axonal inclusions predominate, with some glial inclusions | Frontal and temporal cortices, medial temporal lobes, midbrain, pons |
Abbreviations: 3R, tau isoform with 3 repeats in the microtubule binding domain; 4R, tau isoform with 4 repeats in the microtubule binding domain; 3R + 4R, mixed 3 and 4 repeat tau isoforms.
A number of different tau‐PET ligands have been developed,1 most of which were originally developed to detect the hyperphosphorylated tau present in AD. Most of the clinical work performed so far has used the [18F]AV‐1451 ligand (originally known as T807).2 Early studies showed that [18F]AV‐1451 had greater selectivity for paired helical filament tau compared to beta‐amyloid, and it was shown to have rapid uptake and washout, cross the blood‐brain barrier, and have minimal white matter binding in transgenic mice.2 Two other ligands that also show high affinity for tau and have been used in clinical studies are the [18F]THK‐53513 and [11C]PBB34 ligands. These ligands have shown strong binding to the tau pathology present in AD in autoradiographic studies.5, 6, 7, 8 This review will discuss findings from studies that have utilized these tau‐PET ligands in patients clinically diagnosed with different parkinsonian disorders, including Parkinson's disease (PD), dementia with Lewy bodies (DLB), PSP syndromes, corticobasal syndrome, multiple system atrophy (MSA), and frontotemporal dementia associated with MAPT mutations. Autoradiographic studies relevant to each disease will be discussed in each section.
Materials and Methods
Literature was searched on PubMed for entries up until November 13, 2017 using search terms to capture tau PET studies (AV‐1451, THK‐5351, PBB3, PET) and the diseases of interest (Parkinson's disease, dementia with Lewy bodies, multiple system atrophy, progressive supranuclear palsy, corticobasal syndrome, corticobasal degeneration, frontotemporal dementia, MAPT mutation, parkinsonism). Only studies published in English and those that reported findings from tau PET imaging in humans were reviewed. A total of 28 manuscripts were identified in the search and fulfilled inclusion criteria (Table 2).
Table 2.
Summary of Tau‐PET Studies in Patients With Parkinsonism
| Reference | Tau ligand | Disease groups | Analysis methods | Results |
|---|---|---|---|---|
| Parkinson's disease and dementia with Lewy bodies | ||||
| Hansen 201617 | [18F]AV‐1451 | 17 PD, 16 controls | Volume of distribution for subtantia nigra. SUVR for basal ganglia, referenced to cerebellar cortex. PVC− | PD < C = substantia nigra |
| Gomperts 201615 | [18F]AV‐1451 | 7 DLB, 9 PD, 4 PDD, 4 PD‐MCI, 29 PiB‐ controls | SUVR referenced to cerebellar grey matter. PVC+ and PVC− |
DLB > C = inferior/lateral temporal lobe, precuneus (PVC+/PVC−) PD‐impaired > C = inferior/lateral temporal lobe, precuneus (PVC+/PVC−) DLB > PD = inferior temporal gyrus (PVC+/PVC−) and precuneus (PVC+ only) PD‐impaired > PD = inferior temporal gyrus (PVC+ only) |
| Kantarci 201718 | [18F]AV‐1451 | 19 DLB, 19 AD, 95 controls | SUVR referenced to cerebellar crus. PVC+ and PVC− |
DLB > C = inferior/lateral temporal lobe, precuneus, posterior cingulate and occipital cortex (PVC+ and PVC−) DLB > AD = no findings AD > DLB = findings throughout the cortex, but relatively sparing occipital and sensorimotor cortices |
| Hansen 201716 | [18F]AV‐1451 | 26 PD (9 with MCI), 23 controls |
SUVR, referenced to cerebellar cortex. PVC+, PVC− |
PD > C = no findings (PVC+/PVC−) PD < C = substantia nigra (PVC+/PVC−) |
| Coakeley 201781 | [18F]AV‐1451 | 6 PSP, 6 PD, 10 controls | SUVR in substantia nigra referenced to cerebellar cortex |
PD < C = substantia nigra PSP < C = substantia nigra PD < PSP = no findings |
| Hansen 201750 | [18F]AV‐1451 | 27 PD (16 on MAO‐B inhibitor, 11 not on inhibitor) | SUVR, references to cerebellar cortex. PVC− | No differences observed between PD patients on MAO‐B inhibitors and those not on MAO‐B inhibitors |
| Progressive supranuclear palsy | ||||
| Ishiki 201740 | [18F]THK‐5351 | 3 PSP, 13 AD, 9 controls | SUVR referenced to cerebellar cortex | PSP > C and AD = globus pallidus and midbrain |
| Coakeley 201739 | [18F]AV‐1451 | 6 PSP, 6 PD, 10 controls | SUVR referenced to cerebellar cortex. PVC+ |
PSP > C = no findings PD > C = no findings |
| Hammes 201782 | [18F]AV‐1451 | 1 PSP‐RS, 19 controls | Z scores, PVC− | PSP > C = bilateral globus pallidus, midbrain |
| Whitwell 201738 | [18F]AV‐1451 | 10 PSP‐RS, 10 AD, 50 controls | SUVR referenced to cerebellar crus grey matter. PVC+ and PVC− |
PSP > C = globus pallidus, dentate nucleus of the cerebellum, thalamus and midbrain (PVC+ and PVC−), and supplementary motor area, precentral cortex, frontal inferior opercularis, caudate nucleus and middle frontal gyrus (PVC+) PSP > AD = midbrain, dentate nucleus of the cerebellum, thalamus and the pallidum (PVC+ and PVC−) AD > PSP = all cortical regions (PVC+ and PVC−) |
| Cho 201734 | [18F]AV‐1451 | 14 PSP, 15 PD, 15 controls | SUVR referenced to cerebellar cortex, PVC− |
PSP > C = putamen, globus pallidus, subthalamic nucleus, and dentate nucleus PSP > PD = substantia nigra, putamen, globus pallidus, subthalamic nucleus, and dentate nucleus PD > C = no findings PD < C = substantia nigra |
| Smith 201737 | [18F]AV‐1451 | 11 PSP (9 PSP‐RS, 2 PSP‐P), 11 controls | SUVR referenced to cerebellar grey matter. PVC− | PSP v > C = globus pallidus, putamen, thalamus, midbrain, |
| Passamonti 201735 | [18F]AV‐1451 | 19 PSP‐RS, 15 AD, 13 controls |
BPND, referenced to superior grey matter of cerebellum. PVC+ and PVC− |
PSP > C = putamen, globus pallidus, thalamus, midbrain, and dentate nucleus of the cerebellum (PVC+ and PVC−) PSP > AD = midbrain AD > PSP = frontal, parietal, lateral temporal, and occipital cortices as well as the hippocampus and other medial temporal lobe regions |
| Ishiki 201740 | [18F]AV‐1451 | 3 PSP‐RS, 13 AD, 9 controls | SUVR referenced to cerebellar cortex. PVC− |
PSP > C = globus pallidus, midbrain, with some evidence in precentral, superior parietal, posterior cingulate, occipital lobe PSP > AD = globus pallidus and midbrain AD > PSP = middle and inferior temporal, fusiform and parahippocampal gyri |
| Perez‐Soriano 201741 | [11C]PBB3 | 6 PSP (5 PSP‐P, 1 PSP‐RS), 3 SNCA duplication carriers, 1 MSA‐P, 6 controls | Binding potential, referenced to cerebellar white matter. PVC− |
