

Abstract
Secale L. is a small but important genus that includes cultivated rye. Although genetic diversity of cultivated rye is high, patterns of genetic diversity in the whole genus, and potential factors affecting the distribution of genetic diversity remain elusive. The population structure and distribution of genetic variation within Secale, and its correlation with taxonomic delimitation, cultivation status or spatial distribution in relation to geography and climate zones were analyzed in this study. A collection of 726 individual plants derived from 139 different accessions representing Secale cereale, S. vavilovii, S. strictum, and S. sylvestre were investigated using SSR analysis and sequence diversity analysis of a nuclear EST region. Our results indicated that perennial S. strictum subspecies are genetically divergent from annual forms of the genus. Existence of two distinct clusters within the annual taxa was observed, one corresponding to samples from Asia, and a second to those outside of Asia. No clear genetic structure was observed between different annual species/subspecies, indicating introgression between these taxa. The analysis of cultivated rye revealed that landrace populations from the Middle East have the highest genetic diversity, supporting the idea of the area being the center of origin for cultivated rye. Considering high adaptive potential of those populations, Middle Eastern landraces should be regarded as genetic resources reservoirs for new niches and future breeding programs.
Introduction
Sustainable food production is a vital environmental issue, in the context of global climate change. Elevated temperatures and accompanying alterations in precipitation regimes are expected to decrease yields significantly. At the same time, global requirement for food is expected to increase by 60% by 2050. The adaptive capacity of plant populations under stress conditions are positively related to the degree of genetic diversity maintained in those populations [1]. Genetic diversity of modern varieties (cultivars) of crop plants is quite low due to genetic erosion stemming from domestication syndrome and modernization bottlenecks. On the other hand, wild relatives of crop plants and unimproved varieties known as ‘landraces’ are genetically diverse [2,3] and contain many adaptive alleles in their gene pools.
Secale L. is a small but economically important taxon, which includes such wild relatives and landraces, comprising cultivated rye, containing annual, perennial, self-incompatible or self-compatible, cultivated, weedy and wild taxa [4]. Although the genus is regarded as the typical representative of Mediterranean flora and Southwest Asia, specifically Turkey, Lebanon, Syria, Iran, Iraq, and Afghanistan are the main centers of its distribution [5]. Taxonomy of the genus Secale is still not without contention, due to disagreements on the delimitation of species and intraspecific taxa, the out-crossing nature of many species, and lack of hybridization barriers between species and subspecies. Hence, regarding the taxonomical classification and phylogeny of Secale species, several hypotheses were proposed [5–8]. Recent molecular findings [9–11] are consistent with Frederiksen and Petersen`s [7] hypothesis that the genus is represented by three species: S. sylvestre, S. strictum and S. vavilovii. There is a general agreement on S. strictum, the perrenial species being the ancestral form [10,11]. The first species that diverged from S. strictum was proposed to be S. sylvestre [9,12,13]. S. strictum subsp. africanum is considered to have diverged from S. strictum during the early Pleistocene and evolved independently [13]. S. cereale and S. vavilovi, on the other hand, are considered to be evolutionarily the youngest species [12,14]. Although S. vavilovi was classified as a distinct species based on morphological differences [6,8,15], recent molecular studies revealed no clear difference between S. vavilovii and S. cereale [10,11,13,16], and suggested that S. vavilovii should be ranked as a subspecies within the cereale group.
For the ancestry of cultivated rye, S. cereale subsp. cereale, different researchers have different opinions as well: S. vavilovii [17,18], S. ancestrale [19], and S. strictum [20] were suggested to be the progenitor of cultivated rye. Similarly, the exact geographical center of origin for cultivated rye is not known, but south-western Asia was proposed to be most probably the center of origin [5,18]. Although production of cultivated rye declined worldwide during the 20th century, it has long been an important crop in Northern and Central Europe, especially in the cooler parts that is not suitable for cultivation of other cereals [21]. Rye has a cross-pollinating reproductive system and thus its levels of intraspecific diversity are high compared to self-pollinating grains [22]. Although, cultivated varieties of cereal rye has been experiencing extensive genetic bottleneck [23] due to strong selection pressure, like many other cereal crops, significant proportion of genetic diversity is maintained in landraces [24,25] and wild and weedy forms [26,27]. Furthermore, these populations contain potentially useful traits such as resistance to diseases, adaptability to biotic and abiotic stress [28,29]. Therefore, considering that the wild and weedy forms may crossbreed with cultivated rye [30], these taxa, along with the landraces, constitute gene pools for desirable genes, and can be regarded as genetic resource reservoirs for new niches and future breeding programs of wheat, triticale and other crops [22]. Hence understanding genetic structuring of the genus Secale and distribution of genetic diversity within the genus is extremely important. In this vein, with this study, we investigated wild, weedy, landrace and cultivated varieties of rye using Simple Sequence Repeats (SSRs) and sequence diversity analysis of a nuclear Expressed Sequence Tag (EST) region in order to obtain further insights about taxonomy, phylogeny, and genetic structure of Secale species. Specifically we evaluated the correlation of genetic structure with taxonomic delimitation, cultivation status or spatial distribution in relation to geography and climate zones [31] at a global scale.
Materials and methods
Plant material and DNA extraction
In this study, a total of 726 samples belonging to 139 different accessions of cultivated varieties, landraces, weedy and wild populations of Secale were investigated from 45 different countries (S1 Table). Among these, 584 samples from 100 accessions of S. cereale, 46 samples from nine accessions of S. vavilovii, 89 samples from 23 accessions of S. sitrictum, two samples from two accessions of S. sylvestre, and five hybrid samples were used (S1 Table). In terms of cultivation status, 137 genotypes belonged to wild accessions, 51 genotypes to weedy accession, 343 genotypes to landrace accessions and 195 genotypes to cultivated accessions.
The accessions were provided by United States Department of Agriculture Germplasm Resources Information Network (USA), and Leibniz Institut für Pflanzengenetik und Kulturpflanzenforschung (Germany). Two accessions were collected in farms from Turkey, in 2010. In order to confirm taxonomic delimitations of accessions, seeds were planted in trial fields from December 2010 to June 2011, and the samples were regularly evaluated for certain phenotypic characters during all developmental stages following [7]. Total DNA was extracted according to the method described by Doyle and Doyle [32].
SSR analysis
Initially, 20 nuclear SSR primers previously used in the genus Secale [33,34] were screened in eight individual plants, representing four Secale species in terms of PCR amplification success and peak profiles. Among these, a set of ten microsatellite primers yielding good PCR products and scorable peaks were selected (REMS1187, REMS1254, REMS1323, REMS1264, REMS1205, REMS1238, REMS1160, REMS1303, REMS1259 and SCM 180) and used for the analysis of 721 samples (S2 Table). All PCR reactions were performed as described by Khlestkina et al. [33] and Saal and Wricke [34]. Amplification success was checked and successful PCR products were read on an ABI 3100 capillary sequencer with GS400HD size standard (Applied Biosystems).
The alleles were automatically binned using FlexiBin [35] and checked manually. The genotyping errors stemming from null alleles, large allele dropout or the scoring of stutter peaks that can potentially lead to deviations from Hardy–Weinberg proportions were detected using Micro-checker version 2.2 [36]. Based on the results, three markers (REMS1303, REMS1259 and SCM 180) were and seven SSR markers (REMS1187, REMS1254, REMS1323, REMS1264, REMS1205, REMS1238, REMS1160) were used for the subsequent analyses. The mean polymorphism information content (PIC) was calculated for each marker using MolKin v.3.0 software [37].
We analyzed the whole data set, consisting of 721 samples, excluding the hybrids, in three different categories on the basis of (1) taxonomic identity, (2) cultivation status and (3) climatic conditions of geographical origin. In the first category, all of the genotypes were pooled into 11 groups based on their taxonomic identity, at the species and subspecies level, in order to understand the distribution of genetic diversity in different taxonomic groups. In the second category, all genotypes were grouped as wild, weedy, landrace and cultivated varieties, to evaluate the effect of cultivation status on the distribution of genetic diversity. In the third category, all genotypes excluding two samples of unknown geographical origin were assigned to 18 climate subgroups belonging to five main climate groups, as determined by Köppen-Geiger classification system, which is based on classifying the mean climate conditions on geographic areas around the globe using different climatic variables [31], in order to understand whether climatic conditions of geographic origins affect distribution of genetic diversity.
