

Abstract
Multiple sclerosis (MS) is one of the most important, disabling, and prevalent neurological disorders of young adults. It is a chronic inflammatory and neurodegenerative disease when autoreactive B and T cells have downstream effects that result in demyelination and neuronal loss. Anti-inflammatory disease-modifying therapies do have proven efficacy in delaying disease and disability progression in MS. While the progress in MS treatments has already improved the prognosis and quality of patients’ lives overall, there are some clear shortcomings and unmet needs in the current MS treatment landscape. The most promising means of MS treatment is selective immune reconstitution therapy (SIRT). This therapy is given in short-duration courses of immunosuppression, producing durable effects on the immune system and preventing nervous tissue loss. This review discusses the mechanisms of action and the data of clinical trials of cladribine tablets as an example of SIRT in MS. The clinical benefits of cladribine tablets in these studies include decreased relapse rate and disability progression with large reductions in lesion activity, and protection against brain volume loss. Whether all of these neurological findings are direct results of lymphocyte depletion, or if there are downstream effects on other, unknown, neurodegenerative processes are yet to be determined, but these clearly point to an interesting area of research.
Keywords: multiple sclerosis, therapy, selective immune reconstitution, cladribine
Multiple sclerosis (MS) is driven by both inflammatory and neurodegenerative processes
MS is one of the most prevalent neurological disorders in the world, and the scale of the problem appears to be gradually increasing.1,2 Globally, the number of patients with MS is estimated to have increased from 2.1 million in 2008 to 2.3 million in 2013, an increase of nearly 9% in a 5-year period.1,2 It is not possible to state whether this represents a true increase in MS incidence, or merely improvements in diagnostic capabilities. Over the same time period, there has been a general improvement in the availability of health resources, including access to neurologists and imaging facilities, and effective disease-modifying therapies (DMTs).1,2 Regardless of the underlying cause of increased prevalence, it is true that the overall global MS burden shows no signs of reduction. Moreover, with a typical age of onset between 20 and 40 years, MS can be a cause of serious disability in young adults particularly if treatment with adequate DMTs is not started on time.3
Fundamentally, MS is a chronic inflammatory and neurodegenerative disease that affects the central nervous system (CNS). The key pathological characteristics of MS are the multifocal lesions and diffuse changes in the brain and spinal cord arising from immune-mediated demyelination and axonal/neuronal damage.3,4 These pathological changes observed in MS are suggested to result, at least to some extent, from autoreactive lymphocytes and monocytes infiltrating the blood–brain barrier (BBB) and destroying myelin, oligodendrocytes, axons, and, to a lesser extent, neurons.4 Diffuse cortical and gray matter degeneration along with demyelinated cortical lesions are not only hallmarks of MS but are also important drivers of pathology and disability.5 The presence of gray matter demyelination and neuroaxonal degeneration is associated with activated microglia, but appears largely independent of BBB disruption and local and diffuse inflammation of white matter.5,6
The underlying etiology of MS is still unclear, although it is likely to have at least some genetic component.7–9 However, genetic risk factors probably account for less than a third of the heterogeneity of MS, with environmental risk factors accounting for the rest.10,11 Regardless of the underlying etiology, it is clear that the immune system plays a central role in MS pathogenesis and, certainly in early MS, autoimmune processes are a key driver of disease course.11,12
Within the CNS compartment, the autoreactive B and T cells have downstream effects that result in inflammation, demyelination, and neuronal loss (Figure 1).11–13 Importantly, there is evidence that B-cell development and maturation can be sustained in the CNS over time.14,15 Immunoglobulin (Ig) oligoclonal bands, a characteristic immunodiagnostic abnormality of MS, are detectable in the cerebrospinal fluid (CSF) of patients with MS for many years, even with treatment, suggesting the persistence of Ig producing B cells in the CNS.14
Figure 1.

Immunopathogenesis of multiple sclerosis and associated immune targets.
Abbreviations: APC, antigen presenting cell; HLA, human leukocyte antigen; IFN, interferon; IL, interleukin; MHC, major histocompatibility complex; MMP, matrix metalloproteinase; NO, nitric oxide; ROS, reactive oxygen species; TCR, T-cell receptor; Th, T helper cell; Treg, regulatory T cell.