PSP > C = putamen, midbrain, pallidum SNCA > C = GP, putamen, thalamus, ventral striatum, SN, peduncolupontine nucleus, occipital lobe MSA > C = frontal lobe, GP, midbrain, parietal lobe, putamen, temporal lobe, SN, thalamus, ventral striatum |
| Schonhaut 201736 | [18F]AV‐1451 | 33 PSP, 26 PD, 46 controls | SUVR referenced to cerebellar grey matter, PVC+ and PVC− |
PSP > C = globus pallidus, putamen, thalamus, subthalamic nucleus, midbrain, and dentate nucleus of the cerebellum (PVC+ and PVC−) PSP > PD = globus pallidus, putamen, subthalamic nucleus, midbrain, and dentate nucleus of the cerebellum (PVC+ and PVC−) PD < C = substantia nigra |
| Corticobasal syndrome | ||||
| Maruyama 20134 | [11C]PBB3 | 1 CBS (PiB‐ve) | SUVR referenced to cerebellum. PVC− | Increased signal observed in neocortex, basal ganglia, thalamus, midbrain |
| Josephs 201644 | [18F]AV‐1451 | 1 CBD | SUVR referenced to cerebellar crus grey matter. PVC− | Increased signal observed visually in putamen, pallidum, thalamus, precentral cortex, rolandic operculum, supplemental motor area, and left Broca's area |
| McMillan 201652 | [18F]AV‐1451 | 1 CBD | SUVR, referenced to cerebellar grey matter (excl. deep grey nuclei). PVC+ | Increased signal observed visually in bilateral substantia nigra, globus pallidus, and midbrain, and then with more uptake in bilateral frontal and posterior temporal lobes, midbrain and pons on 10 month follow‐up |
| Smith 201764 | [18F]AV‐1451 | 8 CBS, 11 PSP, 31 AD, 17 controls | SUVR referenced to cerebellar grey matter. PVC+ |
Two CBS patients showed patterns of tau and beta‐amyloid uptake consistent with AD CBS(Aβ−) > C = asymmetric precentral and postcentral cortex, superior parietal lobe, corticospinal tract, putamen and globus pallidus CBS(Aβ−) > PSP = subcortical white matter underlying motor PSP > CBS(Aβ−) = not reported CBS(Aβ−) > AD = subcortical white matter underlying motor cortex and basal ganglia AD > CBS(Aβ−) = temporoparietal cortical regions |
| Cho 201762 | [18F]AV‐1451 | 6 CBS, 20 controls | SUVR referenced to cerebellar cortex. PVC− | CBS > C = asymmetric putamen, globus pallidus and thalamus, precentral grey and white matter, midbrain, dentate nucleus of the cerebellum |
| Kikuchi 201763 | [18F]THK‐5351 | 5 CBS, 8 AD, 8 controls | SUVR referenced to cerebellar grey matter. PVC− |
CBS > C = asymmetric precentral cortex, postcentral cortex, superior frontal, superior parietal, globus pallidus CBS > AD = precentral gyrus, right globus pallidus AD > CBS = parahippocampal gyrus and inferior temporal cortex |
| Xia 20172 | [18F]AV‐1451 | 1 CBS(Aβ+), 77 controls | SUVR, referenced to cerebellar grey matter. PVC− | CBS > C = bilateral but asymmetric primary and association sensorimotor (perirolandic) cortices |
| Multiple system atrophy | ||||
| Cho 201769 | [18F]AV‐1451 | 4 MSA‐P, 30 controls | SUVR, referenced to cerebellar cortex. PVC− | MSA‐P > C = bilateral posterior putamen, contralateral anterior putamen |
| Frontotemporal dementia with MAPT mutations | ||||
| Lowe 20165 | [18F]AV‐1451 | 3 MAPT (2 S305N, 1 N279K) | Visual assessment. PVC− | Low levels of diffuse increased signal in the two S305N mutation carriers, predominantly in white matter. Increased signal in basal ganglia of N279K carrier |
| Bevan Jones 201680 | [18F]AV‐1451 | 1 MAPT (IVS10 + 16), 12 controls | BPND referenced to superior cerebellar grey matter. PVC− | MAPT > C = inferior temporal lobe, temporal pole |
| Smith 201679 | [18F]AV‐1451 | 3 MAPT (R406W), 5 AD, 4 controls | SUVR referenced to cerebellar cortex. PVC‐ | MAPT > C = temporal pole, hippocampus, inferior temporal gyrus, frontal lobes |
| Spina 201778 | [18F]AV‐1451 | 1 MAPT (V337M), 20 controls | SUVR referenced to cerebellar grey matter. PVC− | MAPT > C = frontal pole, orbitofrontal cortex, inferior temporal lobe, insula, anterior cingulate, prefrontal cortex, lateral temporal lobe, striatum |
Analysis methods includes description of how the tau‐PET data was analyzed, which reference regions were used and whether partial volume correction (PVC) was utilized.
Abbreviations: Aβ, beta‐amyloid; AD, Alzheimer's disease; BPND, non‐displacable binding potential; CBS, corticobasal syndrome (clinical term); CBD, corticobasal degeneration (pathologically confirmed); MAPT, microtubule associated protein tau; MSA‐P, parkinsonian variant of multiple system atrophy; PD, Parkinson's disease; PDD, PD dementia; PD‐MCI, PD with mild cognitive impairment; PSP‐RS, progressive supranuclear palsy Richardson's variant; PSP‐P, progressive supranuclear palsy parkinsonism variant; ROI, region‐of‐interest; SUVR, standard uptake value ratio.
Parkinson's Disease and Related Disorders
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by tremor, stiffness, slowness, impaired balance, and shuffling gait. It is characterized neuropathologically by the deposition of alpha‐synuclein‐immunopositive Lewy neurites and Lewy bodies; and hence, at first glance, does not appear to be a useful target for tau‐PET imaging. Autoradiographic studies have consistently found no binding of [18F]AV‐1451 to alpha‐synuclein pathology in either Lewy body disease5, 6, 7 or PD.7 However, patients with PD can develop cognitive impairment and dementia. Patients that develop dementia a year or more after the onset of motor symptoms are often diagnosed with Parkinson's disease dementia (PDD), while those that develop dementia closer to the onset of motor symptoms, or before the onset of motor symptoms, are often diagnosed with Dementia with Lewy Bodies (DLB).9 Pathological studies have shown that coexistent AD pathology, characterized by both beta‐amyloid and hyperphosphorylated 3R/4R tau deposition occurs in both PDD10, 11, 12, 13 and DLB, occurring in up to 38% of PDD12 and 25% of DLB14 cases. In contrast, coexistent AD pathology is rare in PD in the absence of cognitive impairment.10, 12, 14 Given the fact that the tau ligands show strong binding to the tau pathology present in AD,5, 6, 7, 8 tau‐PET imaging provides a potentially valuable opportunity to detect the presence of AD‐related tau pathology during life and to investigate the influence of tau pathology on disease course and patient outcomes.