In addition to these three categories, patterns of genetic diversity in cultivated rye i.e. S. cereale subsp. cereale genotypes (both landrace and cultivated varieties were analyzed separately. In this analysis, a total of 533 genotypes from 83 different accessions originating from various geographical regions, representing 10 main gene pools (Africa, Australia, Europe, Balkans, Caucasus, East Asia, South and Central Asia, Middle East, North America and South America) were used. These samples were analyzed using the same seven microsatellite primers, as described above.
Allelic frequencies were tested for the deviations from Hardy–Weinberg equilibrium (HWE) using an exact test with a Markov chain (10000 steps) and 1000 dememorisation steps in Genepop version 4.0.10 [38,39]. Linkage disequilibrium was also tested between all loci using Genepop version 4.0.10 [38,39]. Genetic diversity parameters were computed for each group using GenAlEx v6.4 [40]. The sample sizes in different accessions/regions used in the study were different from each other. Therefore, to compensate for this sampling bias that may lead to inaccurate comparisons of allelic richness between loci, allelic richness (RS) and private allele richness (PR), independent from sample size were computed by a rarefaction method as implemented in HP-RARE version 1.0 [41]. In addition to these genetic diversity parameters, the overall gene diversity (HT), the within-population genetic diversity (HS), the amount of gene diversity among populations (DST), and the coefficient of genetic differentiation between populations (GST) were calculated with FSTAT version 2.9.3 [42]. To avoid any misinterpretation stemming from sampling bias, DST, HT and GST values were also calculated independently of sample size, using the same program. Pairwise FST values between each population were calculated using GenAlEx version 6.4 [40] Population structure was also analyzed using a Bayesian clustering algorithm, as implemented in STRUCTURE version 2.3.3 [43]. Admixture model of ancestry and correlated allele frequency were allowed. The LOCPRIOR model was also applied using population information as a prior, to assist clustering [44]. The length of the burn-in was set to 30,000, and data were collected over 300,000 Markov Chain Monte Carlo (MCMC) replications in each run (K = 1–5). The optimum number of clusters (K), was determined as described by Evanno et al. [45]. Each individual with an ancestry value equal to or larger than 0.7 was assigned to the corresponding cluster, while the individuals with a smaller ancestry value were considered to have mixed ancestry following Coulon et al. [46]. The correspondences of obtained groups were evaluated for taxonomic identity, cultivation status, geographical origin, and climatic zones, as mentioned above. Finally, an Unweighted Pair Group Method with Arithmetic Mean (UPGMA) tree was constructed using Poptree2 [47] based on Nei’s genetic distance (DA) [48] with 10,000 bootstrap iterations.
Sequence diversity analysis of nuclear EST markers
Varshney et al. [49] had shown that existing barley nuclear expressed sequence tag (EST)-derived DNA markers could be employed in sequence diversity analysis in rye. Four of these markers were tested (S2 Table) and GBS0551, which gave the best results, was selected and used in this study. A total of 61 samples representing four species of Secale and five hybrid samples were included in the analysis. The PCR reactions were performed as described by Varshney et al. [49]. The amplified fragments were commercially sequenced at Macrogen Europe and the sequences were edited visually and aligned using Sequencher version 4.5 (Gene Code Corp). However, the discrimination of the alleles of heterozygote samples, especially with multiple differences was not straightforward. Therefore, these sequences were edited by DNAsp version 5.0 [50] using the coalescent-based Bayesian algorithm of PHASE software [51] that resolves haplotype phases and infers haplotypes correctly. A maximum-likelihood (ML) tree was constructed using MEGA 5 [52], and the reliability of the phylogenetic relationships was tested by bootstrapping (1000 replicates).
Results
Informativeness of the SSR markers
The number of alleles per locus ranged between 9 and 22 with an average value of 14. Polymorphism information content (PIC) values ranged from 0.605 (REMS1264) to 0.882 (REMS1160), with an average value of 0.718 (Table 1).
Table 1. Levels of genetic variability at the seven microsatellite loci.
| Locus | N | Na | Ne | PIC | Ho | He |
|---|---|---|---|---|---|---|
| REMS1187 | 659 | 9 | 3.09 | 0.676 | 0.825 | 0.676 |
| REMS1254 | 541 | 17 | 3.25 | 0.692 | 0.698 | 0.692 |
| REMS1323 | 660 | 22 | 3.93 | 0.745 | 0.85 | 0.746 |
| REMS1264 | 644 | 11 | 2.54 | 0.605 | 0.651 | 0.606 |
| REMS1205 | 600 | 11 | 3.14 | 0.681 | 0.733 | 0.681 |
| REMS1238 | 665 | 9 | 3.94 | 0.745 | 0.768 | 0.746 |
| REMS1160 | 576 | 19 | 8.48 | 0.882 | 0.858 | 0.882 |
| Average | 621 | 14 | 4.05 | 0.718 | 0.769 | 0.718 |
N, sample size; Na, number of alleles; Ne, number of effective alleles; PIC, Polymorphism information content; Ho, observed heterozygosity; He, expected heterozygosity; uHe, unbiased expected heterozygosity.
Distribution of SSR genetic diversity in different categories
Genetic allelic patterns were calculated for each group, in the three categories created based on taxonomic identity, cultivation status, and climatic conditions of geographical origin of the samples (Table 2). In the taxonomy based groups observed heterozygosity was higher than the expected heterozygosity in all taxa. Expected heterozygosity was the highest in S. strictum subsp. strictum (0.731) and the lowest in S. cereale subsp. afghanicum (0.579), excluding S. cereale subsp. dighoricum, S. strictum subsp. irmanuso and S. sylvestre that had small sample sizes. The highest and lowest differentiation based on FST was observed between S. cereale subsp. afghanicum and S. sylvestre (FST = 0.181), and S. cereale and S. vavilovii (FST = 0.007), respectively (S3 Table). S. cereale subsp. afghanicum and S. sylvestre were found to be the most divergent from the rest of the taxa analyzed.
Table 2. Mean genetic diversity measures in different panels.
| Taxonomic Identity | Group | N | Na | I | Ho | He |
| S. cereale subsp. afghanicum | 3 | 3.14 | 1.00 | 0.67 | 0.58 | |
| S. cereale subsp. ancestrale | 11 | 5.14 | 1.34 | 0.72 | 0.67 | |
| S. cereale subsp. cereale | 533 | 13.00 | 1.57 | 0.77 | 0.71 | |
| S. cereale subsp. dighoricum | 1 | 1.14 | 0.40 | 0.57 | 0.29 | |
| S. cereale subsp. segetale | 36 | 7.43 | 1.55 | 0.80 | 0.72 | |
| S. strictum subsp. anatolicum | 13 | 5.43 | 1.32 | 0.84 | 0.66 | |
| S. strictum subsp. irmanuso | 1 | 1.29 | 0.40 | 0.57 | 0.29 | |
| S. strictum subsp. kuprijanovii | 6 | 4.14 | 1.25 | 0.94 | 0.67 | |
| S. strictum subsp. strictum | 69 | 8.71 | 1.63 | 0.71 | 0.73 | |
| S. sylvestre | 2 | 2.29 | 0.74 | 0.79 | 0.48 | |
| S. vavilovii | 46 | 7.86 | 1.53 | 0.81 | 0.71 | |
| Cultivation Status | Wild | 137 | 11.29 | 1.68 | 0.78 | 0.74 |
| Weedy | 51 | 8.43 | 1.58 | 0.76 | 0.73 | |
| Landrace | 343 | 12.86 | 1.61 | 0.78 | 0.72 | |
| Cultivar | 190 | 9.29 | 1.44 | 0.76 | 0.68 | |
| Climate Type | Tropical monsoon | 6 | 2.57 | 0.77 | 0.67 | 0.46 |
| Savanna | 20 | 5.00 | 1.28 | 0.88 | 0.67 | |
| Hot semi-arid | 11 | 4.43 | 1.18 | 0.75 | 0.62 | |
| Cold semi-arid | 69 | 9.14 | 1.58 | 0.72 | 0.71 | |
| Hot desert | 9 | 3.86 | 1.05 | 0.72 | 0.57 | |
| Cold desert | 15 | 5.43 | 1.36 | 0.73 | 0.67 | |
| Humid subtropical | 64 | 8.43 | 1.51 | 0.78 | 0.71 | |
| Temperate oceanic | 140 | 9.86 | 1.55 | 0.77 | 0.71 | |
| Hot-summer Mediterranean | 88 | 10.86 | 1.61 | 0.76 | 0.72 | |
| Warm-summer Mediterranean | 78 | 10.00 | 1.62 | 0.76 | 0.73 | |
| Subtropical highland | 9 | 3.71 | 1.08 | 0.82 | 0.60 | |
| Hot-summer humid continental | 22 | 6.43 | 1.48 | 0.85 | 0.71 | |
| Warm-summer humid continental | 71 | 8.14 | 1.53 | 0.77 | 0.71 | |
| Subarctic | 19 | 5.71 | 1.33 | 0.81 | 0.67 | |
| Hot, dry-summer continental | 11 | 5.00 | 1.19 | 0.72 | 0.60 | |
| Warm, dry-summer continental | 68 | 8.71 | 1.53 | 0.81 | 0.70 | |
| Monsoon-influenced humid continental | 15 | 4.86 | 1.19 | 0.75 | 0.61 | |
| Mild tundra | 4 | 3.14 | 1.00 | 0.86 | 0.58 |
N, sample size; Na, number of alleles; I, Information Index Ho, observed heterozygosity; He, expected heterozygosity; uHe, unbiased expected heterozygosity.