Meningeal follicles of B cells have been proposed to be critical for cortical lesions and atrophy in MS, especially in patients with secondary-progressive MS (SPMS).15 Ectopic meningeal B-cell follicles were identified in postmortem brain tissue of 41.4% of patients with SPMS but not in brain tissue of patients with primary-progressive MS.15 The presence of ectopic B-cell follicles was associated with earlier onset of disease, age at death, greater cortical demyelination, microglia activation, cerebral cortex neurite depletion, and irreversible disability.15 A similar prevalence of B-cell follicles (40%) was reported in another study of 143 patients with SPMS.16 B-cell follicles were associated with a greater degree of diffuse meningeal inflammation, correlated with the level of activated microglia and cortical demyelination.16 Again, the presence of B-cell follicles was associated with earlier disease onset, disease progression, disability progression, and age at death.16
Progressive degeneration of both gray and white matters is integral to the natural history of MS, manifesting in atrophy of the brain and the spinal cord.17 Focal lesions are only one aspect of the pathological changes that occur in MS, which also include diffuse tissue damage throughout the CNS, driven by ongoing inflammation and neurodegeneration, leading to cumulative brain atrophy.18,19 The functional disabilities that develop in MS are a consequence of these neurodegenerative processes that occur in all stages of MS.17 Furthermore, the overall loss of brain volume occurs irrespective of the underlying level of inflammation.17
There is ongoing discussion on whether inflammatory or neurodegenerative processes are the primary drivers of disease, and whether inflammatory demyelination is a primary cause or a secondary consequence of the disease.4,17 This question has profound implications for the management of patients with MS. If the inflammatory activity seen in MS is actually a secondary consequence of, or response to, a fundamentally neurodegenerative disease process then treatments must tackle the underlying neurodegeneration. Indeed, there are studies which suggest that neurodegeneration in patients with MS may occur independently of inflammation or even be the primary cause of damage to the CNS.20–23 There are, for example, data that suggest that intracortical demyelination is not necessarily associated with increased lymphocyte infiltration or with changes to the lymphocyte subsets within the cortex.21 Axonal loss can occur independently of demyelination which may be the result of an underlying, so far undescribed, neurodegenerative axonopathy.22 Furthermore, cortical lesions have been observed to involve many fewer lymphocytes, macrophages, and microglia than white matter lesions.23
However, it is likely that there is a complex interdependence between inflammation, neurodegeneration, and disease progression in all stages of MS,24 and that various feedback mechanisms interlink inflammatory and neurodegenerative processes to drive the progression of the disease.24–27 In this review article, we will discuss cladribine tablets, principally an antilymphocytic and, therefore, an anti-inflammatory agent in the context of MS features of both inflammation and neurodegeneration.
Current treatment
There is currently no cure for MS. Rather, the current aims of treatment are to effectively manage the disease, reduce relapse frequency, prevent or delay disability progression, and control magnetic resonance imaging (MRI) activity.28,29 To date, the available treatments have primarily focussed on the inflammatory component of MS. As discussed, the inflammatory component of the disease is better characterized than any putative underlying neurodegenerative mechanism, and the inflammatory pathways present numerous potential therapeutic targets. Interferon β (IFNβ) was the first anti-inflammatory therapy to demonstrate significant efficacy in patients with relapsing-remitting MS (RRMS).30,31
In the two decades since the first approval of IFNβ for RRMS, the treatment landscape has expanded. Presently, first-line treatment involves one of the different formulations of IFNβ, glatiramer acetate (GA), teriflunomide (TF), and dimethyl fumarate (DMF) and, in patients with high disease activity, natalizumab (NTZ), fingolimod (FL), alemtuzumab (ATZ), or, rarely, mitoxantrone.32 Recently, ocrelizumab (OCZ) has been approved in several countries. In cases of disease breakthrough, the next stage of therapy is the horizontal or vertical switch to treatment with another DMT, such as initiating treatment with a high-efficacy second-line therapy such as NTZ or mitoxantrone.32
