

Dear Editor
With great interest have we read the recent paper “Unexplained gastrointestinal symptoms: Think mitochondrial disease” by Thomas P. Chapman and colleagues in Digestive and Liver Disease.[1] The authors present three pedigrees with complex mitochondrial systemic disorders featuring prominent gastrointestinal manifestations, which, in one family, is due to mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). The article highlights the notorious intricacies and entwined misdirections in diagnosing mitochondrial diseases, in general, and MNGIE, in particular. MNGIE is an uncommon multisystemic disorder owing to autosomal recessive, loss-of-function thymidine phosphorylase gene (TYMP) mutations, causing unbalanced cellular nucleoside pools with secondary mitochondrial DNA (mtDNA) instability and ensuing mitochondrial dysfunction.[2] Like Chapman and colleagues, we have also encountered patients, with severe gastrointestinal symptoms due to mitochondrial disorders that are misdiagnosed as eating disorder. Such individuals are mostly referred to gastroenterology clinics, which are often not familiar with rational diagnostic approaches to this disease category, which are typically clinically heterogenous even within families with the same genetic defect. By contrast, MNGIE stands out by virtue of its uniform clinical presentation which includes hallmark GI dysmotility up to full-blown chronic intestinal pseudoobstruction (CIPO). In a large cohort of French CIPO patients, 6.25% had MNGIE while 19% mitochondrial defects.[3] Hirano et al. have elaborated on and defined clinical criteria for MNGIE comprising signs of impaired intestinal motor function, external ophthalmoplegia/ptosis, severe weight loss (due to a combination of defective GI transit and impaired energy metabolism), peripheral neuropathy, which may be subclinical early in the disease, and leukocencephalopathy that is characteristically asymptomatic.[4] Cerebral MRI (which had not been performed in the reported MNGIE pedigree) consistently demonstrates striking diffuse hyperintensities in the cerebral white matter, which is an important diagnostic clue for MNGIE.[5] Absence of diffuse leukoencephalopathy on brain MRI makes TYMP mutations unlikely and indicates alternative diagnoses such as a MNGIE-like disorder due to mutations in the POLG gene, encoding the mtDNA polymerase-γ.[6] Given appropriate clinical and radiology findings, diagnostic confirmation of MNGIE is performed by specialized biochemistry assessment of plasma and/or urine thymidine (dThd) and deoxyuridine (dUrd) levels or by enzymatic assessment of buffy coat thymidine phosphorylase (TP) activity . These tests are technically demanding and performed by a few specialized laboratories. In addition, or alternatively, we recommend diagnosing MNGIE by TYMP sequencing, which allows genetic screening of kindreds. A proposed diagnostic algorithm for MNGIE is shown in the Figure. It is noteworthy that the proposed diagnostic tools are non-invasive as muscle and full-thickness intestinal biopsies (which may show mitochondrial alterations not specific for MNGIE) are not required while laborious mtDNA analysis are not needed and can be performed for research purposes. With the advent of novel treatment options aimed at reducing pathogenic nucleoside levels including allogenic stem cell transplantation and encapsulation of TP within autologous erythrocytes, identification of presymptomatic and/or less-advanced MNGIE individuals is becoming a urgent clinical challenge, given that in advanced disease stages full reversibility of end-organ damage appears unlikely.[7–8]
We believe that the proposed diagnostic algorithm, albeit not evidence-based, might be helpful in demystifying the clinical diagnosis of MNGIE as the most relevant and clinicially discernible mitochondrial disease in gastroenterology.
Figure. Proposed clinical algorithm for MNGIE diagnosis.