The assessment of cultivation status based genetic diversity in 137 wild, 51 weedy, 343 landrace, and 190 cultivated plants showed that expected heterozygosity was the highest in wild accessions (0.735) and the lowest in cultivated varieties (0.675). Comparison of pairwise FST values revealed no significant differentiation between different groups.
In terms of the climate subgroups, the highest expected heterozygosity was observed in the Warm-summer Mediterranean subgroup (0.73), and the lowest in Tropical monsoon climate subgroup (0.46) (Table 2). Pairwise FST comparisons revealed the Tropical monsoon climate subgroup to be the most different from the remaining climate subgroups, with the highest genetic distance when compared to the Mild tundra climate populations (0.19) (S4 Table).
STRUCTURE and UPGMA results
The Bayesian clustering analysis based on the distribution of 98 alleles at seven SSR loci among 721 accessions revealed presence of three separate clusters (Fig 1). The primary division at K = 2 was observed mainly between perennial S. strictum and remaining annual taxa. At K = 3, S. strictum cluster remained fairly intact, while annual taxa (S. cereale and S. vavilovi) grouped within two different clusters. The first cluster contained a total of 25 samples of S. strictum subsp. strictum and one S. strictum subsp. anatolicum sample. Among these, 20 samples originated from Iran, and the remaining samples originated from other parts of the Middle East (Fig 2A). The second cluster included 222 samples of S. cereale (202 samples of S. cereale subsp. cereale, 13 samples of S. cereale subsp. segetale, six samples of S. cereale subsp. ancestrale and one sample of S. cereale subsp. afghanicum). All of the six S. strictum subsp. strictum samples in cluster 2 originated from Russia. Geographical origins of majority of the Secale cereale samples (70.17%) in this cluster corresponded to the Middle East or South-Central Asia (Fig 2B). The third cluster was composed of 66 S. cereale subsp. cereale and three S. vavilovi samples, most of which (86.4%) originated from out of Asia and the Middle East (Fig 2C). The remaining 408 samples could not be assigned to any of these three clusters, and was considered to have mixed ancestry. Except for the first cluster that consisted of wild S. strictum samples, a weak correlation between clustering and cultivation status was noted. The structuring exhibited no significant correlation with major agro-climatic zones as described by Kottek et al [31].
Fig 1. STRUCTURE results at K = 2 to K = 5.

Fig 2. Distribution of samples in different clusters.
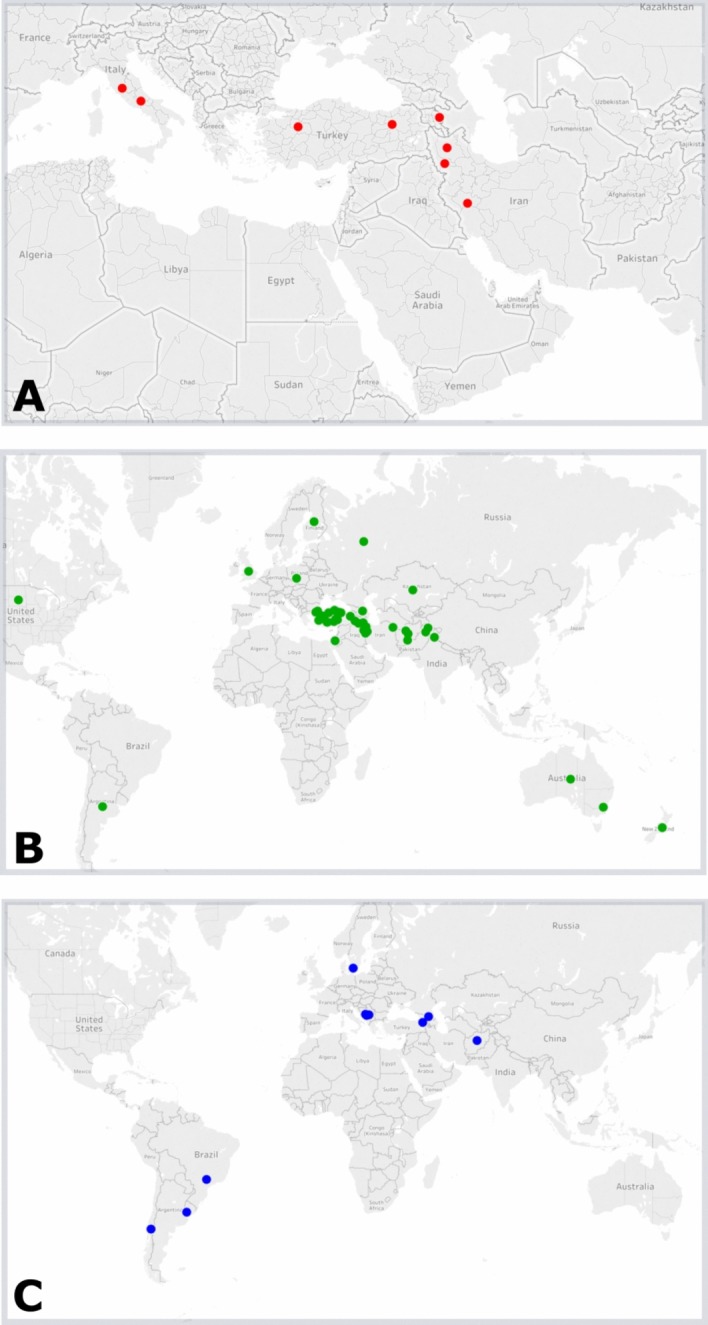
(A) Distribution of samples in cluster 1. (B) Distribution of samples in cluster 2. (C) Distribution of samples in cluster 3. This figure produced using Tableau Public Software.
The UPGMA dendogram constructed using subspecies of S. cereale, subspecies of S. strictum, S. vavilovii and S. sylvestre revealed a clear separation between S. sylvestre and the rest of genus (Fig 3A). S. cereale subsp. afghanicum separated from the other subspecies of S. cereale and S. strictum. S. strictum subsp. kuprijanovii also diverged from the other remaining subspecies at a relatively basal position in the tree topology. S. cereale subsp. cereale, S. cereale subsp. segetale, S. vavilovii, S. strictum subsp. anatolicum and S. strtictum subsp. strictum constituted a group, while S. cereale subsp. ancestrale remained outside of this cluster. S. cereale subsp. cereale and S. cereale subsp. segatale were more closely related to S. vavilovii, rather than their conspecifics S. cereale subsp. afghanicum and S. cereale subsp. ancestrale.
Fig 3. UPGMA dendograms showing the phylogenetic relationship of Secale species based on pairwise DA.

(A) UPGMA dendogram I. (B) UPGMA dendogram II. Bootstrap values are provided on the nodes.
As the STRUCTURE analysis revealed a clear separation between S. strictum subsp. strictum samples originating from Iran, as a next step, these populations were grouped separately (S. strictum subsp. strictum clade 1). The remaining S. strictum populations were also grouped together (S. strictum subsp. strictum clade 2), and the dendogram was rebuilt using these separated groups (Fig 3B). Branching off of S. strictum subsp. strictum clade 2 with a high bootstrap value (99%) revealed its significant divergence. Except for this difference, both trees reflected nearly identical topologies.
