

Summary
Memory is an adaptation to particular temporal properties of past events, such as the frequency of occurrence of a stimulus or the coincidence of multiple stimuli. In neurons, this adaptation can be understood in terms of a hierarchical system of molecular and cellular time windows, which collectively retain information from the past. We propose that this system makes various time scales of past experience simultaneously available for future adjustment of behavior. More generally, we propose that the ability to detect and respond to temporally structured information underlies the nervous system’s capacity to encode and store a memory at molecular, cellular, synaptic and circuit levels.
Keywords: synaptic plasticity, memory encoding, memory consolidation, coincidence, information storage, temporal hierarchy, pattern extraction, phosphorylation, cell signaling, long-term potentiation
Introduction
Synaptic plasticity is generally accepted as the principal implementation of information storage in neural systems (Kandel et al., 2014). It involves relative and/or absolute changes in synaptic strength, established by a wide range of mechanisms and persisting for various periods of time, ranging from seconds and minutes to days, weeks and in the limit, a lifetime.
While “synaptic plasticity” is a relatively unambiguous term applicable to a specific cellular phenomenon, the basic concept of memory is broader, more abstract and thus more open to interpretation. For the purposes of this review, we consider memory as any perturbation in a system, caused by external stimulation, which persists past the cessation of the initial stimulation and alters the system’s responsiveness to subsequent stimulation. Thus memory, in the broadest of terms, is an adaptation to the past. We should point out that this definition of memory can also apply to many perturbations in non-biological systems, including viscoelastic deformation, anomalous diffusion, capacitor voltage changes, and stock market fluctuations (Sun et al., 2011; Westerlund, 1991). However, in biological systems, memory underlies the ability to adapt behavior to experience and thus allows an organism to increase its fitness ontogenetically. As Schacter and colleagues put it, one “remembers the past to imagine the future” (Schacter et al., 2007). Brains can thus be understood as prospective devices.
Why is synaptic plasticity a highly suitable mechanism for the implementation of memory? The primary reason is that changes in synaptic strength are not only stochastic or genetically preprogrammed, but dependent on the previous history of the synapse – in other words, the current state of a synapse is a function of its past states. A pattern of synaptic stimulation (whether by transmitters, modulators, cytokines, or changes in membrane potential) produces a cellular response in either or both the pre- and postsynaptic neurons. This response in turn alters the way subsequent stimulation produces subsequent synaptic responses, i.e. causes a change in synaptic strength. This feedback is what defines memory as an adaptive response.
Various forms and classifications of synaptic plasticity have been characterized. The distinction between short-term, intermediate-term and long-term plasticity is based on the mechanistic requirements for their formation (Goelet et al., 1986). Short-term plasticity, lasting seconds, depends on post-translational modifications of pre-existing proteins. New protein synthesis is required for intermediate-term plasticity lasting minutes to hours (Sutton et al., 2002), and new gene expression for long-term plasticity, which typically lasts for hours or longer. Additionally, plasticity can be expressed either as an increase (potentiation) or decrease (depression) of synaptic strength. Finally, plasticity can be expressed either presynaptically, postsynaptically, or both (Kandel et al., 2014).
While synaptic plasticity has been analyzed in neuroscience literature for decades, its direct empirical connection to memory is often more in the realm of metaphor rather than mechanism, largely because the term “memory” can have many meanings in a wide range of contexts. Yet a deep conceptual understanding of memory is critical for the general field of neuroscience. Ideas such as memory trace, memory acquisition, memory consolidation, or memory retrieval, to name a few, are routinely used to formulate scientific hypotheses. While certainly open to various definitions, they linguistically presuppose the idea of “a memory”, which may be seen as separate from the “memorizer”, who acquires it by “memorizing”. Synaptic plasticity in this paradigm is implied to aid the memorizer in the memorizing procedure. Various mechanisms of synaptic plasticity, such as phosphorylation of ion channels or expression of regulatory genes, are typically said to “contribute”, “underlie”, or “be required for” memory formation, which is seen as a distinct, if elusive, end to the mechanistic process of memory acquisition.
This general approach may be justified if memory is defined solely through its behavioral expression. From a behavioral standpoint, memory can indeed be easily parsed into stages of acquisition, retention, and expression or retrieval. This disassociation of “memory input” and “memory output” in turn necessitates a concept of a memory store, an idea that historically has been central to memory research (Fig. 1). Multistore models of memory (Fig. 1A, a) explain the transition between short- and long-term memory by the transfer of information between physically distinct stores. Depth of processing models (Fig. 1A, b) presume that distinct temporal domains of memory are localized to the same store, but are processed differently (Craik, 1972). Yet seen from a modern cellular and molecular biological perspective, behavioral categorization of memory stages imposes an arbitrary and perhaps unnecessarily restrictive “scale constraint” on what amounts to behavior and therefore memory in the first place. For example, a movement of a limb is typically considered behavior, whereas a movement of an ion across the cell membrane is not, and thus can be part of a “memory mechanism”, but not the “memory” itself if the definition of memory requires the additional constraint of behavioral expression.
Fig. 1. Frameworks for understanding memory.
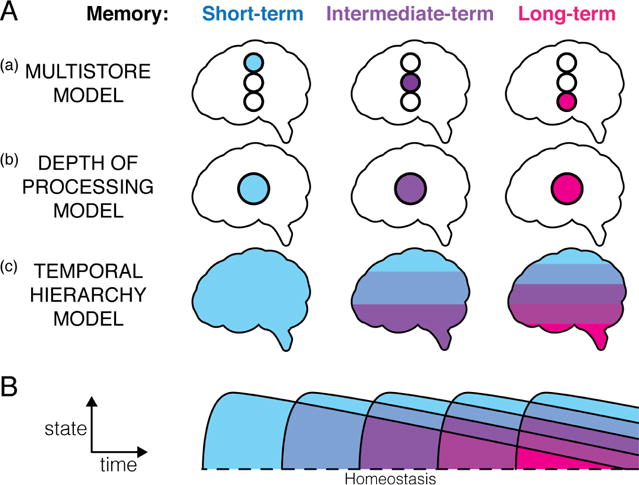
A. Theoretical models explaining the transition between short-term, intermediate-term and long-term memory. (a) In multistore models learned information is progressively passed on between multiple stores utilized for short- or long-term retention. (b) In depth of processing models the same store transitions from short- to long-term retention of information (Craik, 1972). (c) In the temporal hierarchy model advocated in this review, many levels of short- and long-term information simultaneously contribute to ongoing experience at any given time. No particular store can be isolated within the complete biological system that retains information. B. Experience can be seen as a series of temporally limited deviations from homeostasis. The temporal structure of experience, illustrated by coloration, defines the temporal structure of memory shown in A. For example, repeated-trial learning simultaneously retains information from the most recent trial as well as from all combinations of all preceding trials.
In this review, we agree with and extend a broader view that memory cannot be viewed separately from the system that encodes it (Milner et al., 1998), i.e. the “memorizer”. No single object, process, or state represents “a memory”. No “store” can be isolated as a particular substructure within the complete biological system that retains external information. Rather, the structure of memory lies in the temporal domain. Experience can be seen as a series of temporally limited deviations from homeostasis, the timing of which determines the structure of resulting memory (Fig. 1B). Memory is inherently and fundamentally multi-leveled, with each level represented as a distributed combination of distinct physical entities forming a hierarchy of cause and effect (Fig. 1A, c). Instead of treating memory as an end to memorization, we view biological memory, and specifically synaptic plasticity, as functions of a holistic, dynamic, hierarchically structured system that represents the timing of past events. It consists of multiple nested levels of molecular, cellular and higher-order homeostatic perturbations, each with particular temporal properties. Levels of this hierarchical system are linked to each other through both emergence (coincidence detection) and recursion (feedback).
A key feature of this system is its ability to represent temporal particularities of past experience which span milliseconds to years (Fields et al., 2005; Fuster and Bressler, 2012; Loewenstein and Sompolinsky, 2003; Markram et al., 1997; Mons et al., 1999; Tsien, 2000). These physical representations of the past range from single atoms to neuronal populations and states of entire organ systems, which allows the organism to use many past timescales simultaneously to modify ongoing behavior (Fig. 1). Long-term episodic memory is physically represented in countless variables of cortical and hippocampal state and structure and is expressed as autobiographical recall of polymodal experiences. Short-term facilitation (as revealed for example by paired-pulse facilitation) is physically represented in post-translational states of membrane ion channels, expressed as briefly altered membrane conductance, and constitutes memory of the fact that the synapse was used milliseconds ago (Buonomano and Maass, 2009; Zucker and Regehr, 2002). What is in common between these extremes of scale and complexity is the representation of temporal information retained from past events by the organism’s own devices.
We thus place time and temporal patterning at the center of the concept of memory and argue that the nervous system’s extraordinary ability to represent time at multiple time scales is a prerequisite for its unmatched capacity for information storage and, by extension, for its adaptive value.
It is important to stress at the outset that some of the themes that we will discuss in this review have been previously considered in a variety of contexts. This is especially true of several discussions addressing two major intersecting themes: (i) learning and memory (Kandel, 2012; Kandel et al., 2014; Korte and Schmitz, 2016), and (ii) temporal patterning (Buonomano and Maass, 2009; Feldman, 2012; Lynch et al., 2013; McGaugh, 1966; Philips et al., 2013; Smolen et al., 2016). So what is new in the current review? What we hope to accomplish here is to consider memory processing from a novel perspective. Specifically, we will develop the argument that identifying and elucidating specific and unique temporal domains in memory processing falls short of capturing the extremely wide range of nested temporal domains that are simultaneously operating in the service of encoding and storing long term memories. We will propose that this broader consideration of a wide range of scaled and integrated temporal domains can afford unique insights that can both elucidate the mechanistic complexity of memory storage, and suggest novel experimental approaches that have the capacity to significantly advance our understanding of the mechanisms of long-term memory storage in the nervous system.
This review is composed of four primary sections. We first discuss theoretically how the terms “coincidence”, “hierarchy” and “pattern extraction” apply to time representation in neurons, and consider the phrase “time window” as a reference to various homeostatic disturbances limited in time. In the following section, we address the hierarchy of these time windows, which we propose form a nested system of temporal pattern detection (Table 1). We discuss its molecular and cellular mechanics and their connection to synaptic plasticity. Finally, in the next section we discuss synergistic interactions between time windows, both within and across levels of the hierarchy. We conclude in the last section by summarizing the discussion and considering “time” more broadly as the key variable in the functioning of a nervous system.
Table 1. Classification of neuronal time windows.
Descriptions and examples of regulatory inputs related to temporally restricted homeostatic disturbances in the neuron. Similar classification can be applied to signaling in many non-neuronal cells.
| Nature of time window | ON inputs | OFF inputs | ||
|---|---|---|---|---|
| Description | Examples | Description | Examples | |
| First messenger availability | Release of the messenger by upstream effector cells | Firing of presynaptic cell; constitutive or regulated secretion of neurotrophins | Removal of messenger by diffusion or active transport | Reuptake of 5-HT by presynaptic termini |
| Transport or diffusion of the messenger to the receptor | Diffusion of neurotransmitter across synaptic cleft | |||
| Receptor state | Activation of receptor (binding to first messenger) | Dimerization and cross-phosphorylation of RTKs | Deactivation of receptor | Desensitization of GPCRs; glycine binding by NMDAR |
| Activation of receptor-coupled effectors | Dissociation of heterotrimeric G-proteins | Deactivation of receptor-coupled effectors | Guanine exchange in G-proteins facilitated by RGS proteins | |
| Second messenger availability | Production of second messenger | Lipid phosphorylation and cleavage, influx or release of Ca2+, cyclization of ATP or GTP, synthesis of NO, depolarization | Removal of second messenger | Dephosphorylation of lipids, removal of Ca2+ to internal stores, recruitment of DAG to anabolic pathways, hydrolysis of cyclic nucleotides, shunting of membrane potential |
| Stabilization of second messenger | Inhibition of phosphodiesterases, phosphatases, sustained changes in membrane potential | |||
| Diffusion of second messenger to the site of action | Diffusion of Ca2+ to the nucleus; spread of the membrane potential to the trigger zone | Diffusion of second messenger away from the site of action | Diffusion of cGMP away from cationic channels in rod photoreceptor cells; attenuation of membrane potential | |
| Target molecule state | Binding to second messengers or cofactors | Binding of calmodulin (CaM) to Ca2+, binding of AMPK to AMP | Dissociation of second messengers or cofactors | Dissociation of CaM and Ca2+ upon decrease in Ca2+ concentration |
| Covalent modifications | Mediated by ligases, e.g. kinases or ubiquitin ligases. Phosphorylation of ion channels in response to stimulation | Removal of covalent modifications | Mediated by hydrolases, e.g. phosphatases or DNA demethylases. Resetting of the epigenetic code | |
| Intermolecular interactions | Regulated assembly of ribosomes, proteasomes; binding of RISC to miRNA-mRNA complexes | Disassembly of complexes | Depolymerization of cytoskeletal filaments | |
| Target molecule availability | Transport or diffusion of the target molecule to the site of action | Translocation of kinesin-bound GluR2 to dendrites; diffusion of Ca2+/CaM to the nucleus; capture of CaMKII RNA at the synapse | Transport or diffusion of the target molecule away from the site of action | Removal of AMPARs from the postsynaptic density |
| Synthesis of target molecule | Availability and activation of transcription factors in the nucleus or translation factors in the cytosol; favorable epigenetic landscape | Degradation of target molecule | Proteolytic degradation of the PKA regulatory subunit | |
Abbreviations: 5-HT, 5-hydroxytryptamine; RTKs, receptor tyrosine kinases; GPCRs, G-protein-coupled receptors; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NMDAR, N-methyl-D-aspartate receptor; AMPK, 5′-adenosine monophosphate-activated protein kinase; RGS, regulator of G-protein signaling; DAG, diacylglycerol; CaM, calmodulin, CaMKII, Ca2+/calmodulin-dependent protein kinase II, PKA, protein kinase A.
