

Abstract
BACKGROUND:
Gut microbiota, the collective community of microorganisms inhabiting the intestine, have been shown to provide many beneficial functions for the host. Recent advances in next-generation sequencing and advanced molecular biology approaches have allowed researchers to identify gut microbiota signatures associated with disease processes and, in some cases, establish causality and elucidate underlying mechanisms.
CONTENT:
This report reviews 3 commonly used methods for studying the gut microbiota and microbiome (the collective genomes of the gut microorganisms): 16S rRNA gene sequencing, bacterial group or species- specific quantitative polymerase chain reaction (qPCR), and metagenomic shotgun sequencing (MSS). The technical approaches and resources needed for each approach are outlined, and advantages and disadvantages for each approach are summarized. The findings regarding the role of the gut microbiota in the health of patients with cancer and stem cell transplant (SCT) patients (specifically in modulating the development of gut-derived bacterial infections and a posttransplant immune-mediated complication known as graft-vs-host-disease) are reviewed. Finally, there is discussion of the potential viability of these approaches in the actual clinical treatment of cancer and SCT patients.
SUMMARY:
Advances in next-generation sequencing have revolutionized our understanding of the importance of the gut microbiome to human health. Both 16S rRNA gene sequencing and MSS are currently too laborintensive or computationally burdensome to incorporate into real-time clinical monitoring of gut microbiomes. Yet, the lessons learned from these technologies could be adapted to currently used methods (e.g., qPCR) that could then be rigorously tested in the clinical care of these patients.
Gut microbiota, the collective community of bacteria residing within the gastrointestinal tract, have been shown to have many beneficial functions for the host, including enhancing digestion, modulating metabolism, and promoting immune system development (1). These findings were made possible by recent advances in molecular biology and deep sequencing technologies, and also by coordinated research efforts to begin to understand the role of the microbiome in human health, such as the National Institutes of Health Human Microbiome Project, where researchers have characterized the composition and diversity of microbial communities that inhabit major mucosal surfaces of the human body, including nasal passages, oral cavities, skin, gastrointestinal tract, and urogenital tract, and also evaluated the genetic metabolic potential of these communities (2). An explosion of microbiome research has now documented numerous microbiome associations with specific disease processes, while also yielding deep insight into their mechanistic underpinnings. There is mounting evidence that modulating gut microbiota populations (in both preclinical models and human patients) has the potential to be a viable therapeutic modality.
Patients with cancer and stem cell transplant (SCT)4 patients are particularly susceptible to disruptions of gut microbiota communities because of antibiotic exposure (3, 4) and chemotherapy treatment (5). Cytotoxic chemotherapy continues to be the mainstay of cancer and SCT therapy, and the resulting deficits in host immunity (namely, neutropenia and gut mucosal damage) have made these patients highly prone to developing invasive bacterial and fungal infections, many of which originate from the gastrointestinal (GI) tract. Thus, standard clinical practice is to treat these patients with prophylactic or empiric antibiotics (in many cases, multiple antibiotics) for long durations of time (up to several weeks) to prevent these invasive infections. Recent studies have now shown that patients with cancer and SCT patients with large disruptions in gut microbiota communities are at increased risk for developing invasive infections (6) and the posttransplant complication known as graft-vs-host disease (GVHD) (3, 7, 8). In fact, specific antibiotic therapies appear to be linked to the development of these complications (3, 9). Thus, this patient population could potentially benefit from real-time gut microbiota monitoring to either modify current clinical practice (e.g., antibiotic use) or perhaps in the future initiate a specific medical intervention, such as an antibiotic conjugated to a pathogen-specific antibody (10) or genetically engineered bacteria created to outcompete pathogenic bacteria (11).
This review will describe the basic principles underlying methods for profiling and quantifying gut microbiota, show how these approaches have yielded important insights into how changes in gut microbiota populations impact the health of patients with cancer and SCT patients, and provide an overview of how monitoring gut microbiota concentrations might be used to modify the clinical care of these patients.
Methods for Profiling Gut Microbiota
The notion that commensal bacteria provide benefit to the host is a long-standing concept in both science and medicine (12). Initial attempts at dissecting the mechanisms by which specific commensal bacteria confer benefit to the host were hampered by culture-dependent methods that were laborious, time-consuming, and unreliable (because many gut commensal bacteria are difficult, if not impossible, to culture). Over the years, advances in culture-independent molecular biology-based methods have led to innovative approaches for profiling complex communities ofbacteria. For example, sequencing of the 16S rRNA gene was developed nearly 40 years ago as a powerful tool to determine bacterial phylogeny (13). Advanced molecular biology methodologies, such as in situ hybridization and PCR, have also been used to identify and quantify bacterial isolates. More recently, the advent of next-generation sequencing platforms has allowed unbiased approaches to high-resolution characterization of complex microbial communities known as metagenomic shotgun sequencing (MSS). Here, we will focus on 16S rRNA gene sequencing, MSS, and bacterial group/species quantitative polymerase chain reaction (qPCR).
16S rRNA GENE SEQUENCING
16S ribosomal RNA (rRNA) is a component of the 30S small subunit of a prokaryotic ribosome. The 16S rRNA gene has been the focus of phylogenetic studies because it has regions that are highly conserved between different species of bacteria (14) and variable regions that are unique for specific bacterial groups or species. By sequencing a variable region of the 16S rRNA gene (designated as V1—V9), the taxonomic identity of any single bacteria can be ascertained. Unfortunately, amplifying different variable regions can lead to taxonomic bias secondary to technical factors (i.e., primer design, chemical reagents used, and amplification conditions) and sample choice (bacterial composition and environment) (15—17).