Consistent with expectations from autopsy findings, tau‐PET studies using [18F]AV‐1451 have observed no tau uptake in the cortex or basal ganglia in patients with PD that do not have cognitive impairment15, 16, 17 (Table 2). However, it has been shown that PD patients have reduced [18F]AV‐1451 uptake in the substantia nigra,16, 17 a finding that has been interpreted as reflecting loss of neuromelanin due to the progressive loss of dopaminergic neurons observed in PD. This hypothesis is supported by autoradiographic studies that have shown that [18F]AV‐1451 binds to neuromelanin.6
In contrast, increased cortical tau‐PET uptake has been observed in patients with PDD and DLB compared to healthy controls (Table 2). One study observed elevated [18F]AV‐1451 uptake in inferior and lateral temporal gyri and precuneus in PD patients with cognitive impairment (PD‐mild cognitive impairment and PDD) and DLB, although findings were more striking in DLB.15 There was some evidence that the PD patients with cognitive impairment and DLB patients showed greater uptake in these regions compared to PD patients without cognitive impairment, although this was only observed when a partial volume correction was performed on the PET data. They also found that uptake in these regions correlated with general cognitive performance. Uptake in inferior and lateral temporal gyri and precuneus were also elevated in DLB compared to controls in another study; although that study found that the regions of greatest uptake in DLB were actually located in the occipital lobe18 (Fig. 1). The finding of tau uptake in temporal and parietal regions fits well with the typical distribution of tau deposition at autopsy in AD,19 although the finding of high occipital tau uptake may suggest that the distribution of tau pathology in DLB is atypical,18 as an autopsy study has found.20 However, the relationship between these tau‐PET findings and beta‐amyloid PET findings were variable. While one study observed a correlation between severity of tau uptake and severity of beta‐amyloid deposition on PET,18 the other did not.15 Furthermore, both studies observed patients with elevated tau uptake that did not show elevated beta‐amyloid uptake15, 18 (Fig. 1), suggesting that tau can accumulate in the neocortex in the absence of beta‐amyloid. This contrasts to findings in AD where beta‐amyloid and tau are both typically elevated. The authors conclude that it is possible that in these cases tau deposition may precede beta‐amyloid deposition,18 or that soluble beta‐amyloid oligomers and non‐fibrillar amyloid in diffuse plaques could contribute to tau accumulation in these cases.15 Another possibility is that the tau‐PET findings may reflect off‐target binding. A further difference between these findings and those observed in AD was in the severity of tau uptake which was markedly less than that observed in AD at the group‐level, with only a small proportion of patients showing tau uptake in the range observed in AD patients18; some pathological studies have similarly observed a low proportion of PDD patients with AD pathology.13 Furthermore, a recent study did not find any evidence of elevated tau uptake in PD patients with mild cognitive impairment, with only one of nine patients showing any evidence for tau uptake.16 These discrepancies across studies and unexpected findings regarding the relationship with beta‐amyloid deposition point to the need for further tau‐PET studies using larger patient samples, and the critical need for autopsy studies to investigate the neuropathological underpinnings of elevated tau‐PET signal in these PD spectrum disorders.
Figure 1.
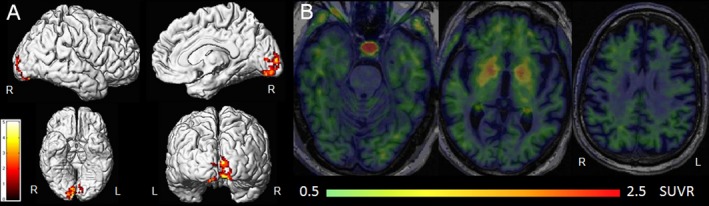
[18F]AV‐1451 findings in dementia with Lewy bodies. Group‐level patterns of tau‐PET uptake in 19 patients with dementia with Lewy bodies compared to controls are shown in (A). Elevated uptake is observed in the occipital lobe. These renders were kindly provided by Dr. Kantarci, Mayo Clinic, and modified from Kantarci et al. 2017.18 B: Mild elevated cortical tau‐PET uptake in a patient with Dementia with Lewy bodies that showed no beta‐amyloid uptake on Pittsburgh Compound B PET imaging.
Parkinson‐plus Syndromes
Progressive Supranuclear Palsy Syndromes
Progressive supranuclear palsy is a tauopathy associated with a predominance of 4‐repeat (4R) tau isoforms21 (Table 1). The most commonly recognized clinical presentation of PSP is Richardson's syndrome (PSP‐RS), in which patients have early and notable gait and postural instability, frequent falls, and abnormal vertical eye movements (supranuclear gaze palsy).22, 23, 24 However, a number of other clinical presentations of PSP have been described,24 including, PSP with predominant parkinsonism (PSP‐P),23 PSP with progressive gait freezing,25 PSP with predominant frontal presentation,26 PSP with a predominant speech/language disorder,27 and PSP with predominant corticobasal syndrome.28 Patients diagnosed clinically with PSP most commonly have an underlying PSP pathology,29, 30, 31, 32, 33 although other pathologies observed include CBD and MSA. The probability that the underlying pathology will be PSP is higher for patients that present with the typical symptoms of PSP‐RS.31 Tau‐PET imaging is particularly important in PSP as it is a pure tauopathy, concordance between syndrome and pathology is relatively good, and it tends to have a relatively young age at onset, which minimizes the probability of other comorbidities. Hence, patients with PSP are important targets for treatment trials testing tau‐modifying drugs. Given that PSP can present with a number of variant clinical syndromes, a diagnostic biomarker for PSP that can be identified early in the disease course is needed.
A number of studies have now assessed tau‐PET imaging using the [18F]AV‐1451 ligand in PSP patients, with the majority investigating patients clinically diagnosed with PSP‐RS (Table 2). A very consistent message has emerged across group‐level studies, with PSP‐RS showing elevated uptake particularly in subcortical structures, including midbrain, dentate nucleus of the cerebellum, thalamus, subthalamic nucleus, globus pallidus and striatum compared to controls34, 35, 36, 37, 38 (Fig. 2). Two studies have found increased uptake in frontal regions, including premotor and precentral regions, although it has been observed predominantly in white matter and it was less striking that the uptake in subcortical regions.36, 38 A few studies looked at how well tau in subcortical regions could differentiate PSP‐RS from controls.34, 37 The globus pallidus provided optimal differentiation in a couple of studies, with sensitivity and specificity of 93% in one study34 and 85% and 92% in the other.36 The globus pallidus also performed well in one other study, although optimum differentiation was achieved with the thalamus.37 These studies therefore suggest that subcortical regions have potential to be useful diagnostic biomarkers in PSP. It should be noted, however, that studies have observed a large degree of overlap between PSP‐RS patients and controls, which makes diagnosis difficult at the individual‐level. This is because off‐target binding in controls is observed in many of the PSP‐related regions (as discussed below). In fact, one study failed to observe any regions of increased [18F]AV‐1451 uptake in a small PSP‐RS cohort.39
Figure 2.