It is beyond the scope of this review to consider all possible temporal interactions observed in neurons. Rather, by considering specific examples, we aim to provide a conceptual framework through which any new pathway contributing to synaptic plasticity can be understood. We posit that memory as a biological process is defined by a hierarchy of homeostatic perturbations that occur on multiple time scales and simultaneously represent the timing of past events during ongoing experience. Although representation of temporal relationships at the microcircuit and network levels is beyond the scope of this review, we propose that it may extend and parallel a temporal hierarchy entrenched in molecular and cellular biology of individual neurons, potentially representing a result of a unified evolutionary drive to adapt behavior to temporal regularities of the external world.
Temporal interactions and feedback in synaptic plasticity: theoretical considerations
Coincidence and continuity: an example drawn from NMDA receptors
In order to drive plasticity, events inside and outside of the neuron must co-occur within a defined temporal range. To illustrate this principle of coincidence, we will consider a well-characterized example of the N-methyl-D-aspartate receptor (NMDAR), an important regulator of synaptic plasticity. A key feature of NMDARs is their permeability to Ca2+ ions, which, upon entry into the postsynaptic cell, trigger a multitude of downstream biochemical cascades. However, to permit calcium entry, NMDA receptors must be simultaneously exposed to the neurotransmitter glutamate and membrane depolarization (Markram et al., 1997; Spruston et al., 1995; Yuste et al., 1999). Neither exposure can be seen as a discrete event. Both transmitter availability and membrane depolarization are alterations in states persisting within a specific and limited temporal window. Depolarization that activates NMDARs can arise from several sources. For example, backpropagating action potentials (bAPs) (Blanke and VanDongen, 2009; Spruston et al., 1995; Stuart et al., 1997), which are longer in duration than axonal spikes and allow for a relatively persistent window of depolarization, can be sufficient to cause dissociation of an inhibitory Mg2+ ion from NMDAR (Blanke and VanDongen, 2009). Depolarization can also arise from dendritic stimulation alone, either independently of bAPs or synergistically with them (Blanke and VanDongen, 2009; Enoki et al., 2004; Gordon et al., 2006; Schiller et al., 2000; Schiller and Schiller, 2001). When pairs or trains of EPSPs occur, NMDARs amplify the later EPSPs (Clark and Collingridge, 1996; Collingridge et al., 1988; Nicoll et al., 1992; Thomson, 1997; Thomson et al., 1988), suggesting that ongoing local glutamatergic stimulation of dendrites can continuously recruit NDMAR channels independently of bAPs or dendritic spikes. Backpropagating and/or persistent states of dendritic depolarization act to greatly increase the number of NDMAR channels active and to prolong their activity. Thus, all sources of depolarization converge to open a single temporally defined window during which glutamatergic input can engage NMDAR activation.
This example illustrates that “coincidence” is not, in fact, an all-or-none state when applied to a neuron, a synapse or even a population of molecules. Depending on the temporal properties of depolarization (e.g. whether it arises from bAPs or ongoing local stimulation) and transmitter availability (which in turn depends on the timing of action potentials arriving at the presynaptic neuron as well as the diffusion and uptake of the neurotransmitter), either or both of the criteria for calcium influx through a given NMDAR may be satisfied for various periods of time. For a population of NMDARs, coincidence, and by extension its downstream effect – aggregate Ca2+ influx – is more appropriately described a temporally graded continuum rather than an immediate and discrete state (Gerstner and Kistler, 2002).
Similarly, the biochemical regulation of complex systems such as molecular pathways, gene networks, or cellular ensembles typically proceeds in a temporally graded, continuous manner. To emphasize this continuity, we find it helpful to re-evaluate the traditional molecular biological terminology of “activators”, “inhibitors” or “targets”. “Single-molecule language” reinforces the impression of discrete-state mechanics, whereby enzymes or genes can be either active, or inactive, depending on the upstream signals. In this review, we utilize the terms “ON and OFF pathways” to refer to temporally graded inputs contributing to a given response. Each ON or OFF pathway is an abstraction of many individual activation/inhibition events.
Hierarchy of timing: an example drawn from cAMP signaling
There is no single event, or even single type of event, that causes synaptic plasticity. Instead, the cell possesses a vast repertoire of temporal windows defined by the state and availability of individual molecules or systems of molecules (Buonomano and Maass, 2009). These windows range from the transiently activated state of NDMARs in a depolarized membrane to long-term changes in transcriptional or even epigenetic state of the cell. Any such window is characterized by ON and OFF mechanisms which together determine its timing. These temporal windows are organized into a hierarchical system, which can be effectively illustrated by considering another relatively well understood example, cAMP signaling.
cAMP is produced from ATP by the action of adenylyl cyclases. Canonically, an adenylyl cyclase is activated by a G-protein, which provides the ON mechanism for a time window of elevated cAMP during signaling via G protein-coupled receptors (GPCRs). This window is closed by phosphodiesterases, enzymes that hydrolyze cAMP. Needless to say, the two reactions do not proceed in isolation or sequentially. It is more informative to think of a steady intracellular cAMP level in a state of equilibrium between ON and OFF mechanisms. When adenylyl cyclase is activated, the equilibrium shifts towards a higher level of cAMP. This level persists until activation of adenylyl cyclase subsides, e.g. when the extracellular signal causing its activation is terminated. OFF mechanisms then take over by simple Michaelis-Menten kinetics, and cAMP levels return to the steady state. The transient state of cAMP increase produces a transient state of PKA activation, which increasingly phosphorylates downstream proteins. Thus, a temporal window of elevated cAMP serves as an ON mechanism for a different temporal window, defined by the increased quantity of PKA-phosphorylated proteins. The OFF mechanism for this novel window is provided by dephosphorylation or protein turnover, ultimately returning the levels of phosphorylation to the baseline. A temporal window conditional on a particular pattern of events can therefore itself serve as a condition for novel, emergent temporal windows, establishing a “hierarchy of timing”. As we will discuss below, this hierarchy is not fixed in stone since temporal windows of different levels interact to produce emergent responses.
“Time windows” as temporally restricted homeostatic disturbances
The term “time window” is commonly used to refer to a period of time with a distinct beginning and end, particularly when the state of a system is somehow altered within these given temporal limits (Fulton et al., 2005; Montarolo et al., 1986; Seidenbecher et al., 1997; Tsodyks and Markram, 1997). It is important to underscore that for the purposes of this review we define a “time window” differently from “time period”. Biological molecules do not generally “respond” to time per se (as could be argued for radioactive nuclei). They respond to chemical or physical covariates of time in a dynamic system, such as disturbances in levels of second messengers or effector proteins. In order to be meaningful (i.e., to carry information), these covariates must have self-limiting dynamics. For example, in order for transient increases of intracellular cAMP to serve a signaling purpose, a delayed mechanism to return cAMP to an equilibrium level must be present. It is these temporally self-limiting disturbances in cellular homeostasis we will refer to as “time windows”. Absolute time is therefore only one of the variables in “molecular time”.
This definition of a “time window” also includes higher-order temporal features of homeostatic disturbances, such as amplitude and rates of onset or decay. This is important since first- or second-order kinetics can carry temporal information themselves. For example, a rapid, high and short rise in postsynaptic calcium is required for induction of LTP, whereas a slow, low and long-lasting rise is needed for LTD (Evans and Blackwell, 2015). It is difficult to disambiguate individual roles for the variables of (i) time, (ii) rate of increase and (iii) peak value of calcium, all of which contribute to the instantaneous intracellular concentration of calcium, the ultimate factor responsible for the plasticity-driving function of calcium influx. By artificially buffering calcium concentration at a constant level, the effect of timing can be disambiguated from the effect of peak value and rates of onset/decay. However, effector molecules do not discriminate between calcium concentrations produced by slow or fast kinetics – the only variable each individual molecule “takes into account” is instantaneous calcium concentration. Expression of LTP or LTD is therefore an emergent response to a particular pattern of calcium fluctuation, a product of integrating coincidence of activity of many effector molecules over the time-course of calcium changes. Higher-order temporal variables such as rates and amplitudes can be ultimately reduced to the timing of upstream events.
To summarize, “molecular time” is measured in relative temporal windows, which are themselves a function of the state of their respective ON and OFF pathways (Fig. 2). Importantly, “ON” and “OFF” do not denote binary switches. We will use these terms simply to indicate pathways that contribute to either the onset or decay of the state underlying the time window.
Fig. 2. Hierarchy of time windows.
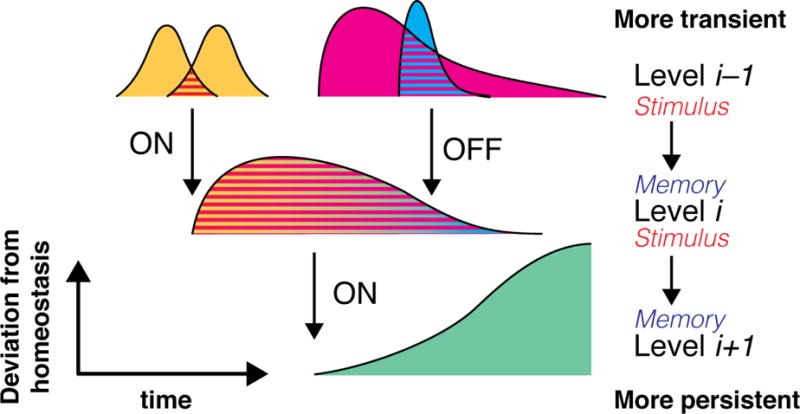
A “time window” is a temporally self-limiting event that constitutes a deviation from homeostasis. Specific patterns of time windows at level i−1 (e.g. overlap between time windows of first messenger availability) contribute ON or OFF influences towards a time window at level i (e.g. a time window of second messenger availability), converting transient stimuli into a more persistent cellular response which can influence the cell’s response to subsequent stimulation. This novel time window represents a form of memory, an adaptive response to the past. This timed response, typically in combination with other inputs/time windows, can in turn contribute ON or OFF influences towards a time window at level i+1 (e.g. a time window of increased protein phosphorylation). Thus, a memory at level i is at the same time a stimulus contributing to a more persistent memory at level i+1.
Pattern extraction as temporal emergence
A final term that requires consideration is “pattern extraction”. In abstract terms, any process is characterized by a particular temporal profile of its occurrence, and therefore mere responsiveness of a system to such timed processes is trivial. Pattern extraction, on the other hand, is defined here as an emergent response to multiple time windows that by themselves do not have the capacity to create the response. To use an everyday example, a single tap on a touchscreen device produces a response to an event, whereas a double-tap (that is, two taps separated by a specific, limited time period) produces a response to a pattern, which the device extracts from the operator’s actions. This response is emergent, since it is derived from the responses to individual taps but not simply a sum of the two.
As discussed above, a transient intracellular calcium elevation is an emergent response to a temporal overlap of membrane depolarization and transmitter binding by NMDAR. As we will show below, such emergent responses themselves constitute time windows that are used for pattern extraction at higher hierarchical levels, which typically correspond to longer time periods and more stable changes in synaptic strength.
Emergent responses produced within the hierarchy typically feed back onto the previous levels of detection and alter their subsequent responsiveness. For example, a transient NMDAR- dependent calcium elevation may cause more lasting changes in the composition of the membrane receptors, resulting in stronger depolarization following the same level of neurotransmitter. Since depolarization contributes one of the ON inputs for NMDAR activation, it alters the time window of subsequent calcium entry upon repetition of stimulation, increasing its probability of interaction with other time windows in the cell, thus producing even more lasting responses. Since we have defined memory as a persistent perturbation caused by past stimulation and altering the effect of future stimulation, it follows that biological memory is fundamentally hierarchical in nature.
Neuronal time windows: all good things must come to an end
First messenger availability and receptor state
First messengers include any extracellular signals that evoke a specific response in a target cell. These may include neurotransmitters, cytokines, growth factors, hormones and even extracellular ions such as calcium (Hofer and Brown, 2003). These molecules typically transduce their effects in the cell by binding to extracellular domains of membrane-spanning receptors, although some membrane-permeant molecules such as nitric oxide, arachidonic acid, and glucocorticoids, all of which have been implicated in synaptic plasticity, exert their effects intracellularly (O’Dell et al., 1991; Pavlides et al., 1996). Environmental factors such as heat (Caterina et al., 1997), light (Kuhn and Dreyer, 1972), or stretch (Vandorpe et al., 1994) can also be viewed as first messengers if they elicit a specific (i.e., receptor-driven) response.
Second messenger availability
The term “second messenger” is usually applied to a small molecule whose main function is to communicate functional states between intracellular biomolecules. For example, cyclic nucleotides allow transmission of dopamine receptor activation to PKA activation (Otmakhova and Lisman, 1998; Stoof and Kebabian, 1981). Depolarization or hyperpolarization of the membrane can be thought of as special cases of second messenger signaling. Fluctuations in instantaneous polarization are generally triggered by first messengers (neurotransmitters) opening ligand-gated ion channels, but additionally by voltage-gated channels that respond to changes in membrane polarization. Thus, the extent of membrane depolarization in a given spatial domain can be viewed as conceptually equivalent to a given concentration of a second messenger. The advantage of “voltage signaling” over conventional chemical signaling is its unparalleled speed, which is particularly useful for communicating signals rapidly between distant parts of a cell.