The same methodologic approach has been used to identify the relative abundance of different bacteria within a complex microbial community. For example, genomic DNA (gDNA) can be extracted from a human stool specimen, of which the majority (approximately 99%) is bacterial gDNA, including 16S rRNA gene DNA. A 16S rRNA gene variable region is amplified from each sample using a composite forward primer and a reverse primer containing a unique nucleotide barcode that is used to tag PCR products from respective samples. Samples are then pooled and sequenced simultaneously (15). Whereas many of the original 16S rRNA gene sequencing experiments (such as those in the Human Mi- crobiome Project) were performed on the Roche 454 sequencing platform, more recent published reports have used sequencing platforms readily used by individual research laboratories (e.g., Illumina MiSeq) (18). The Roche 454 platform uses pyrosequencing chemistry (use of the enzymes ATP sulfurylase and luciferase) and can provide read lengths of up to 700 bp, whereas the Illumina platform uses reversible dye terminator chemistry and can provide single read lengths of up to 300 bp and paired-end reads of up to 600 bp (Fig. 1).
Fig. 1. Overview of 16S rRNA gene sequencing.
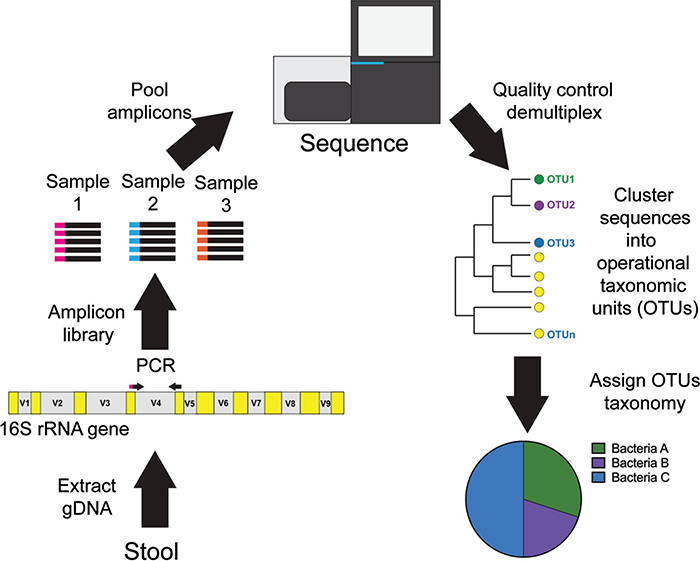
Genomic DNA is extracted from a stool sample. The 16S rRNA gene variable region (e.g., V4) is amplified from each sample using a composite forward primer and a reverse primer containing a unique 6- to 12-base barcode that is used to tag PCR products from respective samples. Amplicon libraries are pooled together and sequenced simultaneously. 16S rRNA gene sequencing data undergoes quality control and filtering and is demultiplexed. Sequences are clustered into phylotypes termed operational taxonomic units (OTUs). Representative sequences of each OTU are then interrogated against a reference database of validated 16S rRNA gene sequences. Relative abundances of bacterial taxa can then be calculated and presented in graphical representation (e.g., pie-chart).
16S rRNA gene sequencing data requires sophisticated computational methods and bioinformatics pipelines. Currently, numerous open-source software programs are available [i.e., Mothur (19), QIIME (20)] to analyze these data sets. Briefly, sequences are clustered first into phylotypes termed operational taxonomic units according to (a) their similarity to previously annotated sequences in a reference database (21) or (b) a de novo approach, based purely on their similarity (22). In either case, representative sequences of each operational taxonomic unit are then interrogated against a reference database of validated 16S rRNA gene sequences (23—25) to provide taxonomic identification. Relative abundances of bacterial taxa (ranging from phylum- to species-level resolution) can then be calculated and presented in graphical representations, most commonly histograms or pie-chart diagrams (Fig. 1).
METAGENOMIC MICROBIAL PROFILING
Whole metagenome shotgun analysis (i.e., MSS) is accomplished by unrestricted sequencing of the genome of all microorganisms present in a given sample. High- quality gDNA is extracted from a tissue sample (e.g., stool) (26), quantified, and undergoes massive parallel sequencing—allowing the sequencing of thousands of organisms simultaneously. Currently, both desktop sequencing systems (e.g., Illumina MiSeq or NextSeq) and high-throughput sequencing systems (e.g., Illumina HiSeq) can perform MSS; whereas the former can be purchased and used by individual research laboratories, the latter provides greater depth of coverage and is typically relegated to core facilities or commercial vendors (Fig. 2). With the capability to obtain high sequence coverage, MSS can detect very low abundance members of the microbiota community that may be missed using other methods (27). Of note, a more recent sequencing platform using single-molecular, real-time sequencing developed by Pacific BioSciences (and commonly referred to as PacBio sequencing) can also be used for microbial metagenomics. Compared with the Illumina platform, PacBio sequencing provides much longer read lengths (average > 10 kb and maximum >60 kb) but also has higher error rates and lower throughput capability (28).
Fig. 2. Overview of MSS.