Nuclear sequence diversity of the genus Secale
The general topology of the maximum-likelihood (ML) tree constructed using a 667 bp fragment of nuclear sequences in 61 samples (GenBank accession numbers: MH421898-MH421958), representing four species in the genus Secale, and five hybrid samples showed that there were two main lineages (Fig 4). However, these groups did not correspond to taxonomic or spatial delimitations. S. vavilovii accessions were dispersed within S. cereale subspecies in both groups. The two S. sylvestre samples clustered together in a subgroup, rather than forming a separate linage. S. strictum subspecies clustered together forming two and one subgroups within the first and second lineage, respectively. Furthermore, a separated subgroup was recovered within the first linage containing S. cereale and S. vavilovii samples from South and Central Asia, except for a S. cereale subsp. cereale sample originating from Argentina. In the second group, no clear relationships were recognized between the genealogy of Secale species and their geographic origin.
Fig 4. Maximum likelihood phylogenetic tree based on the nuclear GBS0551 region.

Microsatellite based genetic diversity and population structure of the cultivated rye (Secale cereale subsp. cereale)
In the genetic diversity analysis conducted exclusively in landrace and cultivated rye samples, the total number of alleles and private alleles were 96 and 26, respectively (Table 3). The highest mean corrected allelic richness was found in the Middle East populations (3.68), and lowest in the Africa population (2.76). Private allelic richness independent of sample size was also the highest in the Middle Eastern populations (0.35), and lowest in South America and South-Central Asia populations (0.14). The observed heterozygosity levels were higher than expected heterozygosity levels in all geographical regions. The mean expected heterozygosity was highest in the Middle East gene pool (0.72), and lowest in the African gene pool (0.51). Shannon’s information index (I) was again highest in the Middle Eastern populations (1.62), and lowest in African population (0.85). The corrected total genetic diversity (HT′) in cereal rye showed variations from region to region, and found to be highest in the Middle East (0.74) and Caucasus (0.71), and lowest in East Asia (0.62). Intra-population genetic diversity (HS), the measure of average differences within populations was found to be the highest in the Middle East and Caucasus populations (0.70), and lowest in North American populations (0.59).
Table 3. Levels of genetic variability at seven microsatellite loci for cultivated rye.
| Region | N | NA | RS | Ne | I | Ho | PA | PR | He | Hs | HT | HT′ | DST | DST′ | GST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Africa | 6 | 3.14 | 2.76 | 2.17 | 0.85 | 0.65 | 0.00 | 0.18 | 0,51 | Nd | Nd | Nd | Nd | Nd | Nd |
| Australia | 32 | 5.86 | 3.13 | 3.38 | 1.25 | 0.74 | 0.29 | 0.20 | 0,63 | 0.64 | 0.65 | 0.66 | 0.01 | 0.01 | 0.01 |
| Balkans | 62 | 7.14 | 3.44 | 3.69 | 1.43 | 0.80 | 0.29 | 0.21 | 0,68 | 0.63 | 0.70 | 0.71 | 0.07 | 0.08 | 0.10 |
| Caucasus | 14 | 5.29 | 3.53 | 3.24 | 1.34 | 0.80 | 0.00 | 0.34 | 0,68 | 0.70 | 0.71 | 0.73 | 0.02 | 0.03 | 0.02 |
| East Asia | 31 | 5.71 | 3.19 | 2.95 | 1.22 | 0.73 | 0.00 | 0.21 | 0,58 | 0.62 | 0.62 | 0.62 | 0.00 | 0.01 | 0.01 |
| Europe | 36 | 6.71 | 3.46 | 3.58 | 1.42 | 0.80 | 0.00 | 0.21 | 0,68 | 0.65 | 0.68 | 0.69 | 0.03 | 0.04 | 0.05 |
| Middle East | 240 | 12.43 | 3.68 | 4.06 | 1.62 | 0.77 | 2.86 | 0.35 | 0,72 | 0.70 | 0.74 | 0.74 | 0.04 | 0.04 | 0.06 |
| N. America | 22 | 5.71 | 3.32 | 3.20 | 1.30 | 0.77 | 0.14 | 0.17 | 0,65 | 0.59 | 0.64 | 0.67 | 0.05 | 0.08 | 0.08 |
| South America | 43 | 6.00 | 3.42 | 3.29 | 1.36 | 0.82 | 0.00 | 0.14 | 0,67 | 0.65 | 0.68 | 0.69 | 0.03 | 0.04 | 0.05 |
|
South and
Central Asia |
47 | 6.43 | 3.20 | 3.27 | 1.29 | 0.74 | 0.14 | 0.14 | 0,63 | 0.64 | 0.66 | 0.66 | 0.01 | 0.02 | 0.02 |
N, sample size; NA, number of alleles; RS, allelic richness; NE, number of effective alleles; PR, number of private alleles independent of sample size; I, Shannon’s information index; HO, observed heterozygosity; HE, expected heterozygosity; PR, private allelic richness; HS, the within population genetic diversity; HT’, the total genetic diversity independent of sample size; DST′ among-populations genetic diversity independent of sample size; GST′ the coefficient of genetic differentiation independent of sample size.
The comparison of coefficient of among-populations genetic diversity independent of sample sizes (DST′), and coefficient of gene differentiation independent of sample sizes (GST′) as the measures of genetic differentiation between populations in each region was found to be highest in North America (DST′ = 0.08, GST′ = 0.12) and the Balkans (DST′ = 0.08, GST′ = 0.11), and lowest in East Asia (DST′ = 0.01, GST′ = 0.01) (Table 3). Furthermore, comparison of pairwise FST differentiation [53, 54] showed the African gene pool to be the most different from remaining gene pools, having the highest genetic distance when compared to the South-Central Asian populations (FST = 0.96) (S5 Table). The genetic differentiation among other gene pools was insignificant.
STRUCTURE analysis showed the presence of two separate clusters, with the first one composed of 333 samples, 72.02% of which were landraces that originated from the Middle East, and South and Central Asia. Except for two samples, all of the Australian cultivars clustered in this group. The second cluster was composed of 136 samples, mainly originating from Europe, Balkans and South America. The proportion of Middle Eastern and south Central Asian samples in this group was only 6.25%.
In the PCA analysis conducted to explore pattern of relationship between cultivated rye populations from different geographical regions with the microsatellite data, the first, second and third components explained 45.44%, 23.37% and 12.54% of the variance, respectively. First and second components of the PCA analysis revealed two clusters (Fig 5A), while three distinct clusters were observed based on the first and third components (Fig 5B). The first cluster was dominated by samples from the Middle East, whereas the other clusters contained samples from diverse geographical areas. PCA clustering did not reflect cultivation status of the samples.
Fig 5. PCA analysis of S. cereale subsp. cereale samples with different geographical origins.

(A) 1st and 2nd components. (B) 1st and 3rd components.
Discussion
Distribution of SSR diversity in Secale
In this study, genetic diversity within Secale was evaluated using seven SSR markers. A world-wide collection of 721 samples belonging to 11 taxonomic units included wild, weedy, landrace and cultivated materials from diverse climatic zones. All of the SSR markers employed in the study had (PIC) values higher than 0.6 and are considered to be highly informative. The genetic diversity ofSecale at a global scale was relatively high compared to other crops like sorghum [55] and maize [56], which can be attributable to the outcrossing nature of many species in the genus Secale, and its wind-pollinated reproduction.
In our study, relative genetic diversity co-varied with the cultivation status: the highest diversity was observed in wild accessions, followed by weedy and landrace accessions, and lowest in cultivated varieties. Furthermore, wild and landrace populations had private alleles which were not detected in the cultivated gene pools, indicating that these forms offer a richer source of alleles, and high potential for crop improvement. High genetic diversity and presence of private or rare alleles in wild and weedy forms can be explained by the lack of a domestication bottleneck, see below.
Genetic clustering
Existence of three distinct clusters in the STRUCTURE analyses of the whole genus indicated the presence of three different gene pools: (1) perennial S. strictum subsp. strictum, (2) annual taxa that originated from Asia (Middle East and South-Central Asia), (3) annual taxa that originated from outside of Asia (mainly Europe). The clear separation between perennial S. strictum and the annual form has been shown previously [11,57,58] and can be explained by restricted gene-flow between annual and perennial taxa, possibly due to the differences in life-history traits such as timing of reproduction.