Most signaling pathways in eukaryotic cells show at least some degree of compartmentalization. Thus for example, cAMP availability does not provide a single time window, but a family of time windows defined by distinct subcellular locations, corresponding ON/OFF pathways, and downstream effects. In both cultured Aplysia sensory neurons and neurons of the intact lobster stomatogastric ganglion, neuromodulatory stimulation with 5-HT produces a rapid and transient elevation of cAMP primarily in fine neurites. After prolonged stimulation however, cAMP diffuses throughout the dendritic tree and eventually to the cell body (Bacskai et al., 1993; Hempel et al., 1996). This diffusion is significant since cAMP and PKA can have different functions in distinct subcellular compartments (Liu et al., 2004). Hypothetically, the “escape” of cAMP from fine neurites can be explained by synergy of ongoing cAMP production with the saturation of relevant sub-membrane OFF pathways, such as phosphodiesterase activity. Indeed, various PDE isoforms can sculpt cAMP gradients through highly compartmentalized hydrolysis (Houslay, 2010). This example illustrates that “molecular time” defined by second messenger availability and employed for information storage is a product of many competing non-linear influences with their own “temporal agenda”.
State of the target molecule
Many intracellular protein ensembles exist in a state of equilibrium, or stable non-equilibrium (steady state) (Henzler-Wildman and Kern, 2007; Nooren and Thornton, 2003). At the structural level, cytoskeletal filaments, ribosomes, proteasomes and many other quaternary and supramolecular complexes are continuously assembled and disassembled. Metabolic pathways are similarly controlled within a homeostatic system that has been likened to a cellular economy of supply and demand (Hofmeyr, 2008; Hofmeyr and Cornish-Bowden, 2000). Post-translational modifications must be reversible in order to serve a regulatory function. Correspondingly, opposing enzymatic pathways exist for modifications with phosphate, ubiquitin and ubiquitin-like modifiers, methyl and acetyl groups, ADP-ribose, O-GlcNAc and other regulatory modifications occurring in the cytosol and nucleoplasm. Structural and enzymatic interactions are tightly linked and often depend on each other. Together, they represent a vast variety of molecular states that can be used to generate time windows. Many of these states are controlled by the time windows of binding to upstream cofactors and second messengers.
Because post-translational modifications are not encoded in the genome, and because proteins (as opposed to first and second messengers, as well as RNA and DNA) are the primary mediators of biological function, post-translational regulation is ideally suited for implementing experience-dependent changes of cellular function. It is therefore not surprising that most persistent states that underlie intracellular temporal computations are largely defined by the post-translational modifications of proteins and protein ensembles.
Availability of the target molecule
It is well established that various forms of long-term memory and long-term potentiation require RNA and/or protein synthesis for induction and consolidation (Davis and Squire, 1984; Flexner et al., 1963; Martin et al., 1997; Nader et al., 2000). In both cases, the requirement is typically temporally restricted, setting up critical temporal windows during which gene expression or protein synthesis must take place in order for lasting changes in the synaptic state to persist (Bourtchouladze et al., 1998; Montarolo et al., 1986; Pearce et al., 2017).
These translational and transcriptional temporal windows are not easily separated for three primary reasons. First, they are often, although not always, causally and temporally coupled. The onset of transcription typically determines the onset of translation, although additional factors (e.g. transport of RNA, or recruitment of translation machinery) synergistically contribute regulation. Second, both transcription and translation can be regulated at the protein level. For example, degradation by the ubiquitin-proteasome system can control both the availability of proteins and, indirectly, the availability of RNA by targeting transcription factors in the nucleus (Muratani and Tansey, 2003). Finally, RNA and protein synthesis in eukaryotic cells proceeds in distinct subcellular locations and requires highly regulated bidirectional trafficking between the nucleus, soma, and dendrites to ensure functional consequences of both RNA and protein synthesis in neurons (Poon and Jans, 2005).
For these several reasons, it may be more informative to refer to temporal windows of RNA and protein availability at a given subcellular location, taking into account all relevant temporally defined constraints. By definition, availability of a protein is dependent on, but not fully accounted by, the availability of its RNA.
Regulation of availability at a given location can also be achieved by redistribution of pre-existing proteins from other locations. Translation-dependent and translation-independent regulation of local protein availability can converge. For example, local availability of Ca2+/calmodulin-dependent protein kinase II (CaMKII) at the postsynaptic density (PSD) increases following high-frequency stimulation (HFS) (Shen and Meyer, 1999). This is achieved immediately by redistribution of existing kinase but is subsequently supported by local protein synthesis (Wu et al., 1998) and subunit exchange (Stratton et al., 2014). Induction of LTP is associated with rapid increases in the spine content of αCaMKII mRNA (Havik et al., 2003). In cultured mouse hippocampal neurons, this was observed within 10 min after HFS (Kao et al., 2010). Additionally, HFS causes redistribution of polyribosomes from the dendritic shafts into spines, observed 2 h after stimulation (Bourne et al., 2007; Ostroff et al., 2002). Thus, both translation-independent and translation-dependent mechanisms of regulating CaMKII availability converge at the PSD with characteristic temporal profiles.
Indeed, the interaction of initial, rapid-onset, protein synthesis-independent time windows of local protein availability with delayed, slow-onset, translationally and transcriptionally controlled temporal windows of global protein and RNA availability is a core mechanism for nuclear-to-cytoplasmic signaling underlying “tag and capture” models for synaptic plasticity (Barco et al., 2002; Bramham and Messaoudi, 2005; Redondo and Morris, 2011). For example, NMDAR calcium transients promote actin polymerization, which recruits microtubule entry into dendritic spines. This has been proposed to allow entry of molecular cargo distributed from the cell body into potentiated spines (Merriam et al., 2013), either constitutively, or as a consequence of previous stimulation. The essence of “tag and capture” is coincidence of a slow global alteration in homeostasis, caused by previous stimulation, with a fast local disturbance, triggered by ongoing activity.
More broadly, any form of neuronal plasticity involves conversion of a transient response (narrow time window) into a more persistent response (wide time window) by means of synergy with other responses, both transient and persistent (Fig. 2). Such conversion is not restricted to a single level, but instead occurs hierarchically, allowing for the progressive generation of more and more persistent responses to recurrent patterns of stimulation. A time window at a given level of the hierarchy can be seen simultaneously as “memory” of levels below and “stimulus” for levels above (Fig. 2). In the next section, we discuss several examples of such hierarchical conversion of time windows.
Synergy of time windows: transient-to-persistent conversion
Memory traces in the nervous system are widely believed to be stored as relative strengths of synaptic connections (Martin et al., 2000). Although debate still exists as to whether synaptic plasticity is sufficient to explain memory encoding and storage (Ryan et al., 2015), most studies support the view that a broader statement is warranted: storage of information in the nervous system is a function of neuronal states. Synaptic plasticity is one example of functionally significant alterations in the states of neurons, whereby changes in two synaptically coupled neurons result in net change in the probability of signal transmission between them. There are however other ways to alter the state of a neuron (with potential effects on information storage and transmission), which are not necessarily confined to specific synapses. For example, many forms of learning produce cell-wide changes in membrane components such as ion pumps and channels, which collectively alter cellular excitability and can profoundly influence the behavior of a neuronal network (Mozzachiodi and Byrne, 2010; Yizhar et al., 2011).
What determines the state of a neuron? At each given moment its state is a product of both currently active outside factors and pre-existing cellular conditions. These in turn are a product of previous genetic, maternal, developmental, epigenetic and/or environmental factors that had influenced the fate of the neuron from embryogenesis to synaptic plasticity in maturity. Aside from genetic and maternal factors, which are shared by all neurons in a mature organism, any other variable in the neuronal state is determined by forces acting from outside of the cell. This includes stimulation by any developmental factors; stimulation by other neurons, including synaptic connectivity; organism-wide signaling by the endocrine system, and many other external signals that elicit a response in a neuron or its precursor. The continuum of development and synaptic plasticity is highlighted by the fact that many mechanisms (e.g., the requirement for growth factor signaling) are shared among these processes (Bonhoeffer, 1996; Patterson and Nawa, 1993; Schinder and Poo, 2000).
Ontogenetic changes in neuronal states and hence synaptic plasticity can therefore be ultimately traced back to extracellular signals that neurons receive. These signals are typically transient, whereas changes that they elicit in the target cell can persist over a wide range of temporal domains. The key feature of temporal analysis by the neuron is therefore its ability to convert transient signals into persistent responses. This is achieved by nested synergistic interactions between time windows, starting from the those set by first messenger availability, followed by intracellular time windows defined by second messengers and target molecules. To better illustrate how such nesting of transient time windows contributes to the generation of persistent responses, we will primarily focus on the relatively well-characterized signaling pathway centered around 5-HT, cAMP, PKA, CREB and C/EBP.
First messenger synergy
We have considered a “first messenger” as any biologically active substance or physical force eliciting a controlled change in the state of a cell. How do first messenger time windows interact with one another? In some cases, the first messenger that triggers a memory is the same first messenger whose responsiveness is altered. Such is the case of typical hippocampal LTP induced by Schaffer collateral tetanization (Dudek and Bear, 1993). In this case, multiple identical rapidly repeated bouts of glutamate elevation in the vicinity of CA1 dendrites alter the response to subsequent repetitions of the same bouts of glutamate. In other cases, one first messenger can influence the responsiveness to other first messengers. For example, during sensitization of the Aplysia sensorimotor synapse, repeated bouts of 5-HT cause long-term facilitation by increasing neurotransmitter release from the presynaptic sensory neuron (Brunelli et al., 1976), a phenomenon called heterosynaptic plasticity. In this case, 5-HT alters the cellular response to mechanical stimulation of the skin, which for a sensory neuron can be seen as equivalent to a distinct first messenger.
These plasticity-inducing effects do not accumulate in a linear additive fashion. Instead, the neuron responds to particular temporal patterns of stimulation. Given sufficient temporal spacing, repeated stimuli are treated as single stimuli since the cell returns to homeostasis before the onset of the repetition. On the other hand, with no spacing at all the stimuli simply combine additively into a single stimulus of increased amplitude. “Massed” stimuli, however, do not generally produce the same effects as spaced stimuli, a difference that is consistently observed on multiple time scales of behavioral learning and its cellular analogs (Kornmeier and Sosic-Vasic, 2012; Smolen et al., 2016). Thus, neuronal learning depends on the specific temporal relationships between the time windows of first messenger availability. In other words, neurons learn by extracting temporal patterns from the history of their stimulation. The pattern can be as simple as high-frequency stimulation. In other cases, more complex temporal relationships between stimuli are extracted. One example is spike time-dependent plasticity (STDP), where the relative timing of a bAP and a synaptic input determines whether and to what extent the synapse will weaken or strengthen (Song et al., 2000). Functionally, STDP “rewards” connections that are “causal” to postsynaptic firing, and “punishes” the ones that are not (Abbott and Nelson, 2000). Since bAPs are themselves ultimately a result of external stimulation, STDP can still be viewed as an interaction between the timing of first messenger time windows.
Interactions between first messenger time windows are the ultimate source of information for a neuron. Yet first messengers themselves do not interact with one another directly. They elicit responses in the cell that occur with a novel temporal profile, typically persisting after the decay of the first messenger. These novel cellular time windows interact with responses elicited by other first messengers, both ongoing and carried over from previous stimulation. The simplest example is non-linear summation of depolarization caused by coincident synaptic inputs (Canepari et al., 2007; Costanzo et al., 1999; Wolf et al., 1998). Neurotransmitter binding throughout the dendritic arbor produces EPSPs and IPSPs which interact in a distinct temporal manner depending on the biophysical properties of the dendrite such as resistance, capacitance and the presence of voltage-gated currents (Curtis and Eccles, 1960; Davies and Collingridge, 1996; Ferster and Jagadeesh, 1992). Neurotransmitter timing windows therefore converge to produce emergent time windows of membrane depolarization at a given location. A similar example is provided by the interaction between depolarization and NMDAR activation/calcium influx, described above. To generalize, the most basic first messenger integration occurs at the level of second messengers, including depolarization as a special case.
Second messenger synergy
Second messengers represent a fundamental point of convergence for signaling by many distinct extracellular ligands. A striking example is olfaction. Mammalian genomes contain ~500–1000 olfactory receptor genes, subject to extensive genetic recombination (Buck, 1996; Hasin-Brumshtein et al., 2009). In any given organism this genetic diversity results in a unique repertoire of olfactory GPCRs. All of them however converge on the same second messenger: cAMP.
Second messengers therefore allow neurons to extract the most basic, rapid patterns of external stimulation and “generalize” the actions of multiple first messengers. Ions like calcium and small molecules like cyclic nucleotides provide the cell with a “temporal toolkit” of homeostatically controlled, extensively regulated, highly convergent and highly divergent regulatory nodes. Time windows of second messengers, produced in response to patterns of first messenger availability, elicit changes in the functional state of downstream proteins, also with characteristic temporal profiles often distinct from the second messenger time windows. These novel protein state windows interact with responses elicited by both first and second messengers. For example, Ca2+-dependent adenylyl cyclases display distinct temporal requirements for activation (Shobe et al., 2009; Yovell and Abrams, 1992). These enzymes are activated by the coincidence of a calcium time window with a neurotransmitter time window. In Aplysia, pairing of 5-HT and Ca2+ causes potentiation of cyclase activity if the Ca2+ influx immediately precedes 5-HT binding (Fig. 3b), but not if it follows the 5HT signal (Fig. 3a). FFiring of different presynaptic cells (1) determines the timing of 5-HT and glutamate release (2). The time window of 5-HT availability determines the onset of G-protein activation, which in turn triggers the activation of adenylyl cyclases (4) and thus the time window of increased production of cAMP (5). Glutamate bouts trigger time windows of depolarization (6, see also Section IA), which upon coincidence with ongoing glutamate bouts cause Ca2+ influx via NMDA receptors (7). This contributes to the broadening of EPSPs (widening of the depolarization time window) (8) and to the onset of a “receptive state” of Ca2+-dependent adenylyl cyclases (9). In order to contribute to cAMP production, this state must coincide with subsequent 5-HT-mediated activation (10). Thus temporal interactions between bouts of first messenger (glutamate) lead to a time window of a second messenger (Ca2+), which leads to a time window of a “receptive state” of an enzyme, which then interacts with a time window produced by another first messenger (5-HT) to produce distinct time windows of another second messenger (cAMP). Such “nesting” of time windows allows for progressive extraction and integration of patterns of increasing scale. This specificity of downstream response to a particular order of upstream events resembles the aforementioned “causality extraction” observed in STDP (Abrams et al., 1991; Yovell and Abrams, 1992).