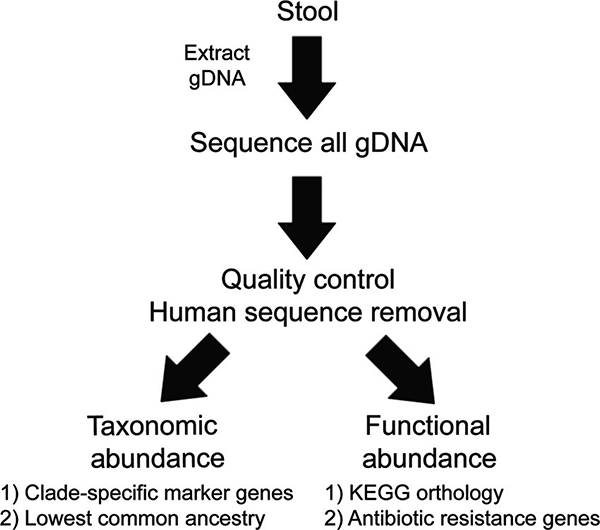
Genomic DNA is extracted from a stool sample, quantified, and assessed for quality. High-quality gDNA then undergoes massive parallel sequencing, allowing the sequencing of thousands of organisms simultaneously. MSS sequencing data undergo quality control and removal of human sequences. For taxonomic identification, unique clade-specific marker genes or lowest common ancestor positioning approaches are commonly used. Functional insight can be ascertained by utilizing the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology database, with each sequence directly mapped to a KEGG orthology representative sequence. Furthermore, antibiotic resistance gene abundance of the entire metagenome can also be determined by mapping to antibiotic resistance gene databases.
The resulting MSS sequencing data, which can consist of up to hundreds of millions of sequences, can then undergo taxonomic and functional analysis. For taxonomic identification, unique clade-specific marker genes (29) or lowest common ancestor positioning approaches have been used. For the former, sequences are queried against a unique clade-specific marker gene marker database. For the lowest common ancestor approach, prealigned sequences are hierarchically classified on a taxonomy tree using a placement algorithm (30) (Fig. 2).
MSS can also provide a direct assessment of the functional attributes of a given microbiome (31). Using the Kyoto Encyclopedia of Genes and Genomes orthol- ogy database, each sequence can be directly mapped to a Kyoto Encyclopedia of Genes and Genomes orthology representative sequence; the sum of sequences belonging to a particular pathway can then be computed (32, 33). Another functional insight that MSS analysis can provide is an assessment of the resistome, the collection of all the antibiotic resistance genes in both pathogenic and non- pathogenic bacteria (34). Given the rising incidence of antibiotic resistance, MSS has been used to better understand the effect of systemic antibiotic treatment on the development of antibiotic resistance in endogenous bacteria (35, 36) (Fig. 2).
In an attempt to move beyond correlating microbiota taxonomic changes with pathophysiology, there is a great focus on trying to identify how metagenomic taxonomic changes relate to microbiome and/or host function, with a particular emphasis on metabolic function. Using a combination of microbial metagenomic sequencing and metabolomic profiling can provide tremendous insight into how taxon-level changes can influence metabolic function and ultimately host function (37). For example, a gut-derived metabolite, trimethylamine N-oxide, directly induces platelet hyperactivity and increases thrombosis potential. Specific gut microbiota taxa are associated with both trimethylamine N-oxide concentrations and thrombosis potential. Antibiotics that deplete these gut microbiota lead to decreases in gut-derived trimethylamine A-oxide and decreased thrombotic risk. Conversely, microbial gut transplantation (into germ-free mice) confers increased thrombosis potential (38). Thus, future microbiome studies that attempt to establish causality will need to move beyond focusing merely on taxa and try to elucidate functional correlates.
BACTERIAL GROUP OR SPECIES qPCR
A limitation of both 16S rRNA gene sequencing and MSS taxonomic profiling is that absolute concentrations of microbiota cannot be determined, only relative abundance. An increase in gut microbiota relative abundance does not necessarily correlate with an increase in total microbiota concentrations. Bacterial group or species- specific qPCR performed on biological samples can provide absolute concentrations. Species-specific genes are often used as targets for bacterial species qPCR (39). In addition, qPCR has been applied to 16S rRNA gene methodology and has been validated for quantification of bacterial groups and specific bacterial species within complex bacterial communities (40—42).
For example, to quantify the concentrations of the bacterial group Enterobacteriaceae (which include gramnegative bacteria such as Escherichia coli, Klebsiella, and Salmonella), Enterobacteriaceae group-specific primers are used to clone a segment of the 16S rRNA gene using a representative strain’s (e.g., E. coli) gDNA (43). This PCR fragment is subcloned into a vector and transformed into E. coli. Varying concentrations of linearized plasmid (circular plasmid can overestimate gene copy abundance) (44) are then used to perform Enterobacteriaceae-specific qPCR, and standard curves are created (Fig. 3A). Unknown samples (e.g., patient stool samples) can then be assessed for Enterobacteriaceae concentrations by performing Enterobacteriaceae qPCR on gDNA extracted from tissue (Fig. 3B). Whereas qPCR measures the number of 16S rRNA gene copies per sample, not actual bacterial numbers or colonyforming units, qPCR values correlate well with colonyforming units (40, 45, 46).
Fig. 3. Overview of bacterial group or species-specific qPCR.