Although it is true that the progress in MS treatments has improved the quality of patients’ lives overall, there are some clear shortcomings and unmet needs in the current MS treatment landscape, beyond the obvious need for a truly curative treatment. Chronic immunosuppression is associated with several major bacterial, viral, fungal, protozoan, and parasitic infections.33 Included in this is the potentially serious complication of progressive multifocal leukoencephalopathy due to John Cunningham virus infection (or activation), most notably reported with NTZ.33 Chronic immunosuppressant use may also increase the risk of certain malignancies and laboratory abnormalities.34,35
The dosing/administration frequency of some MS DMTs represents a high treatment and monitoring burden which, in turn, may lower patients’ adherence.36 Indeed, adherence rates among patients with MS are estimated to vary from 41% to 88%, with nearly a fifth of patients discontinuing DMTs in the first year of therapy.37,38 While poor adherence may directly impact treatment outcomes, this is further compounded by the fact that ~20% of patients experience a suboptimal response to therapy and experience ongoing disease activity.39,40 Improving biomarker panels may, in the future, help to predict treatment response and guide treatment decision, but there are currently no universally accepted or used biomarkers of benign or aggressive MS. Light chains of neurofilaments in serum seem to be the most promising marker of neurodegeneration.41 There are data that suggest that certain inflammatory and degenerative biomarkers in the CSF also correlate with clinical and radiological disease activity, but it is not yet known whether these will have predictive value for treatment decisions.39
Emerging options: maintenance therapy versus selective immune reconstitution therapy (SIRT)
Most of the existing treatment options for patients with MS are maintenance therapies, which are regularly dosed, without interruption. The maintenance therapies for patients with MS include IFNβ, GA, TF, DMF, NTZ, and FL. These treatments may be able to induce remission with no evidence of disease activity for a period of 1–2 years at least. Maintenance therapies may require a long time to establish whether patients are receiving the anticipated treatment benefits. Disease activity rebounds after the treatment is stopped or interrupted; thus, in order to sustain the benefit it needs to be followed with the selected or another product.42
In contrast, the concept of SIRT describes a therapy that is given intermittently to produce durable effects on the immune system, even if given in very short-duration courses.43 Another important difference between maintenance therapy and the SIRT concept is the distribution of risk. With maintenance therapy, the risk of adverse events (AEs) increases with cumulative exposure to the drug. However, given the ongoing nature of treatment, dose adjustments or discontinuations can be strategies to mitigate any serious AE. With SIRT, the risk is essentially frontloaded. The risk of the majority of the most frequent AEs is greatest immediately following treatment administration (eg, infusion reactions or opportunistic infections). However, the risk of some AEs could increase in the posttreatment period without further administration of treatment (eg, autoimmune reactions).
Arguably, the rarely used autologous hematopoietic stem cell transplant (AHSCT) could be considered as an SIRT, although clearly AHSCT is a more intensive and invasive intervention than the purely pharmaceutical SIRT candidates.44 Pharmaceutical interventions for MS that may fit the definition of SIRT include ATZ (12 mg per day for 5 days, followed by a second course 12 months later for 3 days),45 possibly OCZ (initial dose of 300 mg with a second 300-mg dose 2 weeks later; subsequent doses of 600 mg every 6 months),46 and now also cladribine tablets for RRMS (two 5-day courses, 4 weeks apart, repeated after 1 year to give 3.5 mg/kg body weight over 2 years).47–49 In 2017, the European Medicine Agency approved the use of cladribine tablets for patients with highly active RRMS, as defined by clinical or imaging features.49
Cladribine tablets
Pharmacology and mechanism of action
Cladribine (2-chloro-2′-deoxyadenosine) is a synthetic deoxyadenosine analog prodrug (Figure 2).50,51 It was initially developed to treat lymphocytic malignancies, with a specific mechanism of action in mind.52 Based on the observation that deficiency of adenosine deaminase (ADA) leads to the intracellular accumulation of deoxyribonucleotides, cladribine was developed to be resistant to ADA.52,53 The end result is accumulation, after phosphorylation via deoxycytidine kinase (dCK), of 2-chlorodeoxyadenosine 5′triphosphate, leading to failure of DNA synthesis and repair, and apoptosis.51,54,55
Figure 2.