Fig. 3. Nesting of time windows in the regulation of Ca2+-dependent adenylyl cyclases.
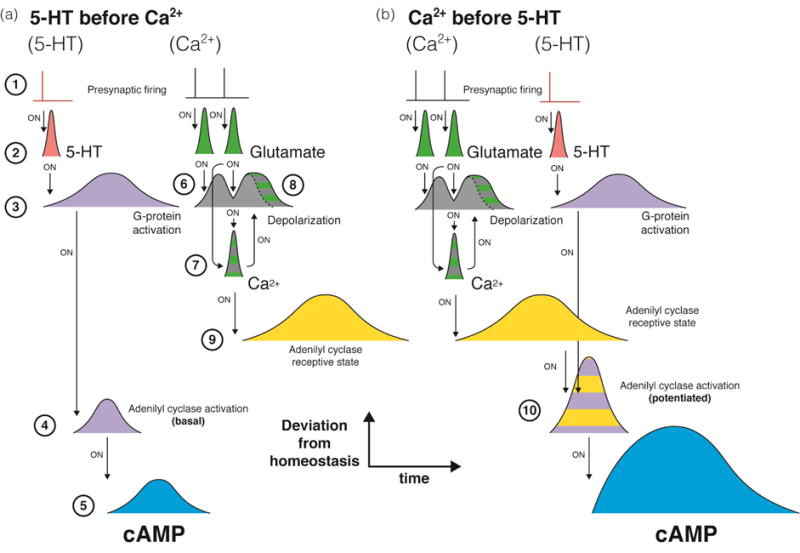
The timing of presynaptic firing (1) determines the onset of transmitter availability (2). For 5-HT, this determines the timing of G-protein activation (3) and consequently, the basal activation of ACs (4), which generate a time window of elevated cAMP (5). Multiple successive glutamate bouts produce relatively sustained depolarization (6), which, by synergizing with ongoing glutamate availability, results in Ca2+ influx via NMDARs (7). This leads to increased postsynaptic depolarization in response to glutamate availability (8) and to enhanced activity of Ca2+-dependent ACs (9). (a) When 5-HT precedes glutamate stimulation, enhanced activity of ACs does not coincide with G-protein activation. (b) When Ca2+ influx precedes 5-HT availability, the activation of ACs by G-proteins and enhancement by Ca2+ coincide (10), resulting in an extended time window of cAMP availability.
Synergy of target molecules: enzymatic interactions
Ultimately, patterns of first and second messenger fluctuations translate into fluctuations of protein states throughout the neuron. Typically, these higher-order fluctuations, “protein state time windows”, persist for a longer time, that is, beyond the time windows of first and second messengers. Biochemically, this persistence can be achieved by two means: (1) feedback loops leading to sustained production of the first/second messenger, or (2) autonomous states, whereby a protein becomes independent of the activating factor. We will discuss each in turn.
First-/second-messenger feedback loops
Persistent neuronal responses can be created by feedback loops involving first messengers. Such responses necessarily involve secretion of the first messenger into the extracellular space, and transmembrane signaling back into the cell. Such autocrine signaling by a secreted messenger can be linked to the production and secretion of that messenger, creating a form of positive or negative feedback. For example, in embryonic hippocampal neurons BDNF serves as a self-amplifying autocrine factor (Cheng et al., 2011). BDNF elevates cAMP and PKA activity, which leads to increased secretion of BDNF and membrane insertion of its receptor TrkB. Exogenous BDNF has also shown to induce dendritic accumulation of BDNF and TrkB mRNAs (Righi et al., 2000), suggesting the existence of a positive feedback loop acting through sustained release of a self-acting first messenger.
Alternatively, persistent feedback loops can be generated by sustained production of second, rather than first messengers. For example, network modeling suggests that PKC is initially activated by Ca2+ and DAG. PKC then indirectly stimulates MAPK, which activates cPLA2, which causes production of arachidonic acid, which activates PKC at the basal level of DAG (Bhalla and Iyengar, 1999). Multiple modes of stimulating PKC are therefore essential for the feedback loop, but ultimately the extended time window of PKC activity is achieved through sustained production of a second messenger.
Autonomy or kinases or kinase ensembles
cAMP/PKA
As discussed in section II, signaling through cAMP and protein kinase A provides a well-characterized example of temporal hierarchy employed in synaptic plasticity (Kandel, 2012). We will now consider this pathway in more detail to illustrate consolidation of transient cellular responses into persistent changes.
PKA consists of two catalytic subunits (C) and two inhibitory regulatory subunits (R) that dissociate upon binding of cAMP (Fig. 4, 2). cAMP (1) is produced by adenylyl cyclase, for example following stimulation of neurons with 5-HT or other neuromodulators such as BDNF in the examples above. This generates a short-term (minutes) time window for PKA activity (Hempel et al., 1996), which is closed by phosphodiesterases that hydrolyze cAMP and allow re-binding of R subunits to the C subunits (3). It is well-established that upon prolonged stimulation, catalytic subunits of PKA translocate to the nucleus, where they phosphorylate CREB1 (cAMP response element-binding protein 1) (4), a transcription factor triggering a vast array of nuclear responses to stimulation (see below). The PKA holoenzyme and the R subunit of PKA are both excluded from the nucleus (Bacskai et al., 1993).
Fig. 4. Persistent PKA activation by positive feedback.
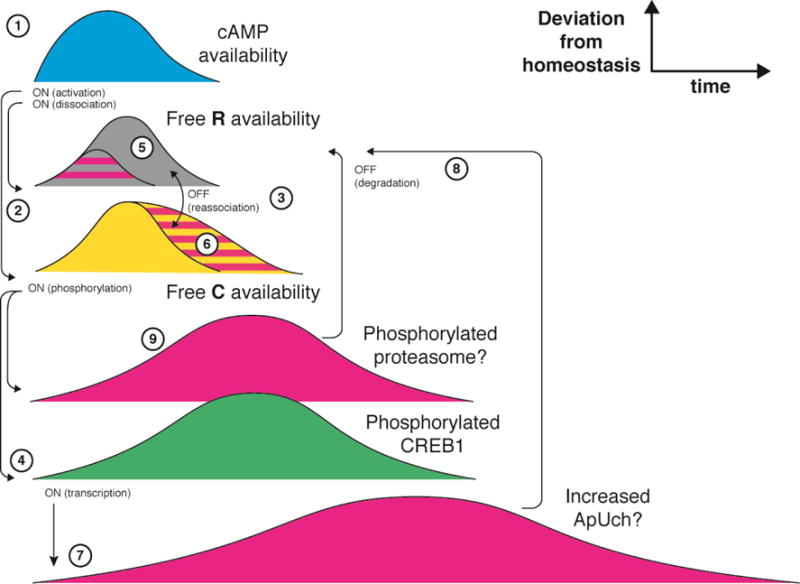
The time window of cAMP availability (1) determines the time window of PKA in a dissociated state (2) (both R and C subunits available in a free form). PKA is deactivated by reassociation of the subunits (3). Availability of free C determines the timing of phosphorylation of its substrates, including CREB-1 (4). This results in enhanced proteasomal activity, either by transcriptional induction of 26S activators such as ApUch (7, 8), or directly by PKA-mediated phosphorylation of the proteasome (9). These time windows of increased proteasomal activity shorten the time of R availability (5) and consequently extend the time window of C availability (6).
Separately, upon prolonged stimulation of sensory neurons with 5-HT, the R subunits are degraded via the ubiquitin-proteasome system (Bingol and Sheng, 2011; Chain et al., 1999; Hegde et al., 1993) (5), which results in prolonged, cAMP-independent activation of PKA (Chain et al., 1999) (6). Both binding to cAMP and dissociation of the holoenzyme are necessary but not sufficient for R subunit degradation. According to a long-standing model, initial dissociation of the holoenzyme requiring the presence of perinuclear cAMP causes CREB1-dependent transcription of many genes, including, in Aplysia neurons, ApUch (7), a deubiquitinating enzyme (DUB) required for proteasomal function which may increase its ability to process ubiquitinated substrates (Hegde et al., 1997) (8). This increases cAMP-dependent degradation of the R subunit and prolongs the activation of PKA in the absence of cAMP (Chain et al., 1999). Although the effect of proteasome-bound DUBs on their overall activity is controversial (Lee et al., 2011; Peth et al., 2009; Peth et al., 2013), other mechanisms may exist that link PKA activity with increased proteasomal function, including the newly discovered route for direct activation of 26S by PKA phosphorylation (Lokireddy et al., 2015) (9).
In either case, the regulation of downstream signaling by PKA involves multiple levels of synergy, whereby a pattern of transient perturbations produces a more lasting change. First, cAMP itself, and specifically perinuclear cAMP acts as a temporal integrator of receptor stimulation, with the ON mechanisms involving the rates of production and the kinetics of diffusion towards the nucleus, and the OFF mechanisms mediated by phosphodiesterases. The resultant time window of increased perinuclear cAMP acts as an ON signal for several other time windows. PKA activation/dissociation/susceptibility to C nuclear translocation and R degradation depend on the cAMP window most directly – that is, both the onset and decay of an altered PKA state are temporally controlled by local cAMP concentrations. Indeed, the extent of eventual translocation is proportional to the peak kinase activity in the vicinity of the nucleus (Bacskai et al., 1993). CREB1 activation as a time window integrates multiple ON inputs and independently controlled OFF inputs (such as dephosphorylation) and thus is temporally dissociable from cAMP elevation (Liu and Graybiel, 1996). Similarly, the dynamics of elevated proteasome activity are linked to but not exclusively controlled by cAMP levels (Upadhya et al., 2006).
The prolonged activation of PKA must therefore integrate multiple partially independent time windows. The increased activity of the proteasome resulting from a previous increase in cAMP synergizes with the current increase in cAMP needed to dissociate the PKA holoenzyme and make the R-subunit available for ubiquitination and proteolysis. This results in a novel, emergent time window – that of a locally decreased R:C ratio, with the direct effect of autonomous PKA activity. The window is probably closed by synthesis of new R subunits.
CaMKII
Many other kinases or kinase ensembles demonstrate conceptually similar mechanisms of establishing persistent states based on transient signals. Perhaps the richest example of second-messenger-induced autonomy is provided by CaMKII, a critical factor in long-term plasticity. This kinase is part of a broader CaMKII/calcineurin system, a powerful temporal integrator intricately tuned to the patterns of transient Ca2+ influx into the cell. Both CaMKII and calcineurin are activated by Ca2+/calmodulin (CaM), but while CaMKII tends to positively regulate synaptic strength (Malinow et al., 1989; Pi et al., 2010), calcineurin typically opposes LTP and is involved in LTD (Mulkey et al., 1994; Winder et al., 1998). While calciumindependent mechanisms for CaMKII activation exist (Erickson et al., 2008; Jalan-Sakrikar et al., 2012), canonically, the timing of incoming calcium spikes is critical in determining the cumulative state of the enzyme. Through self-activation and self-inactivation dynamics discussed in detail elsewhere (Coultrap and Bayer, 2012) (Lisman and Zhabotinsky, 2001) (Li et al., 2012), CaMKII exhibits related, but partially independent time windows of altered total activity, altered autonomy, and altered responsiveness to Ca2+/CaM. At each given moment, each of these time windows interacts with the current concentration of intracellular calcium and “updates” itself. For example, coincidence of ongoing Ca2+ input with already present enhanced responsiveness of CaMKII to Ca2+/CaM leads synergistically to the prolongation of the enzyme’s enhanced activity and autonomy via autophosphorylation at T286 and translocation of the activated enzyme to the PSD (Coultrap and Bayer, 2012). Thus during high-frequency stimulation, total activity and total autonomy of CaMKII gradually rise and extend in time, because CaMKII’s responsiveness to Ca2+/CaM remains high. Consequently, high-frequency stimulation leads to LTP. On the contrary, during low-frequency stimulation, responsiveness of CaMKII to Ca2+/CaM is reduced via inhibitory autophosphorylation at T305/306, which blocks further increases in autonomy and activity (Coultrap and Bayer, 2012). At the same time, calcineurin’s responsiveness to Ca2+/CaM remains unaltered, which leads to a increase in the corresponding OFF input relative to the ON pathways for CaMKII. As a result, total activity of the kinase is reduced in response to further stimulation by Ca2+, which promotes LTD (Li et al., 2012).
PKC/PKM
A distinct mechanism for persistent activation has been demonstrated for atypical (DAG and Ca2+-independent) PKC. In Aplysia, persistent activation of PKC is achieved through calpain-mediated cleavage of a regulatory domain (Bougie et al., 2009; Sutton et al., 2004). This was observed during intermediate-term activity-dependent facilitation of sensorimotor synapses, which involves synaptic potentiation in neurons that receive neuromodulatory input (e.g., 5-HT) coincident with their own activation. In cells that receive 5-HT stimulation alone, only transient activation of PKC occurred through the activity of phospholipase C-coupled receptors. However, neuronal activation in combination with the 5-HT stimulation additionally promoted calcium influx, which activated calpains that cleaved the transiently activated PKC to yield constitutively active product termed PKM. Thus, in Aplysia, the prolonged availability of PKM is achieved through limited proteolysis, which is an emergent result of coincidence between transient events: 5-HT input and neuronal activation.