Standardization of Enterobacteriaceae qPCR assay (A). Enterobacteriaceae group-specific primers are used to amplify a segment of the 16S rRNA gene using a representative strain’s (e.g., E. coli) gDNA. This PCR fragment is subcloned into a vector and transformed into E. coli. Plasmids are recovered from transformed E. coli and sequenced for verification. Varying concentrations of linearized plasmid are then used to perform Enterobacteriaceae-specific qPCR, and standard curves are created. Enterobacteriaceae qPCR on an unknown sample (B). An unknown sample (e.g., patient stool specimen) can then be assessed for Enterobacteriaceae concentrations by performing Enterobacteriaceae qPCR on gDNA extracted from feces. Bacterial numbers are then determined from the previously determined standard curves. qPCR measures the number of 16S rRNA gene copies per sample, not the actual bacterial numbers or colony-forming units. Gene copies per sample are then normalized to both stool gDNA concentration and weight.
Advantages and Disadvantages of 16S rRNA Gene Sequencing, MSS, and qPCR Methods
Each of these approaches to gut microbiota profiling has distinct advantages and disadvantages (Table 1). In general, both 16S rRNA gene sequencing and MSS can provide taxonomic snapshots ofgut microbial communities, with 16S rRNA gene being generally more affordable and less computationally intensive, whereas MSS has the capability of profiling other microbes beyond bacteria and also providing some functional insight. Bacterial group or species-specific qPCR is technically the easiest to perform, both from a laboratory and analysis standpoint. qPCR provides absolute quantitative data but does not provide any metagenomic insight.
Table 1.
Comparison of 16S rRNA gene sequencing (16S), metagenomic shotgun sequencing (MSS), and bacterial group quantitative polymerase chain reaction (qPCR).
| Time required for data production | Potential PCR bias | Microbial populations analyzed | Method of quantification | Functional analysis | Computing requirements | Data storage (per sample) | Estimated cost (excluding labor) | |
|---|---|---|---|---|---|---|---|---|
| 16S | Days to weeks | Yes | Bacteria only | Relative abundance | No | Open-source software that can be run on desktop computers | Megabytes | $10-$50/sample (if batched with a large number of samples) |
| MSS | Weeks to months | No | Bacteria, viruses, fungi | Relative abundance | Yes | Bioinformatic analysis is CPU intensive (requiring computer clusters) | Gigabytes | $200-$800/sample (depending on depth of coverage desired) |
| qPCR | Hours to days | Yes | Bacteria, viruses, fungi | Gene copies | No | Spreadsheet program (e.g., Excel) that can be run on a desktop computer | Bytes to kilobytes | <$1/sample |
Gut-Derived Infections in Cancer and SCT Patients
Patients with cancer and SCT patients are extremely vulnerable to serious bacterial infections, many of which originate from the gut (6, 47). The cytotoxic chemotherapy routinely used to treat cancer and SCT patients markedly suppresses the host’s immune system, resulting in impaired cellular immunity, damaged gut epithelium, and decreased humoral immunity. In fact, when systemic cytotoxic chemotherapy was first being used in the late 1960s, an alarming number of patients developed serious, and often fatal, bacterial bloodstream infections (48). Gram-negative bacterial bloodstream infections were the most common, and 1 gram-negative bacterium, Pseudomonas aeruginosa, was associated with the highest degree of patient morbidity and mortality (49). Ironically, the primary source of these gram-negative bacterial infections in patients with cancer and SCT patients was thought to be the patient’s own GI tract, substantiated by demonstrating the existence of gram-negative bacterial gut colonization preceding the development of gramnegative bloodstream infections using culture-based methods (50). These findings led to sweeping changes in clinical practice whereby initiation of empiric antibiotics for patients with signs of an impending infection (in particular, fever and neutropenia) had a remarkable effect on decreasing the morbidity and mortality associated with these deadly infections (51). Even to this day, treating patients with cancer at risk for infection, specifically those that are febrile and neutropenic, with empiric antibiotics is still standard of care (52). Although countless lives have been saved by this practice, clinicians are now facing an increasing number of antibiotic-resistant bacteria in these patients (9), and there are now data to suggest that widespread antibiotic use may inadvertently be increasing the risk of infection in some patients.
Microbial pathogens that translocate from the GI tract [i.e., Enterobacteriaceae (53), Enterococcus spp. (6,54), and Candida albicans (47)] are typically not abundant in the gut of healthy hosts. More recent studies, using 16S rRNA gene sequencing, have shown that adult SCT patients exhibiting intestinal microbiota domination (defined as >30% relative abundance) with either Enterococcus spp. or Enterobacteriaceae had a significantly increased risk of bacteremia with the gut-dominating bacteria (6). Preclinical models of enteric-derived bacteremia or fungemia have also shown that the risk of invasive infection is directly proportional to the bacterial burden in the gut (45, 54). Thus, the obvious question is why do some patients develop expansion of pathogenic microbes in the gut and subsequently develop invasive infections whereas others do not.
One key factor in determining whether a host can maintain healthy gut microbiota communities is the ability to maintain colonization resistance, the process by which the host’s commensal gut bacteria prevent pathogenic bacteria from colonizing or expanding in the GI tract (12). Colonization resistance was first described >50 years ago and was quickly identified to be relevant to human disease (55). For example, antibiotic therapy disrupts gut commensal microbial communities and fosters the development of infections, such as Clostridium difficile (12). Recently, by using next-generation sequencing gut-profiling techniques (both 16S rRNA gene and MSS) and sophisticated bioinformatics pipelines, 1 specific commensal anaerobic bacterial species, Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium, enhanced resistance to C. difficile infection in a secondary bile acid-dependent manner (56). Likewise, fecal microbiota transplants can restore gut microbial communities and are effective in treating C. difficile infections (57). These data underscore the importance of intact microbial communities in resisting colonization with pathogens.