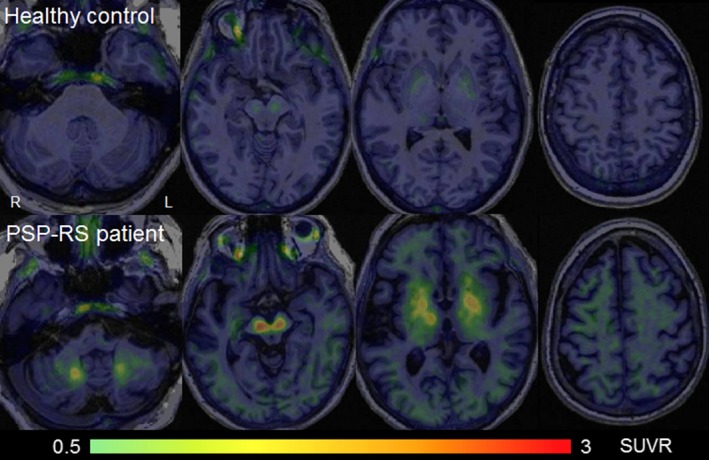
[18F]AV‐1451 tau PET images in a healthy control and PSP‐RS patient. Standard uptake value ratio images are shown for a 59 year old healthy control and a 64 year old PSP‐RS patient. The PSP‐RS patient shows elevated uptake in the dentate nucleus of the cerebellum, midbrain, thalamus, and basal ganglia, with very mild uptake observed in white matter underlying the cortex.
Similar patterns of uptake in globus pallidus and midbrain in PSP‐RS have been observed with [18F]THK‐5351, with weaker evidence for cortical uptake.40 One study using the [11C]PBB3 ligand assessed a cohort of PSP‐P patients.41 Similar to the previously mentioned studies, they identified elevated tau uptake in the putamen, midbrain, and globus pallidus compared to controls. However, they did not identify any elevated uptake in dentate nucleus of the cerebellum or thalamus. While one could hypothesize that these differences may relate to the clinical syndrome, as has been observed with [18F]AV‐1451,36 it is also possible that it may be due to the use of a different tau‐PET ligand, especially since these regions were also not identified in the [18F]THK‐5351 study.40 Studies comparing [11C]PBB3, [18F]THK‐5351, and [18F]AV‐1451, and also comparing PSP variants, will be needed to understand the implication of these findings.
There has been some disagreement across studies in whether tau uptake in subcortical regions correlates with disease severity in PSP, although the majority of studies have not found any correlations.34, 35, 36, 42 A relationship between tau‐PET uptake and disease severity has been observed in PSP‐P.41 There is also some disagreement regarding whether uptake is related to age in PSP, with one study finding a positive correlation between age and uptake in the striatum37 and another finding no correlation at all.34 There is evidence that tau uptake in PSP is not influenced by the presence of beta‐amyloid deposition in the brain.42 Differences across studies may be due to clinical differences in the cohorts or the small samples assessed (most published studies have ~10 patients). Larger studies will be needed to better determine clinical and demographic relationships with tau uptake.
Thus far, it is clear that PSP is associated with patterns of tau uptake on PET imaging that are relatively consistent across studies. However, there are a number of issues regarding the uptake properties of [18F]AV‐1451 that make interpretation of the findings in PSP patients difficult. First, autoradiographic studies have found no,6, 7, 35, 37, 43 or minimal,5, 8, 44 binding of [18F]AV‐1451 to 4R tau. In addition, a study that assessed [18F]AV‐1451 uptake in an autopsy‐confirmed patient with PSP found no correlation between tau‐PET uptake during life and tau burden in the brain at autopsy.45 These results suggest that the affinity of the ligand to 4R tau may be lower than to AD tau, possibly related to differences in tau isoforms or structural differences in tau filaments (Table 1). Autoradiographic studies using the PBB3 and THK‐5351 ligands have been more positive, with specific binding to 4R tau inclusions in PSP observed,40 showing better performance than [18F]AV‐1451.8 Second, studies using [18F]AV‐1451 have shown off‐target binding in healthy controls in many of the regions that are implicated in PSP, including the midbrain and basal ganglia,5 with uptake in these regions appearing to increase with age.34, 37 It has been hypothesized that [18F]AV‐1451 may be binding to iron in the basal ganglia,5, 46 or neuromelanin in the midbrain.6 Binding in basal ganglia has also been observed in healthy control patients with [18F]THK‐535147 and in patients with non‐tau diseases using [11C]PBB3,41 although autoradiographic studies have shown that off‐target binding in these regions is minimal with [11C]PBB3.8 Another issue regarding [18F]THK‐5351 is that recent studies have suggested that it binds to monoamine oxidase B (MAO‐B), and hence that interpretation of PET findings using this ligand will be confounded by the high MAO‐B availability across the brain.48 MAO‐B levels are affected by smoking,49 and hence smoking status could also be a confound. There is some evidence that MAO‐B may not be a confound in [18F]AV‐1451 studies,50 although more work needs to be done to evaluate this issue.
These studies therefore question whether the elevated tau uptake observed in the in vivo studies of PSP using [18F]AV‐1451 reflect true tau deposition. However, this is still unclear and it may be too early to discount [18F]AV‐1451 completely. First, the utility of autoradiographic studies has been questioned, with other PET ligands showing greater sensitivity in in vivo compared to in vitro studies.51 Second, the patterns of tau uptake match well with the post‐mortem distribution of pathology in PSP36 and match well with findings using the other tau ligands that show better performance in autoradiographic studies. Third, two autopsy studies have found good regional correlations between [18F]AV‐1451 uptake during life and tau burden in CBD, another 4R tauopathy.44, 52 One study has demonstrated that there is no neuromelanin in the basal ganglia of PSP patients,35 suggesting at least that neuromelanin does not explain uptake in this region. It is possible that [18F]AV‐1451 may be binding a very small fraction of the total 4R‐tau, resulting in the low signal on PET and the lack of findings on autoradiographic studies. Tau burden in PSP is typically much lower than that observed in AD, which shows striking findings on autoradiography. Further studies are clearly necessary in order to understand the origins of the elevated tau‐PET signal in PSP. It is important to note, however, that regardless of the underlying mechanism, tau‐PET imaging could still provide useful biomarkers in PSP if it performs well in differential diagnosis and if longitudinal measures correlate well with disease progression, akin to say a midbrain volumetric measurement. More work is needed to determine how well tau‐PET performs compared to other more established biomarkers in PSP.53
Corticobasal Syndrome
The term corticobasal syndrome describes a clinical syndrome that is characterized by progressive asymmetric cortical and extrapyramidal dysfunction, including ideomotor limb apraxia, cortical sensory loss, alien‐limb phenomenon, and myoclonus.54 The corticobasal syndrome is, however, pathologically heterogeneous and can be the presenting syndrome of a host of different pathologies including CBD, PSP, AD, and FTLD‐TDP.55, 56, 57, 58, 59 Pathological studies have shown that the most common pathology underlying corticobasal syndrome is CBD, followed by AD, then PSP, and then FTLD‐TDP.33, 55, 60, 61 Studies typically utilize beta‐amyloid PET imaging in order to identify corticobasal syndrome patients that likely have AD pathology; although little data is available on how well beta‐amyloid PET results relate to autopsy findings of AD pathology in corticobasal syndrome. Therefore, tau‐PET imaging has the potential to help investigate tau deposition related to CBD, but also to determine the relationship between beta‐amyloid and tau deposition in corticobasal syndrome.