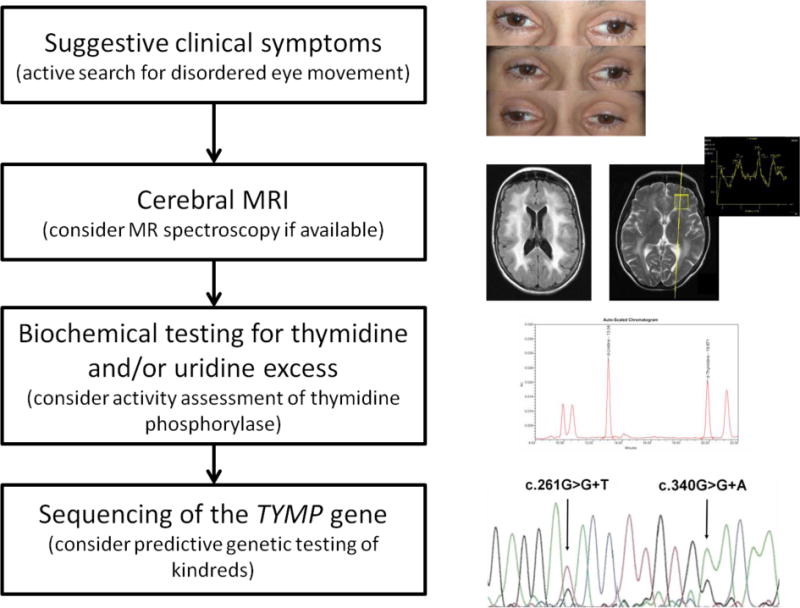
Clinical MNGIE suspicion may arise from variable signs GI dysmotility, severe cachexia, external ophthalmoparesis/ptosis, and (often subclinical) peripheral neuropathy. As a next step, radiology assessment by brain MRI to detect diffuse leukencephalopathy and, optionally, lactate peaks on MR spectroscopy. Biochemical testing for thymidine (dThd) and/or deoxyuridine (dUrd) excess in plasma and/or urine with optional assessment of thymidine phosphorylase (TP) activity. Sequencing of the thymidine phosphorylase (TYMP) gene should be performed to confirm the diagnosis and to identify presymptomatic and asymptomatic mutation carriers in kindreds. (clinical material from the patient reported in [9])
Footnotes
Conflict of Interest: Nothing to declare
Contributor Information
Vincent Zimmer, Department of Medicine II, Saarland University Medical Center, Homburg, Germany.
Michio Hirano, Department of Neurology, Columbia University Medical Center, New York, NY, USA.
Anna Zimmer, Institute of Diagnostic and Interventional Neuroradiology, Saarland University Medical Center, Homburg, Germany.
Frank Lammert, Department of Medicine II, Saarland University Medical Center, Homburg, Germany.
References
- 1.Chapman TP, Hadley G, Fratter C, et al. Unexplained gastrointestinal symptoms: Think mitochondrial disease. Dig Liver Dis. 2013 doi: 10.1016/j.dld.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Zimmer V, Feiden W, Becker G, et al. Absence of the interstitial cell of Cajal network in mitochondrial neurogastrointestinal encephalomyopathy. Neurogastroenterol Motil. 2009;21:627–31. doi: 10.1111/j.1365-2982.2009.01264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amiot A, Tchikviladze M, Joly F, et al. Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction. Gastroenterology. 2009;137:101–9. doi: 10.1053/j.gastro.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 4.Hirano M, Silvestri G, Blake DM, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–7. doi: 10.1212/wnl.44.4.721. [DOI] [PubMed] [Google Scholar]
- 5.Millar WS, Lignelli A, Hirano M. MRI of five patients with mitochondrial neurogastrointestinal encephalomyopathy. AJR Am J Roentgenol. 2004;182:1537–41. doi: 10.2214/ajr.182.6.1821537. [DOI] [PubMed] [Google Scholar]
- 6.Van Goethem G, Schwartz M, Lofgren A, et al. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet. 2003;11:547–9. doi: 10.1038/sj.ejhg.5201002. [DOI] [PubMed] [Google Scholar]
- 7.Hirano M, Marti R, Casali C, et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67:1458–60. doi: 10.1212/01.wnl.0000240853.97716.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bax BE, Bain MD, Scarpelli M, et al. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology. 2013;81:1269–71. doi: 10.1212/WNL.0b013e3182a6cb4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmer V, Zimmer A, Hirano M, et al. Recalcitrant vomiting, disturbed eye movements, and leukoencephalopathy. Mitochondrial neurogastrointestinal encephalomyopathy. Gastroenterology. 2009;137:1581–1861. doi: 10.1053/j.gastro.2009.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]