Until recently, the mechanisms by which commensal gut microbiota maintained or promoted colonization resistance were unknown. Recent studies have shown that commensal gut microbiota, particularly anaerobic bacteria, can induce gut epithelial cells to produce antimicrobial peptides that can kill pathogenic bacteria and fungi (45, 54, 58). When these beneficial commensal anaerobes are depleted, pathogenic microbes expand in the gut, and the likelihood of invasive infections increases. Interestingly, even in the setting of depleted commensal microbiota, inducing gut immune effectors (e.g., antimicrobial peptides) with pharmacologic agents or biological agents (e.g., bacterial lipopolysaccharide or flagellin) can significantly decrease pathogenic microbial colonization in the gut, and dramatically decrease mortality from invasive infections. In preclinical models, a 1 to 2 log-fold reduction in bacterial (54) or fungal gut colonization levels (45) has been shown to be sufficient to significantly decrease dissemination or mortality, suggesting that complete eradication or absence of colonization is not needed to achieve a significant decrease in dissemination. Thus, modulating pathogen GI colonization by manipulating gut microbiota and/or augmenting mucosal immune effectors could be a viable and effective strategy for reducing invasive infections in patients.
Patients with cancer and SCT patients are particularly vulnerable to impairments in colonization resistance, which in part explains the high incidence of gutderived invasive microbial infections in these patients. The major adverse effects of cytotoxic chemotherapy that directly increase the risk of pathogenic microbial translocation from the gut in humans include neutropenia (deficiencies in cellular immunity), mucositis (impaired intestinal barriers), and use of broad-spectrum antibiotics (disruptions in gut microbiota communities) (59, 60). The latter 2 are directly linked to the host’s ability to maintain colonization resistance: Gut mucosal/epithelial damage may inhibit or decrease antimicrobial peptide production, and broad-spectrum depletion of anaerobic bacteria may decrease the microbiota stimulus needed to induce gut-derived antimicrobial peptides. Thus, recent studies now show that broad-spectrum antibiotic therapy in patients with cancer leads to depletion of commensal gut bacteria, expansion of pathogenic bacteria in the gut, and a subsequent increased risk of gut-derived pathogen infections (4, 6, 9, 61).
With the advent of advanced gut microbiota profiling tools such as 16S rRNA gene or MSS, the reality of real-time monitoring of gut microbiota populations in patients merits discussion. Given that increased gut microbial burden (e.g., increased gut abundance of E. coli) precedes and significantly increases the risk of bacteremia (with E. coli) (6) in this patient population, one could argue that frequent monitoring of gut Enterobacteriaceae (the family of bacteria that includes the notable gramnegative pathogens such as E. coli, Klebsiella spp., and Enterobacter spp.) in patients with cancer and SCT patients should be used to identify those patients at risk for developing Enterobacteriaceae bacteremia. Both 16S rRNA gene sequencing and MSS have not been used in the clinical setting because of logistical barriers to implementation, namely, time, cost, and complexity. As noted previously (Table 1), the turnaround time for obtaining actionable data from 16S rRNA gene sequencing and MSS is on the order of weeks to months, whereas results from group- or species-specific microbial qPCR could realistically be provided to a clinician in days. Many clinical microbiology laboratories already offer qPCR to monitor a number of infectious pathogens, such as cytomegalovirus, Epstein—Barr virus, and adenovirus. In fact, routine screening or monitoring patients for systemic viral infections with qPCR tests (e.g., cytomegalovirus qPCR) (62) is standard practice for patients undergoing SCT. Thus, a clinical microbiology laboratory should be able to implement a bacterial qPCR assay. Bacterial qPCR, however, will not provide a snapshot of the entire gut microbiota population; thus, a clinician will be limited to following concentrations from the specific bacterial qPCRs tests offered by the clinical microbiology laboratory.
Yet, although bacterial qPCR assays are potentially a viable means to monitor gut microbiota populations in cancer and SCT patients, the reality is that there is still a lack of conclusive clinical data and proven clinical utility. In contrast, monitoring cytomegalovirus viral loads during SCT has been well-studied and is the standard of care (62). Therefore, additional studies will need to be conducted to determine whether real-time monitoring of gut microbiota abundance can reliably identify those patients at highest risk for developing invasive microbial infections.
Graft-vs-Host Disease in SCT Patients
The success of SCT is limited, in part, by GVHD, a complex immune-mediated process occurring in 20% to 50% of SCT patients (63) and accounting for 15% to 30% of deaths that occur following allogeneic SCT (64). The newly engrafted immune system (graft) attacks the recipient’s (host) body, with the main target organs including the skin, GI tract, and liver. GVHD is primarily a cytotoxic T cell-driven process. Unfortunately, there are no currently accepted or used noninvasive biomarker tests to diagnose GVHD, leaving biopsy and pathologic confirmation as the gold standard. Furthermore, immunosuppressive medications (e.g., steroids) are the primary treatment for GVHD, which is accompanied with a myriad of side effects, including increased infection risk.