Group studies have found a relatively consistent pattern of tau uptake on PET imaging in corticobasal syndrome patients that do not show beta‐amyloid deposition, and hence, they likely do not have AD pathology. These patients show tau uptake, albeit relatively mild, on [18F]AV‐1451 and [18F]THK‐5351 in precentral and postcentral cortex, superior frontal and parietal lobe, and putamen and globus pallidus62, 63, 64 (Table 2and Fig. 3). Uptake was not only observed in the grey matter of these regions, but also in the white matter underlying these regions, with patterns that are typically asymmetric, showing the greatest involvement in the hemisphere contralateral to the affected limb.62, 64 A similar pattern of uptake in neocortex and basal ganglia has been observed in a single patient scanned with [11C]PBB3.4 One study found evidence that [18F]AV‐1451 uptake in pre and postcentral cortex correlated to motor severity, as measured by the Unified Parkinson's Disease Rating Scale,62 suggesting a relationship between tau uptake and disease progression. The weakness of many of these studies, however, is that the underlying pathology of these patients is unclear, other than ruling out AD due to negative beta‐amyloid PET scans.62, 63, 64 In fact, one study found tau‐PET uptake in structures that typically show elevated tau levels in PSP, including thalamus, midbrain, and dentate nucleus of the cerebellum, suggesting the possibility of underlying PSP pathology in at least some of the patients.62
Figure 3.
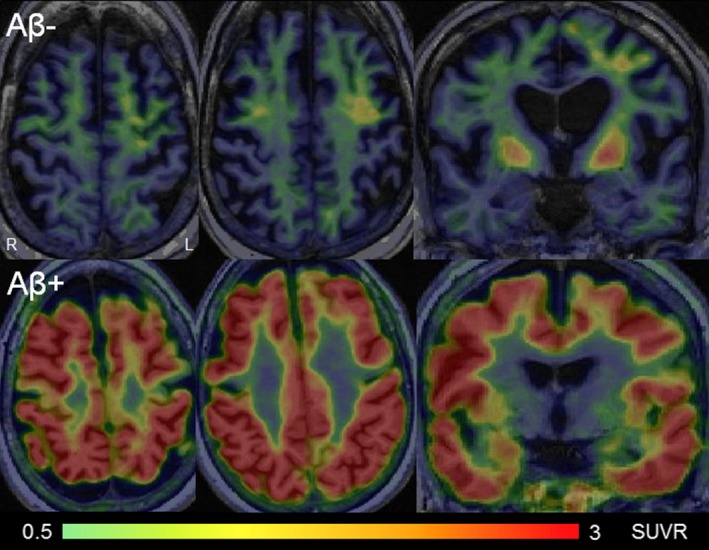
[18F]AV‐1451 tau PET images in corticobasal syndrome. The top panel shows standard uptake value ratio images for a 67‐year‐old corticobasal syndrome patient that showed no beta‐amyloid uptake on Pittsburgh Compound B PET imaging. This patient showed elevated uptake in premotor and motor cortices and the basal ganglia. The bottom panel shows standard uptake value ratio images for a 53‐year‐old corticobasal syndrome patient that showed elevated beta‐amyloid uptake on Pittsburgh Compound B PET imaging. This patient showed widespread and severe tau‐PET uptake throughout the cortex, consistent with underlying Alzheimer's disease pathology.
Tau‐PET findings using the [18F]AV‐1451 ligand have been reported in two patients that had autopsy confirmed CBD.44, 52 One patient presented with a primary progressive apraxia of speech65 later developed agrammatic aphasia and clinical features of corticobasal syndrome and showed elevated tau uptake in the putamen, pallidum, thalamus, precentral cortex, rolandic operculum, supplementary motor area, and left Broca's area.44 The other patient presented with PSP‐RS later developed features of corticobasal syndrome and showed elevated uptake in the bilateral substantia nigra, globus pallidus, and bilateral frontal and posterior temporal cortical regions, along with midbrain and pons.52 Patterns of uptake in these two patients overlapped to some degree with the patterns reported in corticobasal syndrome, with some regional differences that appear to be related to clinical differences between the patients. For example, involvement of the supplementary motor area and Broca's area may be related to the speech and language difficulties in the first patient, as is the case with structural MRI,66 and involvement of brainstem may be related to the PSP‐RS in the second patient. Importantly, both of these studies also showed that the degree of regional tau‐PET uptake across the brain correlated well with the regional distribution of tau pathology at autopsy in these two patients,44, 52 suggesting that the tau‐PET signal is reflecting underlying tau pathology in CBD. The fact that these correlations were observed despite autoradiographic studies finding little or no binding of [18F]AV‐1451 to tau in CBD, may suggest that the ligand is binding a small fraction of the total 4R‐tau burden, as is the case in PSP.44
Tau‐PET uptake has been reported in four corticobasal syndrome patients that showed beta‐amyloid deposition on PET scanning, across three studies.2, 63, 64 Three of these four patients also showed elevated tau uptake in the range one would expect in AD (Fig. 3), increasing confidence in the presence of underlying AD pathology in these patients.2, 64 Two of the three patients showed temporoparietal tau uptake in a pattern similar to AD64 and the other showed tau uptake in primary and association sensorimotor cortex, more akin to the patterns reported in corticobasal syndrome.2 However, in one study, one corticobasal syndrome patient with evidence for beta‐amyloid deposition did not show a visually different pattern or degree of tau‐PET uptake compared to four patients without beta‐amyloid deposition.63 The reason for the discrepancy in topographic pattern of tau uptake across these presumed corticobasal syndrome‐AD patients is unclear, and larger studies will be needed to help determine the relationship between AD pathology and patterns of tau‐PET uptake in corticobasal syndrome.
A number of studies have looked to see how well regional patterns of tau‐PET uptake correlate with regional patterns of grey matter atrophy or cortical thickness reduction in corticobasal syndrome. Generally, strong correlations have been observed between tau‐PET and atrophy in patients that show beta‐amyloid deposition.2, 64 However, findings have been more variable in corticobasal syndrome patients that do not show beta‐amyloid deposition, typically with only weak correlations observed.44, 52, 64 We could hypothesize that this difference is likely related to the degree of tau‐PET uptake; since cases with AD tend to show greater tau‐PET uptake and atrophy, and hence better correlations between metrics. In fact, one study made the observation that patterns of atrophy in beta‐amyloid negative corticobasal syndrome were much more striking than the patterns of tau‐PET uptake.64
Multiple System Atrophy
Multiple system atrophy is a rare neurodegenerative disease characterized pathologically by alpha‐synuclein deposits predominantly in oligodendroglial cytoplasmic inclusions.67 Patients present with a combination of cerebellar ataxia, autonomic symptoms, and features of parkinsonism, such as tremor, rigidity, and problems with muscle coordination and gait.68 Patients with the parkinsonian type (MSA‐P) present with parkinsonism as the most prominent symptom. Tau‐PET imaging using the [18F]AV‐1451 ligand has been reported in four patients with MSA‐P, with all four showing elevated uptake in the putamen compared to controls69 (Table 2). However, given that autoradiographic studies have convincingly demonstrated a lack of binding of [18F]AV‐1451 to alpha‐synuclein in multiple system atrophy,5, 6 the authors conclude that uptake likely reflects off‐target binding in this region. Tau‐PET imaging has also been reported using the [11C]PBB3 ligand in one MSA‐P patient.41 This patient showed widespread increased uptake in the cortex and subcortical structures.41 While intriguing (given that MSA is not a tauopathy), the findings are consistent with an autoradiographic study that observed binding of [11C]PBB3 to glial cytoplasmic inclusions in MSA cases with severe pathology.70 However, further work is clearly needed to understand the pathologic underpinnings of the tau‐PET signal in MSA.