Further separation of annual taxa was based on geographic origin, rather than taxonomic identity. This was also supported by maximum likelihood tree constructed using nuclear sequences, where all of the S. cereale subspecies and S. vavilovii were grouped together. Recently, Hagenblad et al. [11] showed that there was no clear taxonomic structuring among annual forms of the genus. Previous studies have also shown lack of morphological [59] and molecular [8,9,10,57] differences between annual forms (S. vavilovii, S. cereale subsp. ancestrale, S. cereale subsp. afghanicum, and S. cereale subsp. segetale) belonging to different taxa. Genetic similarity between annual wild and weedy forms and cultivated subspecies S. cereale subsp. cereale supports the hypothesis that S. cereale is of relatively recent origin, dating back to only a few centuries ago [60]. It is likely that there was insufficient time for the evolution of isolation mechanisms or barriers between cultivated rye, and its wild and weedy relatives, and hence the lack of structuring among annual taxa can be explained by introgression between sympatric populations of cultivated rye, and wild and weedy forms. As a result, morphological differences between the subspecies cannot be explained by genetic differentiation indicating the lack of nonexistence of the taxonomic boundaries at subspecies level. Interbreeding between different taxa, except for S. sylvestre and subsequent formation of hybrids with high pollen and seed fertility is very common in the Secale genus [6,20,61,62].
The further structuring of annual taxa based on geographic origins of samples suggests that each of the two annual clusters detected in the study originated from two distinct gene pools. Subsequently, the two distinct lineages retrieved in this study were initially separated, probably due to restriction of gene flow because of geographical isolation. Consistent with our findings, Hagenblad et al. [11] also showed that geographic clustering was evident among annual taxa, which is reflected by a separation between Asian and European accessions. Furthermore, Bolibok-Bragoszewska et al. [63] noted divergence of the Near Eastern and European accessions. On the other hand, some other studies reported that genetic structuring among different taxa corresponded to cultivation status [58,64–66], and no geographical structuring was found in these studies. These conflicting results might be stemming from low discriminatory power of markers used in these studies. It should be noted that despite the existence of the clear geographical structuring among annual taxa mentioned above, no significant correlation with major agro-climatic zones was detected. Similarly, Hagenblad et al. [11] also reported a limited correlation between genetic structuring and agro-climatic conditions of the sampling localities in geographically structured rye populations. This can be explained by the observed structure stemming from geographical proximity and related pollen dispersal, rather than ecological and climatic adaptations.
The nominotypic S. strictum subsp. strictum has been previously shown to be significantly different from its subspecies [8,9,10, 67], indicating that it has been evolving independently of other S. strictum subspecies [13]. In our study S. strictum subsp. strictum samples from northwest and west of Iran were divergent from the rest of the S. strictum accessions. In the UPGMA dendogram, the ancestral position of this group was observed, when compared to the rest of S. strictum subsp. strictum samples. The same dendogram also revealed that rest of the S. strictum subsp. strictum samples (i.e. other than the Iranian clade) originating from diverse areas, were genetically more similar to S. cereale accessions. This is compatible with the hypothesis that cultivated rye evolved from S. strictum [17,20,60]. In addition, in the dendogram constructed based on microsatellite data, S. strictum subsp. anatolicum and S. strictum subsp. strictum accessions originating from out of Iran were found to be closer to S. cereale subspecies compared to S. strictum subsp. kuprijanovii samples, and S. strictum subsp. strictum accessions originating from Iran. This suggests existence of gene flow between S. cereale subspecies and S. strictum subspecies originating from outside of Iran.
It was also interesting that S. strictum subsp. strictum samples that originated from Iran were basal to the clade that included the rest of S. strictum subspecies and all of the annual taxa, except for S. cereale subsp. afghanicum and S. sylvestre. This observation is also consistent with previous studies that show S. strictum being the most ancestral species, which the rest of the taxa have originated from[4,6,20, 68,69]. This finding also underpins the hypothesis that Northeastern Turkey and the adjacent area including Armenia and northwestern Iran could be the center of origin for the genus [5,18].
Taxonomical position of S. vavilovii has also been a point of discussion: in some studies, S. vavilovii was considered to be a distinct species close to S. cereale [6,8,15,57,58, 70], while some other researchers postulated that S. vavilovii should be classified as a subspecies of S. cereale [7,9–11,13,67]. In our study, SSR and nuclear sequence diversity analysis did not reveal significant differentiation between S. cereale and S. vavilovii. Therefore, it is concluded that S. vavilovii should be considered as a synonym and a subspecies of S. cereale.
Our study affirmed that S. sylvestre is genetically the most divergent species, which is consistent with the general agreement that S. sylvestre is the first species that diverged from S. strictum during the Pliocene, and is morphologically and genetically the most distinct species [12,13,71].
Genetic diversity and structuring of cultivated rye
Genetic diversity
Cultivated rye is a wind pollinated allogamous species with a highly developed self-incompatibility system. As a result, high genetic diversity has been previously noted not only between different accessions [24,72,73], but also within the same cultivar [9,10,65]. Consistently, our study showed high degrees of genetic diversity in cultivated rye from all over the world. Moreover, due to its high tolerance of different environmental conditions, rye has a global geographic distribution which may also have contributed to its high levels of genetic diversity.
In the scope of the present study, genetic diversity levels of different gene pools were compared. Our results showed that landraces are genetically more diverse, when compared to cultivars. Similar results were previously reported in other studies on rye [23,61]. It is well established that current breeding practices narrows genepool and leads to reduction of genetic diversity [74]. Such reduction in genetic diversity results in loss of many important alleles, and this may have significant negative effects on adaptive capacity of plants. On the other hand, landraces are cultivated by traditional agricultural practices through many generations of selection, and they have become locally adapted to various environments by accumulating new alleles [75]. Therefore, compared to cultivars, the genetic diversity of landraces is high. Our findings highlighted that landraces should be regarded as a source of genetic variation, and should be integrated to rye breeding programs to compensate genetic diversity lost during modern breeding processes.
Second, we analyzed the distribution of genetic diversity in different geographic regions. Genetic diversity of cultivated rye was affected by geographic origins of the samples and found to be higher in the Middle East region (Turkey, Iran and Israel) compared to other regions. Although sample size of the region is larger than the others, to avoid any bias due to sample size, corrected genetic diversity measures (independent of sample size) were also used. The degree of genetic diversity was found to be highest in the Middle East for the corrected parameters, as well. Therefore the obtained results likely reflect real genetic diversity patterns of the region, rather than being a sampling artifact. Vavilov [17] proposed that genetic diversity of crop species on interspecific and intraspecific level is not evenly distributed: the genetic diversity in the center of origins is higher. Based on this assumption, our results indicate that the most likely center of origin for the genus is the Middle East or Caucasus. This is consistent with the idea that all Secale taxa have originated somewhere in the Middle East or South-Central Asia [5,18] that also covers the geographical area known as “Fertile Crescent”, the center of origin for many crop species like wheat, barley, pulses, pea and flaxes [18]. Taking into account that many wild and weedy forms of the genus Secale are found in the area between northeastern Turkey and northwestern Iran, gene flow between wild forms and cultivated forms by introgression is quite possible, resulting in an increase in genetic diversity. High genetic diversity observed in the region can also be explained by most of the populations in this region being landraces, rather than genetically more-or-less uniform cultivars.
Genetic diversity of the accessions originating from Africa and East Asia was comparably low, probably due to a potential genetic bottleneck during introduction of cultivated rye to these regions. Besides, in comparison to South American and European samples, genetic diversity was lower in North America that contains populations from Mexico, USA and Canada. This probably stems from extensive use of genetically uniform cultivars in these regions. On the other hand, genetic diversity of the Balkan group (that contained samples from European part of Turkey (Thrace), Montenegro, Serbia, Macedonia, Yugoslavia, and Bosnia and Herzegovina) and the Caucasus gene pool (that consisted of two accessions from Georgia and East Azerbaijan) was high. Finally, the European gene pool sampled in this study contained accessions from a wide geographical range containing Germany, Switzerland, UK, Poland and Sweden, and their genetic diversity levels were found to be moderately high. Although agricultural systems of many countries in Europe favor the genetically uniform cultivars [76], the relatively high levels of genetic diversity observed could be explained by European cultivars having been developed using different genepools.
Origins of the ryes from different continents
The separation of genotypes originating from Asia (Middle Eastern, and south and central Asian) and from out of Asia (mainly Europe, Balkans and South America) was consistent with previous studies reporting a clear separation between the Middle East and European genepools [11,23,63].