In vertebrates, some atypical PKMs can be synthesized locally at the synapse from separate transcripts without the regulatory domain which normally inhibits basal PKC activity (Bougie et al., 2009; Sacktor, 2012). In this case, instead of regulating autonomy of an enzyme, cells regulate production of an autonomous enzyme. However, temporal synergy of upstream signals remains an essential feature of this evolutionarily novel form of autonomy. Initial induction of PKMζ, one of the isoforms of PKM expressed in neurons, involves at least two signals that must be integrated: (i) local inactivation of Pin1, an inhibitor of translation (Hernandez et al., 2003; Kelly et al., 2007a; Kelly et al., 2007b), and (ii) ongoing phosphoinositide signaling, which stimulates PDK1 required to activate newly synthesized PKMζ (Kelly et al., 2007a; Yao et al., 2013). Such coincidence creates an emergent time window of PKMζ activity. Critically, this time window can persist beyond the lifetime of individual PKMζ molecules, potentially lasting for months. This is explained at least in part by the fact that both the synthesis and phosphorylation of nascent PKMζ are stimulated by active PKMζ itself, ensuring positive feedback (Westmark et al., 2010). This potential for extended autonomy, as well as localized nature of PKMζ induction, has been used to explain its role in LTP maintenance (Jalil et al., 2015). Inhibition of PKMζ, an isoform of PKM displaying such translational regulation, is hypothesized to lead to erasure of long-term memory traces (Ling et al., 2002; Shema et al., 2007), although a related subtype of PKM, PKMi/A, may contribute to these effects (Volk et al., 2013).
It is particularly striking that while the mechanism for generating a persistently active PKM from atypical PKC has shifted in the course of evolution from regulated proteolysis to a multi-protein translational and/or autophosphorylating feedback loop, the role of the extended PKM time window in memory maintenance remained essentially the same. Functionally, PKMs promote synaptic facilitation by recruiting AMPARs to the PSD (Migues et al., 2010) and increasing AMPAR-mediated currents (Ling et al., 2002), as do many other regulatory factors (Anggono and Huganir, 2012). Ancestral PKC may have been initially specialized for establishing and maintaining apical compartments in polarized cells (Jalil et al., 2015). This initial function in distributing membrane proteins to specific locations at the surface of the cell may have been adopted in metazoans for synapse development and glutamate receptor redistribution. This is supported by experimental evidence for PKMζ involvement in synapse maturation (Liu et al., 2009; Yoshii et al., 2011). Further selective pressure may have favored various activity-dependent mechanisms capable of producing sustained activation of PKMζ. The different mechanisms for PKM production observed in invertebrates and vertebrates may therefore both be a consequence of the same evolutionary drive favoring a regulated temporal link between transient extracellular signaling and sustained cellular response, and acting on a protein initially specialized for maintenance of spatial specificity, a definitive feature of signal processing by neurons.
Sustained kinase activation resistant to protein turnover is not limited to PKMs. It is a relatively common feature of signaling networks involving a catalytic cycle. As discussed above, autonomy of CaMKII after its initial induction is sustained by increased local protein synthesis (Wu et al., 1998) and subunit exchange (Stratton et al., 2014), paralleling translational feedback observed for PKMs. Similarly, computational simulations of the MAPK pathway, which is required for LTP (English and Sweatt, 1997), demonstrate a capacity for persistent activity and, to a degree, resistance to external dephosphorylation and turnover (Bhalla and Iyengar, 1999; Smolen et al., 2008). These simulations were carried out by isolating the MAPK cascade from its numerous partners within the interactive signaling network (Kopec and Carew, 2013), many of which can clearly contribute to the onset and decay of sustained catalytic cycles, as in the case of self-sustaining cross-talk between PKC and MAPK (Bhalla and Iyengar, 1999).
To sum up, while protein half-lives typically range from minutes to days, protein states can persist for much longer. This is realized using dynamic molecular systems incorporating stable interactions, post-translational modifications, and/or synthesis of new proteins that assume the state of proteins undergoing turnover. Such self-stabilizing feed-forward loops are typical in synaptic plasticity. On the one hand, they are often inducible by fast and transient stimuli and can therefore act to integrate upstream events into a more persistent state that remains after the decay of its trigger. On the other hand, these persistent states are rarely truly stable, i.e. irreversible. Even if the modification itself is irreversible, its “inactivation” can generally be achieved by turnover, and even if a dynamic system is resistant to turnover, it can be returned to homeostasis by an increase in respective OFF forces such as rates of dephosphorylation or protein degradation. Thus, “persistent states” are in fact extended temporal windows with distinct dynamics of activation and inactivation. Epigenetic remodeling is an extreme example of a temporal window that can persist autonomously (i.e. in the absence of the original stimulus) for a lifetime and even into the subsequent generations.
Synergy of target molecules: structural interactions and transport
The examples described so far amount to lasting alterations in “biochemical state” of pre-existing proteins. Aside from “biochemical” changes (e.g. post-translational modifications, cofactor binding, limited proteolysis etc.) changes in the protein state can involve structural rearrangements (e.g. recruitment into supramolecular assemblies, polymerization, conformation change). The distinction is not a strict one. “Structural states” can occur concurrently with “biochemical states” and serve a similar regulatory function. Biochemical states can also trigger structural states, and vice versa. In both cases, a transient change can lead to the onset or decay of a more lasting change.
Supramolecular complexes
A striking example of transient signals triggering a lasting structural change is provided by the prion-like properties of CPEB (cytoplasmic polyadenylation element-binding protein), which contributes to the maintenance of long-term facilitation (Si et al., 2003a; Si et al., 2003b). CPEB acts as a regulator of local protein synthesis at axon terminals. In the basal state CPEB exists in a largely inactive conformation as a soluble monomer. Signaling downstream of 5-HT and PI3 kinase converts some copies of CPEB to an active conformation that forms aggregates. These aggregates act as prions by recruiting additional monomers and converting them to the activated state. Activated aggregates recruit the poly(A) polymerase machinery, allowing activation of previously dormant local mRNAs including the mRNA for CPEB itself. Newly synthesized CPEB proteins continue to sustain translation by assuming the active conformation long after the initial inducing stimulus decays. A “structural cycle” arises, bearing striking resemblance to the “biochemical” cycle of PKMζ or CaMKII autonomy. In both cases, an initial stimulus produces a translation-dependent, lasting, self-perpetuating state that can transcend degradation of individual components.
A special case of lasting “structural time windows” is cytoskeleton rearrangements. Cytoskeleton dynamics underlie morphological plasticity (e.g., changes in the size, shape, or quantity of synapses or neuronal processes) (Schubert and Dotti, 2007), transport of plasticity-related molecules between different compartments of the cell (Bramham and Wells, 2007; Kapitein and Hoogenraad, 2011), and regulate many essential synaptic factors such as CaMKII (Okamoto et al., 2007) or PKMζ (Kelly et al., 2007b).
Cytoskeleton assembly is typically dynamically regulated. Continuous assembly and disassembly of actin and tubulin polymers constitute the ON and OFF mechanisms for a particular state of cytoskeletal filaments at a particular cellular location. Plasticity-inducing stimuli can contribute to these mechanisms by directly modifying the dynamics of cytoskeleton assembly, as in the case of NMDAR-dependent polymerization of actin at dendritic spines (Saneyoshi and Hayashi, 2012). Similarly, microtubule assembly and invasion into potentiated spines can be triggered by signaling from the synapse, as discussed below (Merriam et al., 2013)
Association of proteins with already present cytoskeletal filaments is also subject to dynamic regulation. Binding to or disassociating from actin or tubulin polymers can regulate the effector protein’s downstream activity, as in the case of CaMKII, which is released from actin filaments to be redistributed into the PSD during LTP induction (Okamoto et al., 2007).
Subcellular transport
A critical feature of synaptic plasticity is its specificity to individual synapses. Local changes in synaptic state are not simply a generalized cellular response, but a result of coordination between the synapse, which retains positional information, and the rest of the cell, which provides the synapse with so-called plasticity-related products (PRPs) (Redondo and Morris, 2011). More generally, an essential feature of neuronal plasticity is coordinated transport of cargo between different compartments of the cell.
Such transport can occur in the absence of cytoskeletal binding, i.e. by diffusion, as in the cases of nuclear translocation of second messengers, regulatory subunit of PKA, or, to some extent, CaM (Deisseroth et al., 1998; Mermelstein et al., 2001). Most forms of transport, however, appear to be facilitated by the cytoskeleton and specific motor proteins such as dynein, kinesin or myosin. In either case, translocation of molecules to a given location can be seen within our conceptual framework as a time window of their availability at that location. This time window, in turn, is a result of upstream temporal integration.
Cytoskeleton-mediated transport
Subcellular transport typically involves coincidence of multiple temporally limited states at the “sending” and “receiving” ends of the transport route. In fact, the very notion of “transport” may be confusing since it evokes the notion of a single goal-directed action, whereas in reality it is a synergistic result of multiple, independently controlled processes. In principle, cytoskeleton-dependent transport of cargo can be regulated at multiple levels: availability and activation of appropriate motors (Goldstein and Yang, 2000), availability and activation of appropriately directed filaments (Goldstein and Yang, 2000; Hammond et al., 2010; Konishi and Setou, 2009; Sirajuddin et al., 2014; Yu et al., 2000), motor-cargo interactions (Morfini et al., 2002; Sato-Yoshitake et al., 1992; Wang et al., 2008) or motor-filament interactions (Guillaud et al., 2008; Yin et al., 2012) (Wang and Schwarz, 2009). In reality, many constitutive and inducible pathways probably cooperate to ensure directionally, temporally and cargo-specific transport of RNA and proteins throughout the neuron.
Temporal pattern extraction is embedded in the functioning of this form of transport since it requires favorable coincidence of multiple time windows of availability and state independently of the mode of regulation. These processes collectively alter the availability of organelles, vesicles, proteins or RNA molecules at given subcellular locations. For example, synaptic tag and capture is a form of PRP transport from soma to dendrite, which involves at least two independent processes: an increase in availability of PRPs in the dendritic shaft, and an increase in their “unloading” at the “tagged” synapse. The former is further controlled by the availability of PRPs at the soma, and by the states of locally available motor proteins and cytoskeleton filaments. All of these phenomena are regulated by the timing of neuronal stimulation. Therefore, synaptic tag and capture can be seen as a lasting cell-wide response to a distinct temporal pattern of transient events occurring at various subcellular locations.
Diffusion
In the case of diffusion, rates of diffusion to and from particular cellular locations determine the time windows for protein availability, which can integrate with the timing of ongoing stimulation to produce synergistic responses. In cultured hippocampal neurons, CaM is rapidly (~15 s) translocated to the nucleus following a brief depolarizing stimulus (Mermelstein et al., 2001). However, the decay of nuclear CaM occurs at a much slower time scale (tens of minutes to hours), probably owing to the kinetics of CaM transport in and out of the nucleus. Accordingly, an initial weak stimulus that did not produce significant CREB1 phosphorylation but caused CaM nuclear translocation, markedly increased the phosphorylation of CREB1 15 s after a second weak stimulus delivered 45 s after the first. This increased phosphorylation was not attributable to augumented Ca2+ influx and did not require further CaM translocation. Given that downstream nuclear signaling by CaMKK and CaMKIV steeply depends on CaM, which is likely to be limiting, such fast “priming” was probably a result of an overlap between a time window of increased nuclear CaM retained from the initial stimulus, and a time window of Ca2+ influx from the second stimulus. Disruption of microtubules or actin filaments did not prevent nuclear translocation of CaM, suggesting simple diffusion as the transport mechanism. However, it probably occurs in complex with other proteins (Thorogate and Torok, 2004), since CaM does not contain nuclear localization motifs and would be expected to be strongly buffered in the cytosol.
Higher-order synergy and cellular states
In the previous sections, we have addressed the conversion by neurons of transient external stimuli into persistent states of pre-existing cellular proteins, achieved through biochemical and structural means including subcellular redistribution. A hallmark of long-term memory, however, is its reliance on new protein and RNA synthesis. It is therefore dependent on translational, transcriptional, and epigenetic time windows, which we and others collectively term a “cellular state” (Burrill and Silver, 2010; Levenson and Sweatt, 2006; Marshall and Bredy, 2016). Cellular states are reducible to the history of individual molecule states in the same way that the states of individual molecules are reducible to the history of external stimulation. Cellular time windows, or time-restricted fluctuations of cellular state, are initiated by relatively transient signaling, e.g. post-translational modifications of transcription factors or histones, translocation of effectors into the nucleus, or assembly of essential functional complexes such as ribosomes. Production of new RNA and proteins is therefore a persistent result of a particular configuration of transient upstream protein states.
Synergy of transcriptional cellular states
Typically, regulation at the transcriptional level is highly convergent and highly divergent. For example, CREB1, a central transcriptional regulator of neuronal plasticity, differentiation and survival, integrates a wide range of intracellular signals and produces an equally varied multitude of cellular responses (Lonze and Ginty, 2002). CREB1 can be phosphorylated at a critical Ser-133 residue by a variety of kinases including PKA, Akt, PKC, PKG, ERK, and CaMKII. These activating pathways can interact additively or synergistically (Johannessen et al., 2004). An additional mechanism for transcriptional synergy is provided by transducer of regulated CREB activity 1 (TORC1) which is activated by coincidence of Ca2+- and cAMP-dependent signals but not by either of the stimuli alone, translocates to the nucleus and activates CREB1 in a Ser-133-independent manner (Kovacs et al., 2007). Therefore, activation of CREB1 integrates multiple upstream, transcription-independent intracellular time windows, and transcription of CRE-containing genes is an emergent response to such integration.
Genes whose transcription is induced by pre-existing transcription factors in response to external stimulation are termed immediate-early genes. The products of these genes are typically induced rapidly and transiently, which would seem to challenge their role in establishing a lasting response to fast stimuli. However, many of the immediate-early gene products are themselves transcription factors, which upon their synthesis become available for further temporal integration. For example, CCAAT enhancer-binding protein (C/EBP) (Fig. 5) is transcription factor encoded by an immediate-early gene and controlled by CREB1 (Alberini et al., 1994). In hippocampal neurons, DNA-binding activities of C/EBP isoforms β and δ are enhanced by increased cAMP or Ca2+ signals (Yukawa et al., 1998) (Fig. 5, 1). Interestingly, CaMKIV activated by the Ca2+ signal induces expression of C/EBP members, but also directly enhances C/EBP-dependent gene transcription of late-response genes (Yukawa et al., 1998) (2). Therefore, C/EBP represents a nested system for temporal integration (Fig. 5). The time window of C/EBP availability is driven at the ON end by CREB1 activation (3), which itself integrates many upstream signaling pathways (1). However, the time window of C/EBP product availability is driven by the synergistic interaction between the timing of C/EBP availability and ongoing stimulation by second messengers and kinases.