Frontotemporal Dementia with Parkinsonism
Parkinsonism can occur in patients with frontotemporal dementia, particularly in patients with genetic mutations in the microtubule‐associated protein tau (MAPT) gene that is located on chromosome 17.71 These patients usually present with the behavioral variant of frontotemporal dementia, characterized by changes in behavior and personality, and an early age onset of disease onset. Multiple pathogenic mutations in MAPT have been identified that have different neuropathological underpinnings.72 Some mutations have a pure 4R tauopathy in the brain, some have a pure 3R tauopathy, and others have a mixed 3R/4R tauopathy with tau aggregates, showing similar confirmation and biochemical composition to neurofibrillary tangles in AD73, 74, 75, 76 (Table 1). Autoradiographic results in frontotemporal dementia with MAPT mutations have varied, likely related to variability across specific MAPT mutations (Table 1). Two autoradiographic studies both found a striking binding of [18F]AV‐1451 to tau in cases with the R406W mutation that has both 3R and 4R tau isoforms.5, 7 However, binding was absent to minimal in cases with the N279K and P301L mutations, which are both associated with 4R tau.5, 43 One of two cases with an exon 10 + 16 mutation showed binding to tau in the frontal and temporal lobes and the other showed no binding.7 The [11C]PBB3 ligand has also been shown to bind to tau in frontotemporal dementia cases with MAPT mutations characterized by 4R tau and in those characterized by 3R tau deposition.8 Patients with MAPT mutations typically show frontotemporal atrophy on MRI, with a particular focus on the temporal lobes.77 The MAPT mutations provide a great target for tau‐PET imaging, particularly as they are pure tauopathies and the familial nature allows assessments in presymptomatic disease stages.
Box 1. Main messages of the review.
Studies using tau‐PET imaging ligands [18F]AV‐1451, [18F]THK‐5351, and [11C]PBB3 have been performed across a wide range of parkinsonian disorders over the last year.
Elevated tau uptake in temporal, parietal, and occipital lobes has been observed in patients with Parkinson's disease that have cognitive impairment and dementia with Lewy bodies, likely related to concomitant Alzheimer's disease.
Progressive supranuclear palsy is associated with mild elevated tau uptake in subcortical structures, including midbrain, dentate nucleus of the cerebellum, thalamus, and globus pallidus.
Patients with corticobasal syndrome with biomarker evidence for Alzheimer's disease tend to show striking tau uptake in the range typically observed in Alzheimer's disease, with milder tau uptake observed in motor cortex in corticobasal syndrome without biomarker evidence for Alzheimer's disease.
Patients with frontotemporal dementia and mutations in the microtubule associated protein tau (MAPT) gene show variable results in tau‐PET studies, with mutations associated with 3 and 4‐repeat tau deposition tending to show high frontotemporal tau uptake, and mutations associated with 4‐repeat tau showing less striking uptake often located in the white matter.
Elevated tau uptake has also been observed in multiple system atrophy, a synucleinopathy.
The results of these clinical tau‐PET studies do not always agree with in vitro autoradiographic studies and therefore more work is needed to understand the biological substrate of elevated tau uptake in in vivo studies, particularly in the 4‐repeat tauopathies and synucleinopathies.
Given the limitations of the currently available ligands, there is a clear need for better and more specific tau PET ligands to study this group of disorders.
Tau‐PET imaging findings with the [18F]AV‐1451 ligand have been reported in a handful of patients with MAPT mutations (Table 2). Two studies assessed patients with mutations characterized by deposition of both 3R and 4R tau; one reported findings in one patient with the V337M mutation78 and the other reported findings in three patients with the R406W mutation.79 Elevated tau uptake in the temporal and frontal lobes and basal ganglia was associated with both of these MAPT mutations, matching patterns of atrophy and the pattern of tau deposition at autopsy in other V337M cases.78 A positive correlation between regional tau‐PET uptake and tau burden at autopsy in one of the R406W patients that died within 2 weeks of the PET scan also demonstrated that the [18F]AV‐1451 findings likely reflect underlying tau.79 The degree of tau‐PET uptake in the R406W patients was also in the range expected in AD, perhaps reflecting the pathological similarities between these diseases.
Tau‐PET findings using [18F]AV‐1451 have also been reported in patients with mutations that are characterized by deposition of 4R hyperphosphorylated tau, namely the S305N, N279K, and intron 10 + 16 mutations. Low levels of uptake were observed in two S305N patients and one N279K patient, with uptake mostly observed in the white matter.5 However, elevated tau‐PET uptake was observed in a patient with the intron 10 + 16 MAPT mutation.80 Tau‐PET uptake was observed in the inferior temporal lobe and temporal pole, although the degree of uptake was relatively mild compared to controls and appeared to focus in the white matter. The pathological explanation for the uptake in the white matter is currently unclear. Therefore, it appears as though tau‐PET imaging may be a useful biomarker in MAPT mutations, although possibly only in those associated with 3R/4R tauopathies.
Differential Diagnosis
Little data is currently available to assess the value of tau‐PET imaging to differentiate different parkinsonism syndromes. A couple of studies have compared PSP (a 4R tauopathy) with PD (a non‐tauopathy), showing that patients with PSP show greater [18F]AV‐1451 uptake in the substantia nigra, putamen, globus pallidus, subthalamic nucleus, and dentate nucleus compared to patients with PD.34, 36 The globus pallidus provided optimum separation of these diseases in both studies, with sensitivity of 93% and specificity of 100% in one study34 and sensitivity of 84.8% and specificity of 92.3% in another multi‐center study.36 There is therefore some evidence that tau‐PET could be useful to differentiate tauopathies from non‐tauopathies, although the biological basis for the signal in PSP is unclear and it is uncertain whether tau‐PET could be diagnostically useful when visually assessed in individual patients. Differences in tau uptake have also been observed between PSP‐RS and corticobasal syndrome in one study, with corticobasal syndrome showing greater uptake on [18F]AV‐1451 in the subcortical white matter underlying motor cortex.64 However, while these early investigations seem promising much more work is needed to validate and investigate differences among parkinsonian disorders in larger studies and it will be important to determine how well tau‐PET performs compared to other imaging metrics.