Based on our findings it can be speculated that each of the two clusters obtained in the study originated from two distinct gene pools. The two main distinctive lineages retrieved in this study were initially separated probably due to restriction of gene flow because of geographical separation. Considering that in crop plants geographical distribution patterns usually reflect prevailing human mediated selection pressures in a particular environment [77], another explanation for this separation could be the cultivated rye having been introduced into new geographical ranges in which climatic and environmental conditions are quite different compared to those in the center of origin. This was possibly followed by anthropogenic selection of adaptable phenotypes to the conditions in those regions, leading to adaptive divergence. The Middle Eastern samples were observed in all three clusters, indicating their potential ancestral position, and supporting the conclusion -based on genetic diversity levels above- that the Middle Eastern populations are the likely progenitors of cultivated rye, and they recently expanded globally due to human mediated distribution and long-distance gene flow. Similarly, Einkorn wheat, emmer wheat, barley and lentil [78] were domesticated in the Middle East, more specifically in the Fertile Crescent and subsequently were radiated to Europe [79] and the rest of the world.
In the context of the study, the origin of the samples collected from outside of Asia and Europe was also investigated. Samples from South America grouped together with European samples into second cluster. This is consistent with the idea that many crop plants dispersed to South America from Europe, after the voyages of Columbus [80]. On the other hand, samples from Australia and North America grouped into the first cluster, indicating that cultivated rye was possibly introduced into these areas from the Middle East or South-Central Asia.
Furthermore, genetic differentiation among geographical regions revealed a significant differentiation between the African gene pool and the remaining gene pools. Considering climatic conditions of the region being relatively unique and that the region is physically separated from remaining gene pools by geographical barriers, it can be concluded that rye became locally adapted to this continent and remained separated. This is consistent with the idea that S. cereale subsp. cereale evolved as an isolated population in Africa [5]. Similarly, based on AFLP data, Chikmawati [73] previously reported that African populations of cultivated rye were genetically more distant when compared to other populations.
Conclusion
The global scale analysis of genetic diversity and phylogenetic relationships of Secale genus show a clear separation between perennial S. strictum subspecies and annual taxa. Further separation of annual taxa belonging to different species or subspecies into two groups was based on geographical origin, rather than taxonomic identity. Separation of the Middle Eastern and South Central Asian accessions from remaining accessions confirmed the previous findings revealing partitioning between Asian and European accessions, and the existence of two different genepools. The lack of any structuring within different species or subspecies belonging to annual taxa can be explained by recent separation of the species or subspecies, insufficient time having passed for the evolution of isolation mechanisms, and consequent continuation of gene flow even between species. In addition, the lack of a clear genetic separation between S. cereale and S. vavilovii led us to conclude that S. vavilovii, rather than being a distinct species, should be classified as a subspecies of S. cereale. The phylogenetic relationships of different species in the genus should be investigated in greater detail using high resolution molecular markers, such as RAD-seq, as well.
The evaluation of genetic diversity of cultivated rye populations led us to conclude that high levels of genetic variation exist in cultivated rye. The highest allelic variation and genetic diversity was found in the Middle Eastern landrace populations. This finding supports the idea that the area could be the center of origin for the genus. Nearly all of the populations examined in Near East are locally adapted landraces that have not been exposed to intense artificial selection pressures. Therefore, in contrast to modern crop varieties that have undergone genetic bottlenecks associated with the process of domestication, resulting in a decrease in genetic diversity, landraces constitute a large pool of genetic variation and contain many interesting traits, like strong tolerance to abiotic and biotic stress [81]. Considering that high genetic diversity in crop plant populations is directly related to adaptive potential of those populations to changing environmental conditions, landraces should be regarded as genetic resources reservoirs for new niches and future breeding programs. From a conservation point of view, the results obtained from the study suggest that an immediate action plan is required for in-situ conservation of the ancestral and highly diverse Middle Eastern landrace populations.
Supporting information
Am, Tropical monsoon climate; Aw, Savanna; BSh, Hot semi-arid; BSk, Cold semi-arid; BWh, Hot desert; BWk, Cold desert; Cfa, Humid subtropical climate; Cfb,Temperate oceanic climate; Csa, Hot-summer Mediterranean climate; Csb, Warm-summer Mediterranean climate; Cwb, Subtropical highland climate; Dfa, Hot-summer humid continental climate; Dfb, Warm-summer humid continental climate; Dfc, Subarctic climate; Dsa Hot, dry-summer continental climate; Dsb, Warm, dry-summer continental climate; Dwa, Monsoon-influenced hot-summer humid continental climate; ET, Mild tundra climate.
(DOCX)
Chr, Chromosome number; Temp, Annealing Temperature (°C); ref., Reference.
(DOCX)
S. c. afg., S. cereale subsp. afghanicum; S. c. anc., S. cereale subsp. ancestrale; S. c. cereale, S. cereale subsp. cereale; S. c. dighor., S. cereale subsp. dighoricum; S. c. segetale, S. cereale subsp. segetale; S. s. anatol, S. strictum subsp. anatolicum; S. s. irm. S. strictum subsp. irmanuso; S. s. kupr., S. strictum subsp. kuprijanovii; S. s. strictum, S. strictum subsp. strictum; S. sylv., S. sylvestre; S. vav., S. vavilovii. Significant P values are indicated in bold.
(DOCX)
Am, Tropical monsoon climate; Aw, Savanna; BSh, Hot semi-arid; BSk, Cold semi-arid; BWh, Hot desert; BWk, Cold desert; Cfa, Humid subtropical climate; Cfb,Temperate oceanic climate; Csa, Hot-summer Mediterranean climate; Csb, Warm-summer Mediterranean climate; Cwb, Subtropical highland climate; Dfa, Hot-summer humid continental climate; Dfb, Warm-summer humid continental climate; Dfc, Subarctic climate; Dsa Hot, dry-summer continental climate; Dsb, Warm, dry-summer continental climate; Dwa, Monsoon-influenced hot-summer humid continental climate; ET, Mild tundra climate.
Significant P values are indicated in bold.
(DOCX)
Significant P values are indicated in bold.
(DOCX)
Acknowledgments
We acknowledge Dr. Emrah Coraman and Pahar Patlar for their support during the data analysis and production of figures. Seed materials were kindly provided by National Small Grains Collection of United States Department of Agriculture and Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany. We acknowledge the financial support of the German Research Foundation (DFG) and the Open Access Publication Fund of Bielefeld University for the article processing charge. We wish to express our sincere gratitude to the reviewers, for extremely valuable suggestions and comments.
Data Availability
All of the 61 DNA sequence data generated in this study are available from the GenBank database (accession numbers MH421898-MH421958).