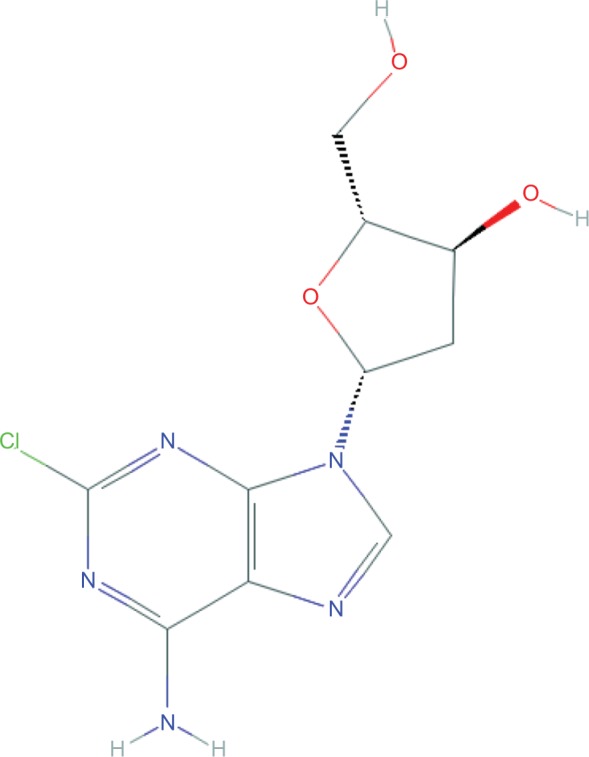
Molecular structure of cladribine
Note: Reproduced from the National Center for Biotechnology Information. PubChem Compound Database; CID=20279.50
Cladribine’s cytotoxic activity is targeted to lymphocytes due to the ratio of dCK (high) to 5′-nucleotidase in these cells, leading to rapid and sustained, but reversible, depletion of both dividing and resting lymphocytes.47,51,54–59 In particular, CD19+ B cells show a large decrease, followed by fairly rapid recovery, with a moderate and more sustained decline in CD4+ and CD8+ T cells.51,57,59 Crucially, cladribine is able to cross the BBB, reaching concentrations in the CSF of ~25% of that reached in the plasma.60 This finding is potentially very important in the context of MS, as it means that cladribine’s therapeutic effect may not be limited to the periphery, but may be acting on lymphocytes within the CNS.
Cladribine tablets clinical development program
Interest in cladribine as a therapeutic option for patients with RRMS began in the 1990s, with some small-scale studies using the parenteral form of cladribine in patients with RRMS as well as progressive forms of MS.55–57,61 Although these studies involved small patient numbers, they did demonstrate that cladribine had an acceptable safety profile and showed promising effects on clinical and MRI outcomes.55–57,61 In response to these initial findings, and reflecting the need for less frequent oral therapies, cladribine tablets were developed as a possible treatment for patients with RRMS.
Over the previous decade, cladribine tablets have been investigated in two large, randomized, double-blind studies with patients with RRMS (the CLAdRIbine Tablets treating MS orallY [CLARITY] and CLARITY Extension studies),47,48 in patients with early MS in the ORAl CLadribine for Early MS (ORACLE-MS) study,62,63 and as add-on to IFNβ in the Phase IIb oral cladribine added oN to IFNβ in patients With Active Relapsing Disease (ONWARD) trial.64 In each study, patients received cladribine tablets in what can be considered an SIRT regimen: two short courses of cladribine tablets (4–5 days each, 4 weeks apart) at the start of the first 48 weeks and two further short courses at the start of the second 48 weeks to give a cumulative dose of either 3.5 mg/kg (the currently approved dose) or 5.25 mg/kg (Table 1).47,48,62–64
Table 1.