It is clear that the patterns of tau‐PET uptake in the pure 4R tauopathies of PSP and corticobasal syndrome differ from patterns reported in AD, with AD showing dramatically more uptake throughout the temporoparietal cortex than either PSP‐RS35, 38 or corticobasal syndrome.63, 64 Conversely, there is also evidence for differences in the opposite direction. Hence, PSP‐RS patients have been shown to have greater uptake in the midbrain,35, 38 dentate nucleus of the cerebellum,38 and thalamus38 than AD. Also, corticobasal patients have been shown to have greater uptake in the motor cortex and basal ganglia than AD.63, 64 These findings reflect the fact that subcortical structures and motor cortex are typically spared in AD. While the differential diagnosis of AD from these atypical parkinsonian disorders is not typically critical, these findings do demonstrate some disease specificity of tau‐PET.
Summary
A number of studies have been published over the last year assessing tau‐PET imaging in patients with parkinsonian disorders, with the vast majority assessing the [18F]AV‐1451 ligand. However, some issues that should be considered (Table 2) are that many, if not all, of these early studies had relatively small patient cohorts. There were also some methodological differences between them, with most studies analyzing the tau‐PET signal as a standard uptake value ratio (SUVR), although some analyzed quantitative dynamic imaging and binding potentials. The use of a cerebellar reference region was consistent across studies. A correction for the presence of atrophy using partial volume averaging was performed in some studies, but not all, although most studies found relatively consistent findings both with and without partial volume correction. How these methodological differences influenced variability across studies is currently unclear and more work is needed to compare analysis, techniques, diseases, and PET ligands.
The general message that emerged from these studies is that tau‐PET imaging using [18F]AV‐1451 shows a striking signal in patients suspected of having underlying AD type tau, and a milder signal in patients with 4R tauopathies such as PSP, corticobasal syndrome, and frontotemporal dementia with MAPT mutations. However, mildly elevated tau‐PET signal was also observed in some alpha‐synucleinopathies, including DLB and MSA‐P. Many of these findings do not concur with autoradiographic studies and there are also limitations due to potential off‐target binding, hence it will be important for future work to help understand the nature of binding in these diseases. Given these limitations, there is a clear need for better and more specific tau PET ligands to study this group of disorders. New tau‐PET ligands that may show higher affinity to 4R tau, and show fewer issues with off‐target binding are currently being developed and may prove valuable in these parkinsonian disorders.
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: NIH, grant numbers: R01‐NS89757, R01‐DC12519, R01‐AG50603, R01‐AG37491, and R21‐NS94684. The author declares that there are no conflicts of interest relevant to this work.
Financial Disclosures for the previous 12 months: The author declares that there are no additional disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Dani M, Brooks DJ, Edison P. Tau imaging in neurodegenerative diseases. Eur J Nucl Med Mol Imaging 2016;43:1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xia C, Makaretz SJ, Caso C, et al. Association of in vivo [18F]AV‐1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 2017;74:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tago T, Furumoto S, Okamura N, et al. Structure‐activity relationship of 2‐arylquinolines as PET imaging tracers for tau pathology in Alzheimer disease. J Nucl Med 2016;57:608–614. [DOI] [PubMed] [Google Scholar]
- 4. Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013;79:1094–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV‐1451 Tau PET in dementia. Acta Neuropathol Commun 2016;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F‐18]‐AV‐1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sander K, Lashley T, Gami P, et al. Characterization of tau positron emission tomography tracer [18F]AV‐1451 binding to postmortem tissue in Alzheimer's disease, primary tauopathies, and other dementias. Alzheimers Dement 2016;12:1116–1124. [DOI] [PubMed] [Google Scholar]
- 8. Ono M, Sahara N, Kumata K, et al. Distinct binding of PET ligands PBB3 and AV‐1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017;140:764–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boller F, Mizutani T, Roessmann U, Gambetti P. Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Ann Neurol 1980;7:329–335. [DOI] [PubMed] [Google Scholar]
- 11. Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J Neural Transm (Vienna) 2002;109:329–339. [DOI] [PubMed] [Google Scholar]
- 12. Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic accumulation of alpha‐synuclein and Abeta in Parkinson disease patients with dementia. Arch Neurol 2012;69:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 2008;115:427–436. [DOI] [PubMed] [Google Scholar]
- 15. Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 2016;73:1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hansen AK, Damholdt MF, Fedorova TD, et al. In vivo cortical tau in Parkinson's disease using 18F‐AV‐1451 positron emission tomography. Mov Disord 2017;32:922–927. [DOI] [PubMed] [Google Scholar]
- 17. Hansen AK, Knudsen K, Lillethorup TP, et al. In vivo imaging of neuromelanin in Parkinson's disease using 18F‐AV‐1451 PET. Brain 2016;139(Pt 7):2039–2049. [DOI] [PubMed] [Google Scholar]
- 18. Kantarci K, Lowe VJ, Boeve BF, et al. AV‐1451 tau and beta‐amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017;81:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 20. Walker L, McAleese KE, Thomas AJ, et al. Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 2015;129:729–748. [DOI] [PubMed] [Google Scholar]
- 21. Dickson DW, Hauw JJ, Agid Y, Litvan I. Progressive supranuclear palsy and corticobasal degeneration In: Dickson D, Weller RO, eds. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2nd ed Chichester, UK: Wiley‐Blackwell; 2011:135–155. [Google Scholar]
- 22. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 23. Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP‐parkinsonism. Brain 2005;128(Pt 6):1247–1258. [DOI] [PubMed] [Google Scholar]
- 24. Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 2007;22:2235–2241. [DOI] [PubMed] [Google Scholar]
- 26. Hassan A, Parisi JE, Josephs KA. Autopsy‐proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase 2012;18:478–488. [DOI] [PubMed] [Google Scholar]
- 27. Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129(Pt 6):1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsuboi Y, Josephs KA, Boeve BF, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord 2005;20:982–988. [DOI] [PubMed] [Google Scholar]
- 29. Josephs KA, Dickson DW. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord 2003;18:1018–1026. [DOI] [PubMed] [Google Scholar]
- 30. Osaki Y, Ben‐Shlomo Y, Lees AJ, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord 2004;19:181–189. [DOI] [PubMed] [Google Scholar]
- 31. Respondek G, Kurz C, Arzberger T, et al. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord 2017;32:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Respondek G, Roeber S, Kretzschmar H, et al. Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord 2013;28:504–509. [DOI] [PubMed] [Google Scholar]
- 33. Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho H, Choi JY, Hwang MS, et al. Subcortical 18 F‐AV‐1451 binding patterns in progressive supranuclear palsy. Mov Disord 2017;32:134–140. [DOI] [PubMed] [Google Scholar]