Funding Statement
Research Fund of Boğaziçi University in Istanbul to RB, Grant No: 09S101, 1903 and 11Y00P2 German Research Foundation (DFG) and the Open Access Publication Fund of Bielefeld University for the article processing charge to OM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Tanksley SD. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science. 1997;277(5329):1063–6. Available from:http://www.sciencemag.org/cgi/doi/10.1126/science.277.5329.1063 [DOI] [PubMed] [Google Scholar]
- 2.Tang S, Knapp SJ. Microsatellites uncover extraordinary diversity in native American land races and wild populations of cultivated sunflower. Theor Appl Genet. 2003;106(6):990–1003. Available from: http://link.springer.com/10.1007/s00122-002-1127-6 10.1007/s00122-002-1127-6 [DOI] [PubMed] [Google Scholar]
- 3.Reif JC, Xia XC, Melchinger AE, Warburton ML, Hoisington DA, Beck D, et al. Genetic diversity determined within and among CIMMYT maize populations of tropical, subtropical, and temperate germplasm by SSR markers. Crop Sci. 2004;44(1):326–34. [Google Scholar]
- 4.Vences FJ, Vaquero F, Garcia P, De Vega MP. Further studies on phylogenetic relationships in Secale: On the origin of its species. Plant Breed. 1987;98(4):281–91. [Google Scholar]
- 5.Sencer HA, Hawkes JG. On the origin of cultivated rye. Biol J Linn Soc. 1980;13(4):299–313. [Google Scholar]
- 6.Khush GS. Cytogenetic and evolutionary studies in Secale. II. Interrelationships of the wild species. Evolution (N Y). 1962;16(4):484–96. [Google Scholar]
- 7.Frederiksen S, Petersen G. A taxonomic revision of Secale (Triticeae, Poaceae). Nord J Bot. 1998;18(4):399–420. [Google Scholar]
- 8.Hammer K. Breeding system and phylogenetic relationships in Secale L. Biol Zent Bl. 1990;109(1):45–50. [Google Scholar]
- 9.Shang HY, Wei YM, Wang XR, Zheng YL. Genetic diversity and phylogenetic relationships in the rye genus Secale L. (rye) based on Secale cereale microsatellite markers. Genet Mol Biol. 2006;29(4):685–91. [Google Scholar]
- 10.Ren TH, Chen F, Zou YT, Jia YH, Zhang HQ, Yan BJ, et al. Evolutionary trends of microsatellites during the speciation process and phylogenetic relationships within the genus Secale. Genome [Internet]. 2011;54(4):316–26. Available from: http://www.nrcresearchpress.com/doi/abs/10.1139/g10-121 10.1139/g10-121 [DOI] [PubMed] [Google Scholar]
- 11.Hagenblad J, Oliveira HR, Forsberg NEG, Leino MW. Geographical distribution of genetic diversity in Secale landrace and wild accessions. BMC Plant Biol. 2016;16(1):23 Available from: http://www.biomedcentral.com/1471-2229/16/23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skuza L., Rogalska S.M., Bocianowski J. RFLP analysis of mitochondrial DNA in the genus Secale. Acta Biol Cracoviensia. 2007;49(1):77–8. [Google Scholar]
- 13.Achrem M, Kalinka A, Rogalska SM. Assessment of genetic relationships among Secale taxa by using ISSR and IRAP markers and the chromosomal distribution of the AAC microsatellite sequence. Turk J Botany. 2014;38(2):213–25. [Google Scholar]
- 14.Chikmawati T, Skovmand B, Gustafsson JP. Phylogenetic relationships among Secale species revealed by amplified fragment length polymorphisms. Genoma. 2005;48:792–801. [DOI] [PubMed] [Google Scholar]
- 15.Cuadrado a, Jouve N. Distribution of highly repeated DNA sequences in species of the genus Secale. Genome. 1997;40(3):309–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9202411 [DOI] [PubMed] [Google Scholar]
- 16.Santos E, Matos M, Silva P, Figueiras AM, Benito C, Pinto-Carnide O. Molecular diversity and genetic relationships in Secale. J Genet. 2016;95(2):273–81. [DOI] [PubMed] [Google Scholar]
- 17.Vavilov NI. Studies on the origin of cultivated plants. Bull Appl Bot. 1926;1–248. [Google Scholar]
- 18.Zohary D, Hopf M. Domestication of Plants in the Old World third ed Univ Press Oxford; 2000;531(12):206. [Google Scholar]
- 19.Zhukovsky PM (1933). La Turguie Agracole: 274–275 Leningrad [Google Scholar]
- 20.Khush, Gurdev S. Stebbins GL. Cytogenetic and evolutionary studies in Secale L: Some new data on the ancestry of S cereale. Am J Bot. 1961;48:723–730. [Google Scholar]
- 21.Grabowski R. Changes in cereal cultivation during the Iron Age in southern Sweden: A compilation and interpretation of the archaeobotanical material. Vol. 20, Vegetation History and Archaeobotany. 2011. p. 479–94. [Google Scholar]
- 22.Schlegel R. Rye (Secale cereale L)—a younger crop plant with bright future In: Sing R. J., Jauhar P, editor. Genetic Resources, Chromosome Engineering, and Crop Improvement: Vol II Cereals. Boca Raton: CRC Press; 2006. p. 365–94. [Google Scholar]
- 23.Targońska M, Bolibok-Brągoszewska H, Rakoczy-Trojanowska M. Assessment of genetic diversity in Secale cereale based on SSR Markers. Plant Mol Biol Report. 2016;34(1):37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matos M, Pinto-Carnide O, Benito C. Phylogenetic relationships among Portuguese rye based on isozyme, RAPD and ISSR markers. Hereditas. 2001;134(3):229–36. [DOI] [PubMed] [Google Scholar]
- 25.Gailīte A, Gaile A, Gaile I, Voronova A, Veinberga I, Kokare A, et al. Genotypic assessment of the Latvian rye (Secale cereale L.) Collection. Proc Latv Acad Sci Sect B Nat Exact, Appl Sci. 2013;67(3):264–7. [Google Scholar]
- 26.Burger JC, Lee S, Ellstrand NC. Origin and genetic structure of feral rye in the western United States. Mol Ecol. 2006;15(9):2527–39. 10.1111/j.1365-294X.2006.02938.x [DOI] [PubMed] [Google Scholar]
- 27.Jenabi T, Saeidi H, Rahiminejad MR. Biodiversity of Secale strictum in Iran measured using microsatellites. Genet Resour Crop Evol. 2011;58(4):497–505. [Google Scholar]
- 28.Schlegel R. Rye–Genetics, Breeding & Cultivation. New York: CRC Press; 2013. [Google Scholar]
- 29.Santos E, Benito C, Silva-Navas J, Gallego FJ, Figueiras AM, Pinto-Carnide O, et al. Characterization, genetic diversity, phylogenetic relationships, and expression of the aluminum tolerance MATE1 gene in Secale species. Biologia Plantarum. 2017;1–12. [Google Scholar]
- 30.Broda Z., Tomkowiak A., Mikołajczyk S., Weigt D., Górski F., Kurasiak-Popowska F. The genetic polymorphism between the wild species and cultivars of rye Secale cereale L. Acta Agrobot. 2016;69:Acta Agrobotanica. [Google Scholar]
- 31.Kottek M, Grieser J, Beck C, Rudolf B, Rubel F. World map of the Köppen-Geiger climate classification updated. Meteorol Zeitschrift. 2006;15(3):259–63. [Google Scholar]
- 32.Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus (Madison). 1990;12:13–5. [Google Scholar]
- 33.Khlestkina EK, Than MHM, Pestsova EG, Röder MS, Malyshev S V., Korzun V, et al. Mapping of 99 new microsatellite-derived loci in rye (Secale cereale L.) including 39 expressed sequence tags. Theor Appl Genet. 2004;109(4):725–32. 10.1007/s00122-004-1659-z [DOI] [PubMed] [Google Scholar]
- 34.Saal B, Wricke G. Development of simple sequence repeat markers in rye (Secale cereale L.). Genome Natl Res Counc Canada Genome Cons Natl Rech Canada. 1999;42(5):964–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10584314 [PubMed] [Google Scholar]
- 35.Amos W, Hoffman JI, Frodsham A, Zhang L, Best S, Hill AVS. Automated binning of microsatellite alleles: Problems and solutions. Mol Ecol Notes. 2007;7(1):10–4. [Google Scholar]
- 36.Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4(3):535–8. [Google Scholar]
- 37.Gutiérrez, Juan, Royo, Luis Alvarez I. Goyache F. MolKin v2.0: A computer program for genetic analysis of populations using molecular coancestry information. J Hered. 2009;6:718–721. [DOI] [PubMed] [Google Scholar]
- 38.Raymond M., Rousset F. GENEPOP (version 12): population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–9. [Google Scholar]
- 39.Rousset F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–6. 10.1111/j.1471-8286.2007.01931.x [DOI] [PubMed] [Google Scholar]
- 40.Peakall R, Smouse PE. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6(1):288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalinowski ST. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5(1):187–9. [Google Scholar]
- 42.Goudet J. FSTAT: a computer program to calculate F-Statistics. J Hered. 2013;104:586–90.23576670 [Google Scholar]
- 43.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9(5):1322–32. 10.1111/j.1755-0998.2009.02591.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol. 2005;14(8):2611–20. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- 46.Coulon A, Fitzpatrick JW, Bowman R, Stith BM, Makarewich CA, Stenzler LM, et al. Congruent population structure inferred from dispersal behaviour and intensive genetic surveys of the threatened Florida scrub-jay (Aphelocoma coerulescens). Mol Ecol. 2008;17(7):1685–701. 10.1111/j.1365-294X.2008.03705.x [DOI] [PubMed] [Google Scholar]