Commensal gut bacteria have long been implicated in initiating and perpetuating GVHD (65). Studies in the 1970s showed that germ-free mice (66) or mice receiving “gut-decontaminating” nonabsorbable antibiotics (a combination of neomycin, streptomycin, bacitracin, and pimaricin) (65) experienced less frequent or attenuated GVHD. Recent studies using next-generation sequencing methods have shown that a depletion of commensal anaerobes, particularly Clostridiales or what has been previously termed as antiinflammatory Clostridia (AIC) (67), and an expansion of Enterococcus spp., occurs in patients who develop GVHD (3, 7, 8, 68). Interestingly, this group AIC bacteria has been shown to ameliorate inflammatory bowel disease (69, 70) and GVHD (71) in mice.
Antibiotics, particularly antibiotics effective in killing beneficial commensal anaerobic bacteria (including AIC), appear to be key drivers of the gut microbiota changes associated with GVHD. The use of specific antianaerobic antibiotics [e.g., clindamycin (72), piperacillintazobactam (3), and imipenem-cilastin (3)] has been associated with the development of GVHD in patients and shown to exacerbate GVHD in mice. Although these specific antibiotics do provide broader coverage, including activity against anaerobic bacteria, infections with anaerobic bacteria are rare (73). The most frequent gutderived pathogens that cause infections in the peritransplant period include gram-negative bacteria (Enterobacteriaceae), gram-positive bacteria (coagulase-negative Staphylococci, Streptococcus viridans group, Enterococcus spp.), and Candida species (73). Thus, understanding how specific classes or types of antibiotics drive gut microbiota changes, and implementing reliable and rapid methods of monitoring gut microbiota changes, could greatly help clinicians make informed choices about which antibiotics to use.
The current gold standard for establishing a diagnosis of GVHD involves biopsy and pathologic confirmation, all of which can take weeks to transpire, thus delaying diagnosis and treatment of GVHD (64). Yet, as stated previously, noninvasive biomarker tests for GVHD are not currently used in practice because there is no clinical consensus on their efficacy or utility. Beginning empiric GVHD therapy, which entails immunosuppression to dampen the inflammatory process, can increase the risk of infection or, in the worst case, exacerbate an infection (i.e., fungal or viral infection). Because of this risk, GVHD therapy is rarely started until a definitive diagnosis has been established. Thus, the idea of monitoring changes in the gut microbiome as a noninvasive biomarker for GVHD is appealing. For example, a bacterial group-specific qPCR could be used to monitor concentrations of AIC on a weekly basis. If AIC concentrations were to fall below a certain threshold (e.g., >3 log-fold decrease from initial levels), this would indicate that the patient could be at high risk for developing GVHD and appropriate GVHD therapy could be started (72). In fact, a study of pediatric patients (n = 15, from 2 different institutions) undergoing SCT showed that patients who developed GVHD showed significant declines in AIC concentrations (sometimes up to 10 log fold), whereas those patients who did not develop GVHD had no more than a 3 log-fold decrease in AIC (72). Furthermore, real-time monitoring of AIC concentrations could also be used to monitor the efficacy of future probiotic therapies that may be used for GVHD and could also provide further confirmation/validation that specific antibiotics are not depleting beneficial commensal bacteria, such as AIC. Again, although the use of bacterial qPCR as a biomarker for GVHD is intriguing, further studies will need to be performed with larger sample sizes from multiple institutions to conclusively determine its efficacy as a biomarker and also to validate whether clinical laboratories can feasibly use this qPCR assay in real time. On a related note, a recent study showed that a gut microbiota-derived metabolite measured in the urine correlated with the abundance of AIC and showed promise as a biomarker assay for GVHD (74).
Summary
The notion that the commensal gut microbiota play a critical role in both infections and GVHD in patients with cancer and SCT patients was first described >40 years ago, but only recently have advances in deep sequencing technology and advanced molecular biology approaches allowed us to begin to understand how and why. The challenge as both clinicians and scientists will be to either adapt these more time- and labor-intensive gut microbiota profiling approaches (i.e., 16S and MSS) into efficient and reliable assays (e.g., qPCR) that could be adopted into clinical practice, or to develop novel approaches that can provide real-time data to clinicians. Thus, in the future, active monitoring of GI microbiota populations, followed by targeted manipulation of the gut immune system (via bacterial products or metabolites and/or chemical compounds) to modulate gut microbiota populations to either prevent or treat infections or GVHD, could become the standard of care for patients with cancer and SCT patients.
Acknowledgments
Research Funding: A.Y. Koh, the Roberta I. and Norman L. Pollock Fund, the US National Institutes of Health (NIH) grant R01AI123163 Centers for Disease Control/National Center for Emerging and Zoonotic Infectious Diseases.
Footnotes
Nonstandard abbreviations: SCT, stem cell transplant; GI, gastrointestinal; GVHD, graft- vs-host disease; MSS,metagenomicshotgunsequencing;qPCR,quantitativePCR; rRNA, ribosomal RNA; gDNA, genomic DNA; AIC, antiinflammatory Clostridia
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contribution to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts ofinterest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Expert Testimony: None declared.