- 35. Passamonti L, Vazquez Rodriguez P, Hong YT, et al. 18F‐AV‐1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain 2017;140:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schonhaut DR, McMillan CT, Spina S, et al. 18 F‐flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann Neurol 2017;82:622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith R, Schain M, Nilsson C, et al. Increased basal ganglia binding of 18 F‐AV‐1451 in patients with progressive supranuclear palsy. Mov Disord 2017;32:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whitwell JL, Lowe VJ, Tosakulwong N, et al. [18 F]AV‐1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord 2017;32:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coakeley S, Cho SS, Koshimori Y, et al. Positron emission tomography imaging of tau pathology in progressive supranuclear palsy. J Cereb Blood Flow Metab 2017;37:3150–3160. 10.1177/0271678X16683695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ishiki A, Harada R, Okamura N, et al. Tau imaging with [18 F]THK‐5351 in progressive supranuclear palsy. Eur J Neurol 2017;24:130–136. [DOI] [PubMed] [Google Scholar]
- 41. Perez‐Soriano A, Arena JE, Dinelle K, et al. PBB3 imaging in parkinsonian disorders: evidence for binding to tau and other proteins. Mov Disord 2017;32:1016–1024. [DOI] [PubMed] [Google Scholar]
- 42. Whitwell JL, Ahlskog JE, Tosaulwong N, et al. Pittsburgh Compound B and AV‐1451 positron emission tomography assessment of molecular pathologies of Alzheimer's disease in progressive supranuclear palsy. Parkinsonism Relat Disord 2017; Dec 13. 10.1016/j.parkreldis.2017.12.016 [Epub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marquie M, Normandin MD, Meltzer AC, et al. Pathological correlations of [F‐18]‐AV‐1451 imaging in non‐Alzheimer tauopathies. Ann Neurol 2017;81:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Josephs KA, Whitwell JL, Tacik P, et al. [18F]AV‐1451 tau‐PET uptake does correlate with quantitatively measured 4R‐tau burden in autopsy‐confirmed corticobasal degeneration. Acta Neuropathol 2016;132:931–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith R, Scholl M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG‐PET, but not AV‐1451‐PET, in progressive supranuclear palsy. Acta Neuropathol 2017;133:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Choi JY, Cho H, Ahn SJ, et al. “Off‐Target” 18F‐AV‐1451 binding in the basal ganglia correlates with age‐related iron accumulation. J Nucl Med 2018;59:117–120. [DOI] [PubMed] [Google Scholar]
- 47. Lockhart SN, Baker SL, Okamura N, et al. Dynamic PET measures of tau accumulation in cognitively normal older adults and Alzheimer's disease patients measured using [18F] THK‐5351. PLoS One 2016;11:e0158460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ng KP, Pascoal TA, Mathotaarachchi S, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F‐THK5351 uptake in the human brain. Alzheimers Res Ther 2017;9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fowler JS, Logan J, Wang GJ, Volkow ND. Monoamine oxidase and cigarette smoking. Neurotoxicology 2003;24:75–82. [DOI] [PubMed] [Google Scholar]
- 50. Hansen AK, Brooks DJ, Borghammer P. MAO‐B inhibitors do not block in vivo flortaucipir([18F]‐AV‐1451) binding. Mol Imaging Biol 2017;79:110. [DOI] [PubMed] [Google Scholar]
- 51. Ikawa M, Lohith TG, Shrestha S, et al. 11C‐ER176, a radioligand for 18‐kDa translocator protein, has adequate sensitivity to robustly image all three affinity genotypes in human brain. J Nucl Med 2017;58:320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McMillan CT, Irwin DJ, Nasrallah I, et al. Multimodal evaluation demonstrates in vivo 18F‐AV‐1451 uptake in autopsy‐confirmed corticobasal degeneration. Acta Neuropathol 2016;132:935–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whitwell JL, Hoglinger GU, Antonini A, et al. Radiological biomarkers for diagnosis in PSP: where are we and where do we need to be? Mov Disord 2017;32:955–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800. [DOI] [PubMed] [Google Scholar]
- 56. Hu WT, Rippon GW, Boeve BF, et al. Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 2009;24:1375–1379. [DOI] [PubMed] [Google Scholar]
- 57. Shelley BP, Hodges JR, Kipps CM, Xuereb JH, Bak TH. Is the pathology of corticobasal syndrome predictable in life? Mov Disord 2009;24:1593–1599. [DOI] [PubMed] [Google Scholar]
- 58. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 60. Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whitwell JL, Jack CR Jr, Boeve BF, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology 2010;75:1879–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cho H, Baek MS, Choi JY, et al. 18F‐AV‐1451 binds to motor‐related subcortical gray and white matter in corticobasal syndrome. Neurology 2017;89:1170–1178. [DOI] [PubMed] [Google Scholar]
- 63. Kikuchi A, Okamura N, Hasegawa T, et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F‐THK5351 PET. Neurology 2016;87:2309–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Smith R, Scholl M, Widner H, et al. In vivo retention of 18F‐AV‐1451 in corticobasal syndrome. Neurology 2017;89:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Josephs KA, Duffy JR, Strand EA, et al. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain 2012;135(Pt 5):1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Whitwell JL, Duffy JR, Strand EA, et al. Distinct regional anatomic and functional correlates of neurodegenerative apraxia of speech and aphasia: an MRI and FDG‐PET study. Brain Lang 2013;125:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy‐Drager syndrome). J Neurol Sci 1989;94:79–100. [DOI] [PubMed] [Google Scholar]
- 68. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cho H, Choi JY, Lee SH, Ryu YH, Lee MS, Lyoo CH. 18 F‐AV‐1451 binds to putamen in multiple system atrophy. Mov Disord 2017;32:171–173. [DOI] [PubMed] [Google Scholar]
- 70. Koga S, Ono M, Sahara N, Higuchi M, Dickson DW. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to alpha‐synuclein pathology. Mov Disord 2017;32:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 72. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 2015;41:24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spillantini MG, Crowther RA, Goedert M. Comparison of the neurofibrillary pathology in Alzheimer's disease and familial presenile dementia with tangles. Acta Neuropathol 1996;92:42–48. [DOI] [PubMed] [Google Scholar]
- 74. Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology 1992;42:120–127. [DOI] [PubMed] [Google Scholar]
- 75. Mott RT, Dickson DW, Trojanowski JQ, et al. Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 2005;64:420–428. [DOI] [PubMed] [Google Scholar]
- 76. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, et al. Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science 1998;282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 77. Whitwell JL, Jack CR Jr, Boeve BF, et al. Atrophy patterns in IVS10 + 16, IVS10 + 3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology 2009;73:1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau‐PET and pathology correlations. Neurology 2017;88:758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Smith R, Puschmann A, Scholl M, et al. 18F‐AV‐1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016;139(Pt 9):2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bevan Jones WR, Cope TE, Passamonti L, et al. [18F]AV‐1451 PET in behavioral variant frontotemporal dementia due to MAPT mutation. Ann Clin Transl Neurol 2016;3:940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Coakeley S, Cho SS, Koshimori Y, et al. [18F]AV‐1451 binding to neuromelanin in the substantia nigra in PD and PSP. Brain Struct Funct 2017. [Epub] [DOI] [PubMed] [Google Scholar]
- 82. Hammes J, Bischof GN, Giehl K, et al. Elevated in vivo [18F]‐AV‐1451 uptake in a patient with progressive supranuclear palsy. Mov Disord 2017;32:170–171. [DOI] [PubMed] [Google Scholar]