Treatment arms in CLARITY, CLARITY Extension, and ORACLE-MS
| Study details | Study arms | ||||
|---|---|---|---|---|---|
| CLARITY47 | |||||
| Treatment arms | Placebo | CT 3.5 mg/kg | CT 5.25 mg/kg | ||
| N | 437 | 433 | 456 | ||
| CLARITY Extension48 | |||||
| Treatment received in CLARITY | Placebo | CT 3.5 mg/kg | CT 3.5 mg/kg | CT 5.25 mg/kg | CT 5.25 mg/kg |
| Treatment received in CLARITY Extension | CT 3.5 mg/kg | Placebo | CT 3.5 mg/kg | Placebo | CT 3.5 mg/kg |
| Cumulative study dose | PC 3.5 mg/kg | CP 3.5 mg/kg | CC 7.0 mg/kg | CP 5.25 mg/kg | CC 8.75 mg/kg |
| N | 244 | 98 | 92 | 186 | 186 |
| ORACLE-MS62 | |||||
| Treatment arms | Placebo | CT 3.5 mg/kg | CT 5.25 mg/kg | ||
| N | 206 | 206 | 204 | ||
| ORACLE-MS: McDonald 2010 reanalysis63 | |||||
| Treatment arms | Placebo | CT 3.5 mg/kg | CT 5.25 mg/kg | ||
| n still FCDE/CIS with new criteria | 134 | 138 | 121 | ||
| n with MS under new criteria | 72 | 68 | 83 |
Abbreviations: CC, cladribine tablets in CLARITY and CLARITY Extension; CIS, clinically isolated syndrome; CLARITY, CLAdRIbine Tablets treating multiple sclerosis orallY; CP, cladribine tablets in CLARITY and placebo in CLARITY Extension; CT, cladribine tablets; FCDE, first clinical demyelinating event; MS, multiple sclerosis; ORACLE-MS, ORAl CLadribine for Early MS; PC, placebo in CLARITY and cladribine tablets in CLARITY Extension.
CLARITY and CLARITY Extension
CLARITY was a large, multicenter, randomized, double-blind, placebo-controlled study of the safety, tolerability, and efficacy of cladribine tablets versus placebo in patients with RRMS (N=1326).47 Patients were randomized to either placebo, cladribine tablets 3.5 mg/kg, or 5.25 mg/kg (cumulative doses over 96 weeks).47 Both doses of cladribine tablets showed significantly decreased annualized relapse rates (ARR) and higher proportions of patients remaining relapse-free, compared to placebo (all comparisons vs placebo, p<0.001).47 Similarly, both doses of cladribine tablets significantly reduced the risk of disability progressing, as measured by a 3-month sustained increase in Expanded Disability Status Scale (EDSS) score, compared to placebo (p≤0.03).47
Patients who completed the CLARITY study were eligible to enter the CLARITY Extension study, a 96-week study which evaluated the safety and efficacy of cladribine tablets in the longer term.48 After a variable treatment gap between the two studies, enrolled patients who had received placebo during CLARITY were assigned to treatment with cladribine tablets 3.5 mg/kg, while patients who had received either of the active doses in CLARITY were re-randomized to either placebo or cladribine tablets 3.5 mg/kg (Table 1).48 During CLARITY Extension, the ARR was similar across the treatment groups, as was the proportion of relapse-free patients, demonstrating that the benefits of treatment with cladribine tablets were maintained for up to 4 years, even without retreatment.48 However, patients who had received placebo in the initial CLARITY study had a higher EDSS score at entry into CLARITY Extension and over the course of the study, and exhibited a higher ARR (p<0.0001) and a shorter time to first relapse compared to patients who had received either dose of cladribine tablets in CLARITY.48 Treatment with cladribine tablets in CLARITY, followed by placebo in CLARITY Extension, produced a durable clinical response (as measured from the end of CLARITY to the end of the Extension period), with no disease rebound after discontinuation of active treatment.48
ORACLE-MS
The use of cladribine tablets has also been investigated in the 2-year ORACLE-MS study, a randomized, double-blind, placebo-controlled study in patients (N=616) with early-stage disease who were at high risk of converting to clinically definite MS (CDMS), as defined by the Poser criteria and the McDonald 2005 criteria.62,63 Patients with a first clinical demyelinating event (FCDE)/clinically isolated syndrome (CIS) were randomized 1:1:1 to placebo and cladribine 3.5 or 5.25 mg/kg (Table 1).62 ORACLE-MS was terminated early due to a subsequently reversed decision to not continue the development of cladribine tablets, which does complicate the analysis of the data.62 Nevertheless, at study termination, cladribine tablets significantly reduced the risk of conversion to CDMS (Poser criteria), compared to placebo (hazard ratio [HR] for 3.5 mg/kg: 0.33, HR for 5.25 mg/kg: 0.38; both p<0.0001).62 The risk reductions were similar when considering conversion to CDMS as defined by the McDonald 2005 criteria.62 Again, the clinical benefits of cladribine tablets were reflected by improvements, compared with placebo, in MRI outcomes, with both doses associated with fewer new or persisting T1 gadolinium enhancing (Gd+) lesions, new or enlarging T2 lesions and combined unique (CU) lesions per patient per scan (all p<0.0001).62