- 47.Takezaki N, Nei M, Tamura K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol Biol Evol. 2010;27(4):747–52. 10.1093/molbev/msp312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nei M, Chesser RK. Estimation of fixation indices and gene diversities. Ann Hum Genet. 1983;47(3):253–9. [DOI] [PubMed] [Google Scholar]
- 49.Varshney RK, Beier U, Khlestkina EK, Kota R, Korzun V, Graner A, et al. Single nucleotide polymorphisms in rye (Secale cereale L.): discovery, frequency, and applications for genome mapping and diversity studies. Theor Appl Genet. 2007;114(6):1105–16. 10.1007/s00122-007-0504-6 [DOI] [PubMed] [Google Scholar]
- 50.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 51.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002929707614244 10.1086/319501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-79957613599&partnerID=40&md5=d616d40ac2af53bae65b25bb4fd5320d 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nei M. Genetic Distance between populations. Am Nat. 1972;106(949):283–92. Available from: http://www.journals.uchicago.edu/doi/10.1086/282771 [Google Scholar]
- 54.Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Vol. 89, Genetics. 1978. p. 583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuevas, Hugo E. Prom LK. Assessment of molecular diversity and population structure of the Ethiopian sorghum [Sorghum bicolor (L.) Moench] germplasm collection maintained by the USDA–ARS National Plant Germplasm System using SSR markers. Genet Resour Crop Evol. 2013;60(6):1817–30. [Google Scholar]
- 56.Liu K, Goodman M, Muse S, Smith JS, Buckler E, Doebley J. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics. 2003;165(4):2117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bustos a De, Jouve N. Phylogenetic relationships of the genus Secale based on the characterisation of rDNA ITS sequences. Plant Syst Evol. 2002;235:147–54. [Google Scholar]
- 58.Chikmawati T, Skovmand B, Gustafson JP. Phylogenetic relationships among Secale species revealed by amplified fragment length polymorphisms. Genome. 2005;48(5):792–801. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16391685%5Cnhttp://www.nrcresearchpress.com/doi/abs/10.1139/g05-043 10.1139/g05-043 [DOI] [PubMed] [Google Scholar]
- 59.Frederiksen SE, Petersen G. Morphometrical analyses of Secale (Triticeae, Poaceae). Nord J Bot. 1997;17:185–98. [Google Scholar]
- 60.Vavilov NI. On the origin of cultivated rye. Bull Appl Bot Genet Plant Breed. 1917;10:561–90. [Google Scholar]
- 61.Khush GS. Cytogenetic and evolutionary studies in Secale: Secale vavilovii and its biosystematic status. Zeitschrift für Pjantmz Dichtung. 1963;50:34–43. [Google Scholar]
- 62.Khush GS. Cytogenetic and evolutionary studies in Secale: Cytogenetics of weedy ryes and origin of cultivated rye. Econ Bot. 1963;17:60–71. [Google Scholar]
- 63.Bolibok-Br Goszewska H, Targo Ska MG, Bolibok L, Kilian A, Rakoczy-Trojanowska M. Genome-wide characterization of genetic diversity and population structure in Secale. BMC Plant Biol. 2014;14(1):184 Available from: http://www.biomedcentral.com/1471-2229/14/184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Persson K, von Bothmer R. Assessing the allozyme variation in cultivars and Swedish landraces of rye (Secale cereale L.). Hereditas. 2000;132(1):7–17. [DOI] [PubMed] [Google Scholar]
- 65.Persson K, von Bothmer R. Genetic diversity amongst landraces of rye (Secale cereale L.) from northern Europe. Hereditas. 2002;136(1):29–38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12184486 [DOI] [PubMed] [Google Scholar]
- 66.Chikmawati T, Miftahudin M, Skovmand B, Gustafson JP. Amplified fragment length polymorphism-based genetic diversity among cultivated and weedy rye (Secale cereale L.) accessions. Genet Resour Crop Evol. 2012;59(8):1743–52. [Google Scholar]
- 67.Cuadrado A, Jouve N. Evolutionary trends of different repetitive DNA sequences during speciation in the genus Secale. J Hered. 2002;93(5):339–45. [DOI] [PubMed] [Google Scholar]
- 68.Zohary D. Origin of south-west Asiatic cereals: wheats, barley, oats and rye In: Davis PH, editor. Plant Lye of South-west Asia. Edinburgh: The Botanical Society of Edinburgh; 1971. p. 235–63. [Google Scholar]
- 69.Jones JDG, Flavell RB. The mapping of highly-repeated DNA families and their relationship to C-bands in chromosomes of Secale cereale. Chromosoma. 1982;86(5):595–612. [Google Scholar]
- 70.Hammer K. Skolimowska E. Knupffer H. Vorarbeiten zur monographischen Darstellung von Wildpflanzensortimenten: Secale L. Kulturpflanze. 1987;35:135–87. [Google Scholar]
- 71.Stutz HC. On the origin of cultivated rye. Am J Bot. 1972;59:59–70. [Google Scholar]
- 72.Ramirez L., Pisabarro G. Studies on enzymatic polymorphism in rye (Secale cereale L) cultivars. Genetica. 1986;69:209–12. [Google Scholar]
- 73.Chikmawati T. 2003. Phylogenetic relationships in Secale and PCR-based EST mapping in wheat.
- 74.Falconer DS, Mackay TFC. Introduction to quantitative genetics. Vol. 4, Introduction to quantitative genetics. 1996. p. 43 Available from: http://www.amazon.com/dp/0582243025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lanter S. Barcaccia G. Molecular marker based analysis for crop germplasm preservation In: Ruane J, Sonnino A, editors. The role of biotechnology in exploring and protecting agricultural genetic resources. Rome: FAO; 2006. p. 55–66. [Google Scholar]
- 76.Hammer K, Arrowsmith N, Gladis T. Agrobiodiversity with emphasis on plant genetic resources. Vol. 90, Naturwissenschaften. 2003. p. 241–50. 10.1007/s00114-003-0433-4 [DOI] [PubMed] [Google Scholar]
- 77.Hawtin G, Iwanaga M, Hodgkin T. Genetic resources in breeding for adaptation. Euphytica. 1996;92(1–2):255–66. [Google Scholar]
- 78.Zohary D. The mode of domestication of the founder crops of Southwest Asian agriculture In: Harris DR, editor. The Origins and Spread of Agriculture and Pastoralism in Eurasia. London: UCL Press; 1996. p. 142–158. [Google Scholar]
- 79.Harris SA. No Title. In: Molecular systematics and plant evolution 1st ed London: Taylor and Francis; 1999. p. 211–28. [Google Scholar]
- 80.Ceccarelli S. Evolution, plant breeding and biodiversity. J Agric Environ Int Dev. 2009;103(1/2):131–45. [Google Scholar]
- 81.Zeven AC. Landraces: A review of definitions and classifications. Euphytica. 1998;104(183643):127–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Am, Tropical monsoon climate; Aw, Savanna; BSh, Hot semi-arid; BSk, Cold semi-arid; BWh, Hot desert; BWk, Cold desert; Cfa, Humid subtropical climate; Cfb,Temperate oceanic climate; Csa, Hot-summer Mediterranean climate; Csb, Warm-summer Mediterranean climate; Cwb, Subtropical highland climate; Dfa, Hot-summer humid continental climate; Dfb, Warm-summer humid continental climate; Dfc, Subarctic climate; Dsa Hot, dry-summer continental climate; Dsb, Warm, dry-summer continental climate; Dwa, Monsoon-influenced hot-summer humid continental climate; ET, Mild tundra climate.
(DOCX)
Chr, Chromosome number; Temp, Annealing Temperature (°C); ref., Reference.
(DOCX)
S. c. afg., S. cereale subsp. afghanicum; S. c. anc., S. cereale subsp. ancestrale; S. c. cereale, S. cereale subsp. cereale; S. c. dighor., S. cereale subsp. dighoricum; S. c. segetale, S. cereale subsp. segetale; S. s. anatol, S. strictum subsp. anatolicum; S. s. irm. S. strictum subsp. irmanuso; S. s. kupr., S. strictum subsp. kuprijanovii; S. s. strictum, S. strictum subsp. strictum; S. sylv., S. sylvestre; S. vav., S. vavilovii. Significant P values are indicated in bold.
(DOCX)
Am, Tropical monsoon climate; Aw, Savanna; BSh, Hot semi-arid; BSk, Cold semi-arid; BWh, Hot desert; BWk, Cold desert; Cfa, Humid subtropical climate; Cfb,Temperate oceanic climate; Csa, Hot-summer Mediterranean climate; Csb, Warm-summer Mediterranean climate; Cwb, Subtropical highland climate; Dfa, Hot-summer humid continental climate; Dfb, Warm-summer humid continental climate; Dfc, Subarctic climate; Dsa Hot, dry-summer continental climate; Dsb, Warm, dry-summer continental climate; Dwa, Monsoon-influenced hot-summer humid continental climate; ET, Mild tundra climate.
Significant P values are indicated in bold.
(DOCX)
Significant P values are indicated in bold.
(DOCX)
Data Availability Statement
All of the 61 DNA sequence data generated in this study are available from the GenBank database (accession numbers MH421898-MH421958).