Fig. 5. Integration of multiple temporal scales within a time window.
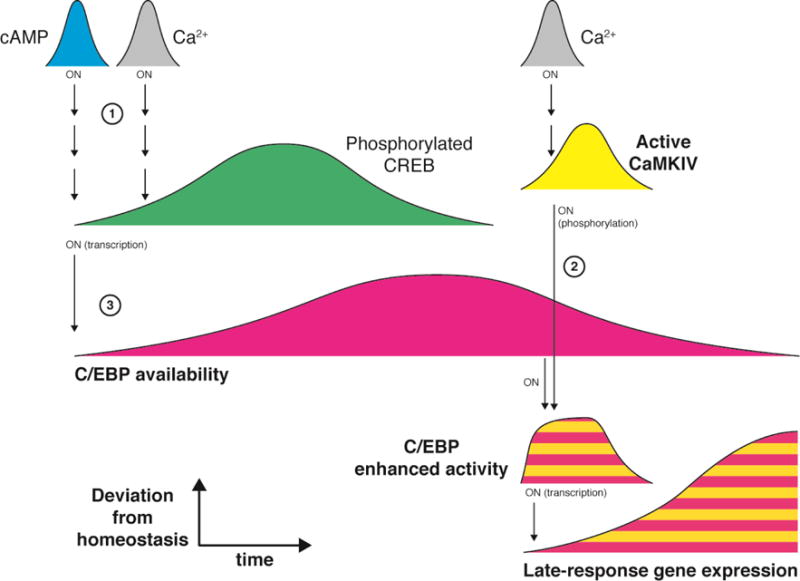
C/EBP is a immediate- early transcription factor whose expression is stimulated by CREB-1 (3), which integrates many signaling pathways including those promoted by cAMP and Ca2+ (1). CaMKIV, a nuclear kinase stimulated by Ca2+ signaling cascades, can directly activate C/EBP (2), promoting late-response gene expression. Thus, C/EBP action is dependent on Ca2+ signaling at multiple temporal domains.
The dichotomy of immediate-early and late-response genes likely represents the tip of the iceberg in the hierarchical “nesting” of transcriptional control. As illustrated by C/EBP, nested transcriptional time windows, in addition to cascading onto each other, can additionally share regulatory mechanisms, like cAMP or Ca2+ signals. This suggests a flexible mode of regulation, whereby entire cascades can be simultaneously altered at multiple levels. “Transcriptional nesting” is further convoluted by positive feedback, as in the case of Aplysia CREB1, which binds to the promoter of its own gene sustaining its transcription (Liu et al., 2008). In this case, the time window of increased CREB expression serves as its own ON input.
An interesting elaboration of this nested system is employed in self-sustaining transcriptional loops such as those underlying circadian rhythmicity. For example, in mammalian tissues the transcription factors CLOCK-BMAL1 positively regulate the expression of the so-called clock- controlled genes (CCGs) (Gallego and Virshup, 2007). These include genes encoding the proteins CRY and PER, which are produced during each circadian cycle and act to repress CLOCK-BMAL1-mediated transcription including their own. Because of that, daily CRY and PER accumulation in the cell is self-limiting. A delayed mechanism involving phosphorylation, ubiquitination and degradation then reduces CRY and PER protein levels (Eide et al., 2005; Yoo et al., 2013), re-enabling their transcription and a new circadian cycle. Thus, a time window of decreased CRY and PER proteins (time window A) contributes to the ON pathway of increased CCG expression (time window B), which contributes the OFF pathway for time window A by resynthesizing CRY and PER. Instead of a clear hierarchy of upstream and downstream time windows, in this case the interactions between levels of temporal analysis nest into one another.
Negative feedback in general is typical for many transcriptional networks. The distinct and functionally critical feature of the CLOCK-BMAL1-CRY-PER system is the delay between time windows B and A, i.e. the delay in CRY/PER degradation (Gallego and Virshup, 2007). If there were no delay, the reciprocal ON and OFF pathways would equilibrate, and no oscillations would be observed in an unperturbed system, as is the case with typical transcriptional networks regulated by negative feedback. However, the delay in the degradation of CRY/PER (that is, the opening of time window A) leads to a delay in the expression of CCGs (the opening of time window B), which leads to a further delay in resynthesizing CRY and PER (the closing of time window A) and consequently, a delay in inhibition of CCG transcription (the closing of time window B). Equilibrium is thus never reached, resulting in bidirectional circadian “swings”. This core circuit is self-sustaining, however many external inputs can contribute to the onset or decay of the corresponding time windows and thus influence circadian rhythmicity. For example, melatonin, by inhibiting the proteasome, can interfere with CRY/PER degradation (Vriend and Reiter, 2014, 2015), thus prolonging the period of the cycle.
In summary, transcriptional states are not merely a single level, but rather a class of levels in the hierarchy of time windows defined by ongoing gene expression. Levels within this class can interact through nesting, whereby downstream transcriptional time windows of increasing persistence are supported by upstream, typically more transient transcriptional time windows. The specific hierarchy of downstream and upstream time windows, however, can be difficult to define, as illustrated by the circadian rhythm example.
Synergy of epigenetic cellular states
An extreme example of protein state persistence leading to an emergent cellular state is histone modification. Even though acetylation and methylation of individual histones are relatively transient, and histones themselves are turned over within days, the epigenetic landscape as a whole can persist for years and in some cases be inherited (Barth and Imhof, 2010; VerMilyea et al., 2009). The contribution of these lasting, potentially lifelong epigenetic changes to memory formation and maintenance has been first recognized about a decade ago (Chwang et al., 2006; Levenson et al., 2004). The specifics of experience-dependent epigenetic remodeling, e.g. the exact ON and OFF pathways that contribute to epigenetic time windows, remain largely unknown. Globally, however, histone modification in postmitotic neurons has been shown to be triggered by neuronal activity and required for certain forms of learning and memory across tasks and brain regions (Zovkic et al., 2013). For example, distinct H3K9me2 and H3K4me3 patterns were induced in the hippocampus and entorhinal cortex following contextual fear conditioning, and the inhibition of H3K9me2 in the entorhinal cortex, but not in the hippocampus, enhanced memory formation (Gupta-Agarwal et al., 2012). DNA methylation has also been implicated in the regulation of transcription of critical memory-related genes. For example, methyl-CpG binding protein 2, which in the hypothalamus influences transcription of thousands of genes, was shown to associate with CREB1 at the activated, but not at repressed targets of this transcription factor (Chahrour et al., 2008).
Even before the discovery of activity-dependent histone modifications, epigenetic changes have been proposed as an attractive mechanism for memory storage (Crick, 1984; Holliday, 1999). Within the repertoire of molecular time windows available to neurons, epigenetic states are probably the longest lasting. However, as in the case with other time windows, epigenetic time windows depend on the balance between ON and OFF pathways which can be independently regulated (Zovkic et al., 2013). Multiple signaling cascades, most notably MAPK/ERK (Chwang et al., 2006), have been shown to influence histone modification during memory formation (Zovkic et al., 2013). ERK involvement in epigenetic remodeling is perhaps not surprising given the central role of this signaling node in relatively slow, long-lasting, low-sensitivity integration of many signaling cascades including signaling by growth factors and other developmental and organism-wide transducers (Wu et al., 2001). ERK activation typically requires stronger or longer-lasting stimulation than the rapid activation of kinases such as PKA or PKC, but once engaged, the ERK system is well-suited for generating self-sustaining responses (Smolen et al., 2008).
As in the case of other time windows we have described, epigenetic states feed back onto lower levels of temporal analysis, i.e. influence the ways in which the upstream, more transient time windows interact. For instance, fear conditioning leads to increased methylation and decreased expression of the PP1 gene in the hippocampus 1 h after training (Miller and Sweatt, 2007), and increased methylation and decreased expression of the cortical calcineurin gene 30 days after training (Miller and Sweatt, 2007). Since both phosphatases are widely utilized as OFF inputs for LTP-induced time windows (such as CaMKII phosphorylation described above), epigenetic time windows can contribute long-term OFF influences to the processing of incoming stimulation at multiple levels. ON inputs can be similarly regulated, as evidenced for example by the epigenetic suppression of CREB2 in response to 5-HT in the Aplysia nervous system. CREB2 is a memory suppressor which inhibits CREBI-regulated, immediate-early memory enhancer genes. Thus, the time window of CREB2 promotor methylation, which peaks between 12 and 24 h of 5-HT application, contributes an ON input to the time window of CREB1 target expression.
In summary, although epigenetic influences on information processing are only beginning to be understood, available evidence firmly places epigenetic states within the nested hierarchy of neuronal time windows with its characteristic feedback onto lower levels of temporal analysis.
Conclusions
The biological utility of memory stems from its ability to modify future behavior based on past experience. This requires storage of information. In this review, we have advanced a model that views information processing and storage in neurons as a series of temporal analyses within a dynamic, hierarchical system of cellular perturbations, which we conceptually consider as “time windows”.
The timing of physiological phenomena, especially those occurring at the micro- and nanoscales, can be notoriously difficult to address experimentally. In recent decades, a great deal of progress has been made in the development of real-time techniques such as live-cell imaging and single-molecule tracking, but overall our knowledge stems from experimental evidence with extremely poor temporal resolution. Such commonly employed techniques as inhibitor treatments, overexpression of recombinant enzymes, or fixed-cell imaging all “collapse” many biological effects occurring on various time scales into a single state which is then assayed in a realistic time frame. A notable exception is electrophysiology, with its relatively easy access to the timing of millisecond-scale events. Indeed the high resolution of electrical measurements has provided neuroscience with a temporal toolkit unique in biology. Yet changes in cell membrane potential – the source of all electrophysiological data – are merely one of many perturbations in homeostasis, many of them equally fast, that continuously occur in the cell and in the organism as a whole. The timing of each of them may be just as critical as frequencies of spikes.
Storage of information is storage of relationships between objects or events. Real-world experience is an infinite-parameter space containing too much potential information to realistically process and store. In biological systems, the particularities of experience – that is, interaction with the outside world – must be first abstracted into a manageable number of variables that could be physically represented as an object or state inside the body. For example, the auditory system receives as input the aggregate vibration of air, but must perform a form of Fourier transform before sound information can be processed and stored. The aggregate vibration is split into bins of individual frequencies to which various populations of cells respond specifically. These individual frequencies are not in fact physically observable in the vibrating air. They are an approximation of a real-world phenomenon as a sum of simple, manageable stimuli with various temporal properties. The same could be said about experience as a whole. The organism, by interacting with the environment, “recodes” the features of this interaction as a combination of many homeostatic disturbances operating at various rates. Hormones and growth factors are typically produced in response to slow, global alterations in organismal state such as chronic stress or changes in dietary patterns. Neurotransmitters are released by neurons and absorbed at an extremely fast rate, which allows to encode fast homeostatic perturbations as rates of neuronal firing.
When these signals are received by a given target cell, they trigger cascades of perturbations which follow the corresponding time scales: hormones and growth factors induce slow neuronal changes, while neurotransmitters cause fast disturbances. Thus, the “holistic”, infinite-parameter experience is parsed by the cell, and cumulatively the organism, into a range of manageable variables and time scales, which collectively provide a reasonably useful approximation of the experience. This model of the past is represented by the organism’s internal devices - molecules, cells, and their states. Its only variable physically derived from the external world is the variable of time. The utility of the model therefore depends on the accuracy of generating specific responses to specific temporal properties of external stimuli. Neurons, as the main bearers of information about experience, must be able to represent time.
This does not imply that absolute time is faithfully preserved by some kind of neuronal “clocks”. Instead, cells, and especially neurons, are adapted to generating sustained but temporally restricted responses to different temporal patterns of stimulation. Stimulation is never a discrete event but rather a perturbation in the level of locally available first messenger, occurring with a characteristic temporal profile – a time window. First messenger time windows are integrated by the neuron into responses that change the responsiveness to subsequent first messenger time windows. Neurons therefore “measure” relative timing of time windows, and “recode it” into new time windows. A hierarchy of time windows is established, with homeostatic perturbations at each level typically influencing the effects of perturbations at other levels both above and below.
An experience that lasted a year cannot be useful in the future if it requires a year for behavioral retrieval. Its utility is based on its sustained ability to influence ongoing events occurring on shorter time scales. This consideration highlights the utility of the neuron’s hierarchical system of time windows. Rather than representing and utilizing time scales independently, neurons “nest” them into a single system that holistically determines the cell’s responsiveness to stimulation at each given moment. Nesting of time scales allows cells to immediately take into account both slow and fast perturbations in their state.
A key feature of this nested system is its dynamic nature – a memory can never said to be “final” or set in stone. In effect, ongoing stimulation by first messengers continuously “updates” the prior receptive state of the cell to produce a posterior receptive state. An important consequence of viewing cellular memory in such Bayesian terms is the blurring of the traditional distinction between memory encoding, storage, and retrieval. Encoding of memory amounts to a specific change in the state of a neuron in response to a temporal pattern of first messenger stimulation. Storage of memory amounts to the alteration in responsiveness to subsequent stimulation as a result of this altered state, and retrieval – the altered response itself.