Patents: None declared
References
- 1.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 2011;9:279–90. [DOI] [PubMed] [Google Scholar]
- 2.Human Microbiome Project C.Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shono Y, Docampo MD, Peled JU, Perobelli SM Velardi E, Tsai JJ, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patientsand mice.Sci Transl Med 2016;8:339ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taur Y,Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 2014;124: 1174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zwielehner J, Lassl C, Hippe B, Pointner A, Switzeny OJ, Remely M, et al. Changes in human fecal microbiota due to chemotherapy analyzed by Taqman-PCR, 454 sequencing and PCR-DGGE fingerprinting. PLoS One 2011;6:e28654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 2012; 55:905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holler E, Butzhammer P, Schmid K, Hundsrucker C, Koestler J, Peter K, et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol Blood Marrow Transplant 2014;20:640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenq RR, Ubeda C,Tau Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation.J Exp Med 2012;209:903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bilinski J, Robak K, Peric Z, Marchel H, Karakulska-Prystupiuk E, Halaburda K, et al. Impact ofgut colonization by antibiotic-resistant bacteria on the outcomes of allogeneic hematopoietic stem cell transplantation: a retrospective, single-center study. Biol Blood Marrow Transplant 2016;22:1087–93. [DOI] [PubMed] [Google Scholar]
- 10.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015;527: 323–8. [DOI] [PubMed] [Google Scholar]
- 11.Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, Simpson P, et al. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 2015;526:719–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 2013;13:790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woese CR, Fox GE. Phylogenetic structure ofthe pro-karyoticdomain: the primary kingdoms. Proc Natl Acad Sci U SA 1977;74:5088–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coenye T, Vandamme P. Intragenomic heterogeneity between multiple 16S ribosomal RNA operons in sequenced bacterial genomes. FEMS Microbiol Lett 2003;228:45–9. [DOI] [PubMed] [Google Scholar]
- 15.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA,Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 2011;108 Suppl 1: 4516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soergel DA, Dey N, Knight R, Brenner SE. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J 2012;6: 1440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang B, Wang Y, Qian PY. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics 2016;17:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina Hiseq and Miseq platforms. ISME J 2012;6:1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. Qiime allows analysisof high-throughput community sequencing data. Nat Methods 2010;7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, DeSantis TZ,Andersen GL, Knight R.Accuratetax- onomy assignments from 16S rRNA sequences pro- duced by highly parallel pyrosequencers.NucleicAcids Res 2008;36:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol2011;77:3219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 2014;42:D633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quast C,Pruesse E,Yilmaz P,Gerken J,Schweer T,Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools.NucleicAcids Res 2013;41:D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Bro- die EL, Keller K, et al. Greengenes, a chimera- checked 16S rRNA gene database and workbench compatible with arb. Appl Environ Microbiol 2006; 72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith MB, Kelly C, Alm EJ. Policy: how to regulate faecal transplants. Nature 2014;506:290–1. [DOI] [PubMed] [Google Scholar]
- 27.Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T, et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol 2016;7:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 2015;13: 278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segata N, Waldron L, Ballarini A, Narasimhan V, Jous- son O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 2012;9:811–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res 2011;21:1552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, et al. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotech- nol 2012;30:513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, et al. Metabolic reconstruction for metag- enomic data and its application to the human micro- biome. PLoS Comput Biol 2012;8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Kim MS, Koh AY, Xie Y, Zhan X. FMAP: functional mapping and analysis pipeline for metagenomics and metatranscriptomics studies. BMC Bioinformatics 2016;17:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright GD.The antibioticresistome:then exus of chemical and genetic diversity. Nat Rev Microbiol 2007;5:175–86. [DOI] [PubMed] [Google Scholar]
- 35.Gibson MK, Wang B, Ahmadi S, Burnham CA, Tarr PI, Warner BB, Dantas G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol 2016;1:16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, de Vos WM. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun 2016;7:10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ChooJ M, Kanno T,Zain NM, Leong LE,Abell GC, Keeble JE, et al. Divergent relationships between fecal microbiota and metabolome following distinct antibiotic- induced disruptions.mSphere 2017;2:e00005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016;165: 111–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shannon KE, Lee DY, Trevors JT, Beaudette LA. Application of real-time quantitative PCR for the detection of selected bacterial pathogens during municipal wastewater treatment. Science Total Environ 2007;382: 121–9. [DOI] [PubMed] [Google Scholar]