In fact, a key point advanced in this review is that the behavioral concept of memory has no privileged position within the broader, abstract framework of memory, but is simply one of the higher-order levels at which memory can be considered. Indeed, the dichotomies of experience and behavior, stimulus and response, memory induction and memory retrieval all depend on the chosen frame of reference. Any memory within a given system can be reduced to internal changes in response to external events, but what is external and what is internal to this system is a matter of perspective. In this review, we have employed a clear and convenient boundary, defined by the neuronal cell membrane, between stimuli (events originating outside cells) and responses (events originating inside cells). A behavioral approach to memory similarly considers a system with a clear boundary: an individual organism. In both systems, transient external events induce more lasting internal changes, i.e., both systems have memory. Between the cell and the organism, there are many subsystems that can be similarly said to possess memory, but their boundaries may be less clearly defined. Consider the example of the hippocampal CA3 autoassociative network (Rebola et al., 2017). If seen separately from the rest of the brain, each mossy fiber or perforant path input can be seen as a separate external source of information for the network. Thus, the network as a whole can be said to integrate transient inputs from the dentate gyrus (DG) and entorhinal cortex (ECx) and convert them into more lasting, stable states, which would represent memories of these upstream signals and be reducible to their relative timing (Brandalise et al., 2016; Brandalise and Gerber, 2014; Mishra et al., 2016; Rebola et al., 2017). If the CA3 autoassociative network is viewed as part of the larger hippocampal formation together with the DG, then the DG–CA3 projections must be seen as internal to the system. From this perspective, ECx provides all the primary inputs, the timing of which is integrated into the holistic state of the hippocampus (Diana et al., 2007; Eichenbaum et al., 2007; Yeckel and Berger, 1990), including among other variables the strength of DG-CA3 synapses and the configuration of the CA3 autoassociative network.(Diana et al., 2007; Eichenbaum et al., 2007; Yeckel and Berger, 1990)
A collision of metaphor and mechanism occurs when multiple perspectives are considered at the same time. Hubener and Bonhoeffer express a view similar to ours: that “real” (behavioral) memory, defined by the phases of encoding, storage and retrieval, is part of a broader “continuum” of sensory-driven, experience-dependent changes in the brain (Hubener and Bonhoeffer, 2010). In response, Takeuchi et al. point out that in many cases, there is no isomorphism between lower-level plasticity and higher-level memory: “the function(s) that activity-dependent synaptic plasticity serves will, in our view, depend critically on the neural circuit in which that plasticity is embedded in a non-monotonic manner” (Takeuchi et al., 2014).
In other words, memory is defined not only by what is being memorized, but also by what is memorizing it. As an example, Takeuchi and colleagues (Takeuchi et al., 2014)provide hippocampal encoding of context in episodic memories, which they argue is a function not simply of ongoing experience, but also of the pre-existing representations of context. To resolve this disagreement, we find it useful again to refer to the boundaries of the systems in question. Hubener and Bonhoeffer’s perspective considers the brain holistically, taking into account all its ontogenetic experience-dependent changes, including the initial establishment of context representations, which may have occurred years prior to the experience but still necessarily involved signals from the external world. Takeuchi et al., on the other hand, consider various brain regions independently of one another (i.e., the context representation is seen as an independent input into the hippocampal network), yet still refer to “function” from a behavioral standpoint – that is, role of a given subsystem in the organism’s behavioral output. Viewing experience-dependent changes as a hierarchy, rather than a continuum, may ease this inherent tension between the organismal and cellular perspectives on the nervous system.
In this review, we have restricted our discussion of temporal processing to single neurons. There is no reason to assume though that temporal analysis by the organism or the nervous system is restricted to intracellular computations. The principle of brain computation as a whole has been described as hierarchical abstraction (Ballard, 2015). Information available at lower levels of the hierarchy – states of sensory systems, for example – is hierarchically recoded into representations of increasing generality, which are then used to interpret the incoming lower-order data. We contend that in any such case of “recoding”, the actual information transmitted between levels of analysis is reducible to temporal patterns. “Spiking” behavior of neurons is sometimes likened to a binary code – a code utilizing two alternative values to represent information. More appropriately, spiking should be seen as a code employing a single “value” plus relative time.
To conclude, time is a fundamental variable in neuronal computations. Neurons – and probably nervous systems as a whole – possess a fundamentally multi-leveled system for extraction of temporal information from past experience. The timing of past events is converted into a hierarchy of homeostatic perturbations that influence the effect of future events, thereby altering behavior. The adaptive, temporally regulated functioning of this system is the essence of what is commonly called memory.
Acknowledgments
The authors would like to thank J. B. Byrne and D. V. Buonomano for thoughtful and constructive comments on earlier versions of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Abrams TW, Karl KA, Kandel ER. Biochemical studies of stimulus convergence during classical conditioning in Aplysia: dual regulation of adenylate cyclase by Ca2+/calmodulin and transmitter. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1991;11:2655–2665. doi: 10.1523/JNEUROSCI.11-09-02655.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Ballard DH. Brain Computation as Hierarchical Abstraction. MIT Press; 2015. [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35:618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Blanke ML, VanDongen AMJ. Activation Mechanisms of the NMDA Receptor. In: Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL): 2009. [PubMed] [Google Scholar]
- Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- Bougie JK, Lim T, Farah CA, Manjunath V, Nagakura I, Ferraro GB, Sossin WS. The atypical protein kinase C in Aplysia can form a protein kinase M by cleavage. J Neurochem. 2009;109:1129–1143. doi: 10.1111/j.1471-4159.2009.06045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Sorra KE, Hurlburt J, Harris KM. Polyribosomes are increased in spines of CA1 dendrites 2 h after the induction of LTP in mature rat hippocampal slices. Hippocampus. 2007;17:1–4. doi: 10.1002/hipo.20238. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- Brandalise F, Carta S, Helmchen F, Lisman J, Gerber U. Dendritic NMDA spikes are necessary for timing-dependent associative LTP in CA3 pyramidal cells. Nat Commun. 2016;7:13480. doi: 10.1038/ncomms13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandalise F, Gerber U. Mossy fiber-evoked subthreshold responses induce timing-dependent plasticity at hippocampal CA3 recurrent synapses. Proc Natl Acad Sci U S A. 2014;111:4303–4308. doi: 10.1073/pnas.1317667111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelli M, Castellucci V, Kandel ER. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science. 1976;194:1178–1181. doi: 10.1126/science.186870. [DOI] [PubMed] [Google Scholar]
- Buck LB. Information coding in the vertebrate olfactory system. Annu Rev Neurosci. 1996;19:517–544. doi: 10.1146/annurev.ne.19.030196.002505. [DOI] [PubMed] [Google Scholar]
- Buonomano DV, Maass W. State-dependent computations: spatiotemporal processing in cortical networks. Nat Rev Neurosci. 2009;10:113–125. doi: 10.1038/nrn2558. [DOI] [PubMed] [Google Scholar]
- Burrill DR, Silver PA. Making cellular memories. Cell. 2010;140:13–18. doi: 10.1016/j.cell.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Djurisic M, Zecevic D. Dendritic signals from rat hippocampal CA1 pyramidal neurons during coincident pre- and post-synaptic activity: a combined voltage- and calcium-imaging study. J Physiol. 2007;580:463–484. doi: 10.1113/jphysiol.2006.125005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A. 2011;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Evidence that heterosynaptic depolarization underlies associativity of long-term potentiation in rat hippocampus. J Physiol. 1996;490(Pt 2):455–462. doi: 10.1113/jphysiol.1996.sp021157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Herron CE, Lester RA. Frequency-dependent N-methyl-D-aspartate receptor-mediated synaptic transmission in rat hippocampus. J Physiol. 1988;399:301–312. doi: 10.1113/jphysiol.1988.sp017081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Co-regulation of synaptic efficacy at stable polyneuronally innervated neuromuscular junctions in reinnervated rat muscle. J Physiol. 1999;521(Pt 2):365–374. doi: 10.1111/j.1469-7793.1999.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik FIAL, Robert S. Levels of processing: A framework for memory research. Journal of verbal learning and verbal behavior. 1972;11:671–684. [Google Scholar]
- Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Eccles JC. Synaptic action during and after repetitive stimulation. J Physiol. 1960;150:374–398. doi: 10.1113/jphysiol.1960.sp006393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Collingridge GL. Regulation of EPSPs by the synaptic activation of GABAB autoreceptors in rat hippocampus. J Physiol. 1996;496(Pt 2):451–470. doi: 10.1113/jphysiol.1996.sp021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–59. [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Diana RA, Yonelinas AP, Ranganath C. Imaging recollection and familiarity in the medial temporal lobe: a three-component model. Trends Cogn Sci. 2007;11:379–386. doi: 10.1016/j.tics.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H, Yonelinas AP, Ranganath C. The medial temporal lobe and recognition memory. Annu Rev Neurosci. 2007;30:123–152. doi: 10.1146/annurev.neuro.30.051606.094328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Enoki R, Kiuchi T, Koizumi A, Sasaki G, Kudo Y, Miyakawa H. NMDA receptor-mediated depolarizing after-potentials in the basal dendrites of cA1 pyramidal neurons. Neurosci Res. 2004;48:325–333. doi: 10.1016/j.neures.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, et al. A dynamic pathway for calciumindependent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RC, Blackwell KT. Calcium: amplitude, duration, or location? Biol Bull. 2015;228:75–83. doi: 10.1086/BBLv228n1p75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferster D, Jagadeesh B. EPSP-IPSP interactions in cat visual cortex studied with in vivo whole-cell patch recording. J Neurosci. 1992;12:1262–1274. doi: 10.1523/JNEUROSCI.12-04-01262.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Lee PR, Cohen JE. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell Calcium. 2005;37:433–442. doi: 10.1016/j.ceca.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB, Stellar E. Memory in mice as affected by intracerebral puromycin. Science. 1963;141:57–59. doi: 10.1126/science.141.3575.57. [DOI] [PubMed] [Google Scholar]
- Fulton D, Kemenes I, Andrew RJ, Benjamin PR. A single time-window for protein synthesis-dependent long-term memory formation after one-trial appetitive conditioning. Eur J Neurosci. 2005;21:1347–1358. doi: 10.1111/j.1460-9568.2005.03970.x. [DOI] [PubMed] [Google Scholar]
- Fuster JM, Bressler SL. Cognit activation: a mechanism enabling temporal integration in working memory. Trends Cogn Sci. 2012;16:207–218. doi: 10.1016/j.tics.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8:139–148. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- Gerstner W, Kistler WM. Spiking neuron models: single neurons, populations, plasticity. Cambridge, U.K; New York: Cambridge University Press; 2002. [Google Scholar]
- Goelet P, Castellucci VF, Schacher S, Kandel ER. The long and the short of long-term memory–a molecular framework. Nature. 1986;322:419–422. doi: 10.1038/322419a0. [DOI] [PubMed] [Google Scholar]
- Goldstein LS, Yang Z. Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Gordon U, Polsky A, Schiller J. Plasticity compartments in basal dendrites of neocortical pyramidal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:12717–12726. doi: 10.1523/JNEUROSCI.3502-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, Lubin FD. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci. 2012;32:5440–5453. doi: 10.1523/JNEUROSCI.0147-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin-Brumshtein Y, Lancet D, Olender T. Human olfaction: from genomic variation to phenotypic diversity. Trends Genet. 2009;25:178–184. doi: 10.1016/j.tig.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Havik B, Rokke H, Bardsen K, Davanger S, Bramham CR. Bursts of high-frequency stimulation trigger rapid delivery of pre-existing alpha-CaMKII mRNA to synapses: a mechanism in dendritic protein synthesis during long-term potentiation in adult awake rats. Eur J Neurosci. 2003;17:2679–2689. doi: 10.1046/j.1460-9568.2003.02712.x. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci U S A. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Hempel CM, Vincent P, Adams SR, Tsien RY, Selverston AI. Spatiotemporal dynamics of cyclic AMP signals in an intact neural circuitm. Nature. 1996;384:166–169. doi: 10.1038/384166a0. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Hernandez AI, Blace N, Crary JF, Serrano PA, Leitges M, Libien JM, Weinstein G, Tcherapanov A, Sacktor TC. Protein kinase M zeta synthesis from a brain mRNA encoding an independent protein kinase C zeta catalytic domain. Implications for the molecular mechanism of memory. J Biol Chem. 2003;278:40305–40316. doi: 10.1074/jbc.M307065200. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003;4:530–538. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- Hofmeyr JH. The harmony of the cell: the regulatory design of cellular processes. Essays Biochem. 2008;45:57–66. doi: 10.1042/BSE0450057. [DOI] [PubMed] [Google Scholar]
- Hofmeyr JS, Cornish-Bowden A. Regulating the cellular economy of supply and demand. FEBS Lett. 2000;476:47–51. doi: 10.1016/s0014-5793(00)01668-9. [DOI] [PubMed] [Google Scholar]
- Holliday R. Is there an epigenetic component in long-term memory? J Theor Biol. 1999;200:339–341. doi: 10.1006/jtbi.1999.0995. [DOI] [PubMed] [Google Scholar]
- Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Hubener M, Bonhoeffer T. Searching for engrams. Neuron. 2010;67:363–371. doi: 10.1016/j.neuron.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Jalan-Sakrikar N, Bartlett RK, Baucum AJ, 2nd, Colbran RJ. Substrate- selective and calcium-independent activation of CaMKII by alpha-actinin. J Biol Chem. 2012;287:15275–15283. doi: 10.1074/jbc.M112.351817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalil SJ, Sacktor TC, Shouval HZ. Atypical PKCs in memory maintenance: the roles of feedback and redundancy. Learn Mem. 2015;22:344–353. doi: 10.1101/lm.038844.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Seternes OM, Johansen B, Moens U. Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain. Cell Signal. 2004;16:1187–1199. doi: 10.1016/j.cellsig.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Kao DI, Aldridge GM, Weiler IJ, Greenough WT. Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci U S A. 2010;107:15601–15606. doi: 10.1073/pnas.1010564107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC. Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol Cell Neurosci. 2011;46:9–20. doi: 10.1016/j.mcn.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC. Regulation of protein kinase Mzeta synthesis by multiple kinases in long-term potentiation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007a;27:3439–3444. doi: 10.1523/JNEUROSCI.5612-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MT, Yao Y, Sondhi R, Sacktor TC. Actin polymerization regulates the synthesis of PKMzeta in LTP. Neuropharmacology. 2007b;52:41–45. doi: 10.1016/j.neuropharm.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Setou M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci. 2009;12:559–567. doi: 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- Kopec AM, Carew TJ. Growth factor signaling and memory formation: temporal and spatial integration of a molecular network. Learn Mem. 2013;20:531–539. doi: 10.1101/lm.031377.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmeier J, Sosic-Vasic Z. Parallels between spacing effects during behavioral and cellular learning. Front Hum Neurosci. 2012;6:203. doi: 10.3389/fnhum.2012.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Schmitz D. Cellular and System Biology of Memory: Timing, Molecules, and Beyond. Physiol Rev. 2016;96:647–693. doi: 10.1152/physrev.00010.2015. [DOI] [PubMed] [Google Scholar]
- Kovacs KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Dreyer WJ. Light dependent phosphorylation of rhodopsin by ATP. FEBS Lett. 1972;20:1–6. doi: 10.1016/0014-5793(72)80002-4. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol Cell Proteomics. 2011;10:R110 003871. doi: 10.1074/mcp.R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stefan MI, Le Novere N. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PLoS One. 2012;7:e43810. doi: 10.1371/journal.pone.0043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Liu FC, Graybiel AM. Spatiotemporal dynamics of CREB phosphorylation: transient versus sustained phosphorylation in the developing striatum. Neuron. 1996;17:1133–1144. doi: 10.1016/s0896-6273(00)80245-7. [DOI] [PubMed] [Google Scholar]
- Liu J, Hu JY, Schacher S, Schwartz JH. The two regulatory subunits of aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci. 2004;24:2465–2474. doi: 10.1523/JNEUROSCI.4331-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Fioravante D, Shah S, Byrne JH. cAMP response element-binding protein 1 feedback loop is necessary for consolidation of long-term synaptic facilitation in Aplysia. J Neurosci. 2008;28:1970–1976. doi: 10.1523/JNEUROSCI.3848-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XF, Tari PK, Haas K. PKM zeta restricts dendritic arbor growth by filopodial and branch stabilization within the intact and awake developing brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:12229–12235. doi: 10.1523/JNEUROSCI.2842-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein Y, Sompolinsky H. Temporal integration by calcium dynamics in a model neuron. Nat Neurosci. 2003;6:961–967. doi: 10.1038/nn1109. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Kukushkin NV, Goldberg AL. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015;112:E7176–7185. doi: 10.1073/pnas.1522332112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Babayan AH, Rumbaugh G, Gall CM. Differences between synaptic plasticity thresholds result in new timing rules for maximizing long-term potentiation. Neuropharmacology. 2013;64:27–36. doi: 10.1016/j.neuropharm.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Marshall P, Bredy TW. Cognitive neuroepigenetics: the next evolution in our understanding of the molecular mechanisms underlying learning and memory? NPJ Sci Learn. 2016;1 doi: 10.1038/npjscilearn.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Time-dependent processes in memory storage. Science. 1966;153:1351–1358. doi: 10.1126/science.153.3742.1351. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Deisseroth K, Dasgupta N, Isaksen AL, Tsien RW. Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci U S A. 2001;98:15342–15347. doi: 10.1073/pnas.211563998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci. 2013;33:16471–16482. doi: 10.1523/JNEUROSCI.0661-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- Mishra RK, Kim S, Guzman SJ, Jonas P. Symmetric spike timing-dependent plasticity at CA3-CA3 synapses optimizes storage and recall in autoassociative networks. Nat Commun. 2016;7:11552. doi: 10.1038/ncomms11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons N, Guillou JL, Jaffard R. The role of Ca2+/calmodulin-stimulable adenylyl cyclases as molecular coincidence detectors in memory formation. Cell Mol Life Sci. 1999;55:525–533. doi: 10.1007/s000180050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzachiodi R, Byrne JH. More than synaptic plasticity: role of nonsynaptic plasticity in learning and memory. Trends Neurosci. 2010;33:17–26. doi: 10.1016/j.tins.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nicoll A, Larkman A, Blakemore C. EPSPs in rat neocortical pyramidal neurones in vitro are prolonged by NMDA receptor-mediated currents. Neurosci Lett. 1992;143:5–9. doi: 10.1016/0304-3940(92)90220-2. [DOI] [PubMed] [Google Scholar]
- Nooren IM, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during lTp in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Usman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH, Nawa H. Neuronal differentiation factors/cytokines and synaptic plasticity. Cell. 1993;72(Suppl):123–137. doi: 10.1016/s0092-8674(05)80032-7. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Ogawa S, Kimura A, McEwen BS. Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res. 1996;738:229–235. doi: 10.1016/s0006-8993(96)00776-7. [DOI] [PubMed] [Google Scholar]
- Pearce K, Cai D, Roberts AC, Glanzman DL. Role of protein synthesis and DNA methylation in the consolidation and maintenance of long-term memory in Aplysia. Elife. 2017;6 doi: 10.7554/eLife.18299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peth A, Kukushkin N, Bosse M, Goldberg AL. Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J Biol Chem. 2013;288:7781–7790. doi: 10.1074/jbc.M112.441907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips GT, Kopec AM, Carew TJ. Pattern and predictability in memory formation: from molecular mechanisms to clinical relevance. Neurobiol Learn Mem. 2013;105:117–124. doi: 10.1016/j.nlm.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Otmakhov N, Lemelin D, De Koninck P, Lisman J. Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:8704–8709. doi: 10.1523/JNEUROSCI.0133-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IK, Jans DA. Regulation of nuclear transport: central role in development and transformation? Traffic. 2005;6:173–186. doi: 10.1111/j.1600-0854.2005.00268.x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Carta M, Mulle C. Operation and plasticity of hippocampal CA3 circuits: implications for memory encoding. Nat Rev Neurosci. 2017;18:208–220. doi: 10.1038/nrn.2017.10. [DOI] [PubMed] [Google Scholar]
- Redondo RL, Morris RG. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci. 2011;12:17–30. doi: 10.1038/nrn2963. [DOI] [PubMed] [Google Scholar]
- Righi M, Tongiorgi E, Cattaneo A. Brain-derived neurotrophic factor (BDNF) induces dendritic targeting of BDNF and tyrosine kinase B mRNAs in hippocampal neurons through a phosphatidylinositol-3 kinase-dependent pathway. J Neurosci. 2000;20:3165–3174. doi: 10.1523/JNEUROSCI.20-09-03165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Memory. Engram cells retain memory under retrograde amnesia. Science. 2015;348:1007–1013. doi: 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. Memory maintenance by PKMzeta–an evolutionary perspective. Mol Brain. 2012;5:31. doi: 10.1186/1756-6606-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Hayashi Y. The Ca2+ and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskeleton (Hoboken) 2012;69:545–554. doi: 10.1002/cm.21037. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifuji H, Inagaki M, Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Schacter DL, Addis DR, Buckner RL. Remembering the past to imagine the future: the prospective brain. Nat Rev Neurosci. 2007;8:657–661. doi: 10.1038/nrn2213. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343–348. doi: 10.1016/s0959-4388(00)00217-8. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Reymann KG, Balschun D. A post-tetanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci U S A. 1997;94:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shobe JL, Zhao Y, Stough S, Ye X, Hsuan V, Martin KC, Carew TJ. Temporal phases of activity-dependent plasticity and memory are mediated by compartmentalized routing of MAPK signaling in aplysia sensory neurons. Neuron. 2009;61:113–125. doi: 10.1016/j.neuron.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003a;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003b;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol. 2014;16:335–344. doi: 10.1038/ncb2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen P, Baxter DA, Byrne JH. Bistable MAP kinase activity: a plausible mechanism contributing to maintenance of late long-term potentiation. Am J Physiol Cell Physiol. 2008;294:C503–515. doi: 10.1152/ajpcell.00447.2007. [DOI] [PubMed] [Google Scholar]
- Smolen P, Zhang Y, Byrne JH. The right time to learn: mechanisms and optimization of spaced learning. Nat Rev Neurosci. 2016;17:77–88. doi: 10.1038/nrn.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF. Competitive Hebbian learning through spiketiming-dependent synaptic plasticity. Nat Neurosci. 2000;3:919–926. doi: 10.1038/78829. [DOI] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- Stoof JC, Kebabian JW. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–368. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- Stratton M, Lee IH, Bhattacharyya M, Christensen SM, Chao LH, Schulman H, Groves JT, Kuriyan J. Activation-triggered subunit exchange between CaMKII holoenzymes facilitates the spread of kinase activity. Elife. 2014;3:e01610. doi: 10.7554/eLife.01610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- Sun HG, Chen W, Wei H, Chen YQ. A comparative study of constant-order and variable-order fractional models in characterizing memory property of systems. Eur Phys J- Spec Top. 2011;193:185–192. [Google Scholar]
- Sutton MA, Bagnall MW, Sharma SK, Shobe J, Carew TJ. Intermediateterm memory for site-specific sensitization in aplysia is maintained by persistent activation of protein kinase C. J Neurosci. 2004;24:3600–3609. doi: 10.1523/JNEUROSCI.1134-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ide J, Masters SE, Carew TJ. Interaction between amount and pattern of training in the induction of intermediate- and long-term memory for sensitization in aplysia. Learn Mem. 2002;9:29–40. doi: 10.1101/lm.44802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130288. doi: 10.1098/rstb.2013.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Activity-dependent properties of synaptic transmission at two classes of connections made by rat neocortical pyramidal axons in vitro. J Physiol. 1997;502(Pt 1):131–147. doi: 10.1111/j.1469-7793.1997.131bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM, Girdlestone D, West DC. Voltage-dependent currents prolong single-axon postsy naptic potentials in layer III pyramidal neurons in rat neocortical slices. J Neurophysiol. 1988;60:1896–1907. doi: 10.1152/jn.1988.60.6.1896. [DOI] [PubMed] [Google Scholar]
- Thorogate R, Torok K. Ca2+-dependent and -independent mechanisms of calmodulin nuclear translocation. J Cell Sci. 2004;117:5923–5936. doi: 10.1242/jcs.01510. [DOI] [PubMed] [Google Scholar]
- Tsien JZ. Linking Hebb’s coincidence-detection to memory formation. Curr Opin Neurobiol. 2000;10:266–273. doi: 10.1016/s0959-4388(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci U S A. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya SC, Ding L, Smith TK, Hegde AN. Differential regulation of proteasome activity in the nucleus and the synaptic terminals. Neurochem Int. 2006;48:296–305. doi: 10.1016/j.neuint.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Vandorpe DH, Small DL, Dabrowski AR, Morris CE. FMRFamide and membrane stretch as activators of the Aplysia S-channel. Biophys J. 1994;66:46–58. doi: 10.1016/S0006-3495(94)80749-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerMilyea MD, O’Neill LP, Turner BM. Transcription-independent heritability of induced histone modifications in the mouse preimplantation embryo. PLoS One. 2009;4:e6086. doi: 10.1371/journal.pone.0006086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend J, Reiter RJ. Melatonin as a proteasome inhibitor. Is there any clinical evidence? Life Sci. 2014;115:8–14. doi: 10.1016/j.lfs.2014.08.024. [DOI] [PubMed] [Google Scholar]
- Vriend J, Reiter RJ. Melatonin feedback on clock genes: a theory involving the proteasome. J Pineal Res. 2015;58:1–11. doi: 10.1111/jpi.12189. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund S. Dead Matter Has Memory. Phys Scripta. 1991;43:174–179. [Google Scholar]
- Westmark PR, Westmark CJ, Wang S, Levenson J, O’Riordan KJ, Burger C, Malter JS. Pin1 and PKMzeta sequentially control dendritic protein synthesis. Sci Signal. 2010;3:ra18. doi: 10.1126/scisignal.2000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Wolf E, Zhao FY, Roberts A. Non-linear summation of excitatory synaptic inputs to small neurones: a case study in spinal motoneurones of the young Xenopus tadpole. J Physiol. 1998;511(Pt 3):871–886. doi: 10.1111/j.1469-7793.1998.871bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]
- Yao Y, Shao C, Jothianandan D, Tcherepanov A, Shouval H, Sacktor TC. Matching biochemical and functional efficacies confirm ZIP as a potent competitive inhibitor of PKMzeta in neurons. Neuropharmacology. 2013;64:37–44. doi: 10.1016/j.neuropharm.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Berger TW. Feedforward excitation of the hippocampus by afferents from the entorhinal cortex: redefinition of the role of the trisynaptic pathway. Proc Natl Acad Sci U S A. 1990;87:5832–5836. doi: 10.1073/pnas.87.15.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Feng X, Takei Y, Hirokawa N. Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:5486–5499. doi: 10.1523/JNEUROSCI.0718-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–1105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Murata Y, Kim J, Zhang C, Shokat KM, Constantine-Paton M. TrkB and protein kinase Mzeta regulate synaptic localization of PSD-95 in developing cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:11894–11904. doi: 10.1523/JNEUROSCI.2190-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yovell Y, Abrams TW. Temporal asymmetry in activation of Aplysia adenylyl cyclase by calcium and transmitter may explain temporal requirements of conditioning. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6526–6530. doi: 10.1073/pnas.89.14.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Cook C, Sauter C, Kuriyama R, Kaplan PL, Baas PW. Depletion of a microtubule-associated motor protein induces the loss of dendritic identity. J Neurosci. 2000;20:5782–5791. doi: 10.1523/JNEUROSCI.20-15-05782.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa K, Tanaka T, Tsuji S, Akira S. Expressions of CCAAT/Enhancer- binding proteins beta and delta and their activities are intensified by cAMP signaling as well as Ca2+/calmodulin kinases activation in hippocampal neurons. J Biol Chem. 1998;273:31345–31351. doi: 10.1074/jbc.273.47.31345. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]