- 40.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 2008;76:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartosch S, Fite A, Macfarlane GT, McMurdo ME. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatmenton the fecal microbiota. Appl Environ Microbiol 2004;70:3575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan MP, Kaparakis M, Galic M, Pedersen J, Pearse M, Wijburg OL, et al. Chronic Helicobacterpylori infection does not significantly alter the microbiota ofthe murine stomach. Appl Environ Microbiol 2007;73: 1010–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson- Sjoberg J, Amir E, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 2010;11:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou Y, Zhang H, Miranda L, Lin S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: microalgal pcna as the model gene. PLoS One 2010;5:e9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X,et al. Activation of HIF-1alphaandLL-37 by com- mensal bacteria inhibits Candida albicanscolonization. Nat Med 2015;21:808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Medina E, Fan D, Coughlin LA, Ho EX, Lamont IL,Reimmann C, et al. Candida albicans inhibits Pseudomonas aeruginosa virulence through suppression of pyochelin and pyoverdine biosynthesis. PLoS Pathog 2015;11:e1005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, Gobara S, et al. Candida colonisation as a source for candidaemia. J Hosp Infect 2009;72:9–16. [DOI] [PubMed] [Google Scholar]
- 48.Bodey GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med 1966;64:328–40. [DOI] [PubMed] [Google Scholar]
- 49.Elting LS, Rubenstein EB, Rolston KV, Bodey GP. Outcomes of bacteremia in patients with cancer and neutropenia: observations from two decades of epidemiological and clinical trials. Clin Infect Dis 1997;25: 247–59. [DOI] [PubMed] [Google Scholar]
- 50.Tancrede CH, Andremont AO. Bacterial translocation and gram-negative bacteremia in patients with hema- tological malignancies.J Infect Dis 1985;152:99–103. [DOI] [PubMed] [Google Scholar]
- 51.Ceftazidime combined with a short or long course of amikacin for empirical therapy of gram-negative bacte- remia in cancer patients with granulocytopenia. The EORTC International Antimicrobial Therapy Cooperative Group. N Engl J Med 1987;317:1692–8. [DOI] [PubMed] [Google Scholar]
- 52.Freifeld AG, Bow EJ, Sepkowitz KA, Boeckh MJ, Ito JI, Mullen CA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 2011;52:e56–93. [DOI] [PubMed] [Google Scholar]
- 53.Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis 2014;58: 1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, et al. Vancomycin-resistant enterococci exploit antibiotic- induced innate immune deficits. Nature 2008;455: 804–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bohnhoff M, Miller CP. Enhanced susceptibility to salmonella infection instreptomycin-treated mice.J Infect Dis 1962;111:117–27. [DOI] [PubMed] [Google Scholar]
- 56.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Go- bourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to clostridium difficile.Nature 2015;517:205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoeten- dal EG, de Vos WM, et al. Duodenal infusion of donor feces for recurrent clostridium difficile. N Engl J Med 2013;368:407–15. [DOI] [PubMed] [Google Scholar]
- 58.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006;313:1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pasqualotto AC, Nedel WL, Machado TS, Severo LC. Risk factors and outcome for nosocomial breakthrough candidaemia. J Infect 2006;52:216–22. [DOI] [PubMed] [Google Scholar]
- 60.Rosen GP, Nielsen K, Glenn S, Abelson J, Deville J, Moore TB. Invasive fungal infections in pediatric oncology patients: 11-year experience at a single institution. J Pediatr Hematol Oncol 2005;27:135–40. [DOI] [PubMed] [Google Scholar]
- 61.Galloway-Pena JR, Smith DP, Sahasrabhojane P, Ajami NJ, Wadsworth WD, Daver NG, et al. The role of the gastrointestinal microbiome in infectious complica- tions during in duction chemotherapy for acutemyeloid leukemia.Cancer 2016;122:2186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panagou E,Zakout G,Keshani J,Smith C, Irish D,Mack- innon S, et al. Cytomegalovirus pre-emptive therapy after hematopoietic stem cell transplantation in the era of real-time quantitative PCR: comparison with recipients of solid organ transplants. Transpl Infect Dise 2016;18:405–14. [DOI] [PubMed] [Google Scholar]
- 63.Ball LM, Egeler RM. Acute GVHD: pathogenesis and classification. Bone MarrowTransplant 2008;41 Suppl: S58–64. [DOI] [PubMed] [Google Scholar]
- 64.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus- host disease. Lancet 2009;373:1550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Bekkum DW, Roodenburg J, Heidt PJ, van der Waaij D. Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. J Natl Cancer Inst 1974;52:401–4. [DOI] [PubMed] [Google Scholar]
- 66.Jones JM, Wilson R, Bealmear PM. Mortality and gross pathology of secondary disease in germfree mouse radiation chimeras. Radiat Res 1971;45:577–88. [PubMed] [Google Scholar]
- 67.Piper HG, Fan D, Coughlin LA, Ho EX, McDaniel MM, Channabasappa N, et al. Severe gut microbiota dysbiosis is associated with poor growth in patients with short bowel syndrome. [Epub ahead of print] JPEN J Parenter Enteral Nutr July 12,2016. as doi: 10.1177/0148607116658762. [DOI] [PubMed] [Google Scholar]
- 68.Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal Blautia is associated with reduced death from graft-versus-hostdisease. Biol Blood Marrow Transplant 2015;21:1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridiastrains from the humanmi- crobiota. Nature 2013;500:232–6. [DOI] [PubMed] [Google Scholar]
- 70.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011;331: 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathewson ND,Jenq R,Mathew AV,Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 2016;17:505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simms-Waldrip TR, Sunkersett G, Coughlin LA, Savani MR, Arana C, Kim J, et al. Antibiotic-induced depletion of anti-inflammatory Clostridia is associated with the development of GVHD in pediatricstem cell transplant patients. Biol Blood Marrow Transplant 2017;23: 820–9. [DOI] [PubMed] [Google Scholar]
- 73.Alonso CD, Treadway SB, Hanna DB, Huff CA, Neofytos D, Carroll KC, Marr KA. Epidemiology and outcomes of Clostridium difficile infections in hematopoietic stem cell transplant recipients. Clin Infect Dis 2012;54: 1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weber D, Oefner PJ, Hiergeist A, Koestler J, Gessner A, Weber M, et al. Low urinary indoxyl sulfate levels early after transplantation reflect a disrupted microbiome and are associated with poor outcome. Blood 2015; 126:1723–8. [DOI] [PubMed] [Google Scholar]