The McDonald criteria were updated in 2010 to include MRI findings as a basis for diagnosis of MS, allowing for earlier confirmation of MS than in the previous iteration of the guidelines.65 In light of these changes, ORACLE-MS was reanalyzed as more than a third of the original population would have met the new McDonald criteria for MS at baseline (Table 1).63 Even with the reduced population of patients who were still classified as having FCDE/CIS at study entry, both doses of cladribine significantly reduced the risk of conversion to CDMS (3.5 mg/kg: 63% risk reduction, p=0.003; 5.25 mg/kg: 75% risk reduction, p<0.0001).63 With respect to disability progression, cladribine tablets 3.5 mg/kg reduced the risk of the next attack or 3-month confirmed EDSS by 74%, compared to placebo (74% risk reduction, p=0.0009).63 However, although cladribine tablets 5.25 mg/kg did also reduce the risk compared to placebo (37%), this result was not significant.63
ONWARD
The ONWARD trial was a 2-year, randomized, double-blind Phase IIb study of cladribine tablets 3.5 mg/kg versus placebo, both given as an add-on to IFNβ in patients who experienced at least one relapse on IFNβ in the previous 48 weeks.64 Safety and tolerability were the primary endpoints of ONWARD, with efficacy as a secondary endpoint.64 In patients with active relapsing MS and breakthrough disease despite IFNβ therapy, cladribine tablets 3.5 mg/kg as add-on to IFNβ resulted in a 63% risk reduction for ARR, compared to placebo as add-on to IFNβ (p<0.001).64
Cladribine tablets’ effects on markers of local inflammation in the CNS
In the original CLARITY study, compared to placebo, cladribine tablets 3.5 mg/kg significantly reduced the number of T1 Gd+ lesions (85.7% relative reduction), active (new or enlarging) T2 lesions (73.4% relative reduction), and CU lesions (74.4% relative reduction; all p<0.001).47 Very similar findings were seen for cladribine tablets 5.25 mg/kg.47 Additional analysis of the CLARITY MRI data demonstrated that improvements in MRI outcomes were already evident at the first assessment time point of 24 weeks.66 There were profound reductions, compared to placebo, in the number and volume of MRI lesions observed from as early as 24 weeks, and sustained through to 96 weeks.66 The proportion of patients free from T1 Gd+ lesions, active T2 lesions, and CU lesions at 96 weeks, compared with baseline, was greatly increased after treatment with each dose of cladribine tablets.66
These reductions in active lesions indicate a reduced inflammatory load and predict treatment response in the short term.66,67 Indeed, the MRI findings are consistent with the clinical findings of CLARITY, with treatment benefits appearing as early as 4 weeks and becoming statistically significant after 24 weeks.47,66
When stratified by baseline disease severity, post hoc analyses showed that treatment with cladribine tablets was associated with significant reductions in the mean number of T1 Gd+, active T2, and CU lesions versus placebo regardless of relapse status at baseline (ie, ≤1, 2, or ≥3 relapses; all p<0.001).66 These findings were also supported by a pooled analysis of CLARITY and ONWARD data, which found that the benefits of cladribine tablets on MRI outcomes were consistent, including in patients with indicators of high disease activity (Figure 3).68 This analysis found that cladribine tablets 3.5 and 5.25 mg/kg significantly reduced the cumulative number of new T1 Gd+ lesions bŷ90% and active T2 lesions bŷ75% compared with placebo in patients with active RRMS.68 In line with the MRI findings, the pooled analysis of CLARITY and ONWARD also demonstrated that the mean ARR and time to confirmed EDSS progression were significantly better for patients receiving cladribine tablets versus placebo.69
Figure 3.

Effects of cladribine tablets 3.5 mg/kg versus placebo in patient subgroups on the RR ratio of cumulative new T1 Gd+ lesions (A) and cumulative active T2 lesions (B).
Note: Reproduced from Giovannoni G, Comi G, Montalban X, Hicking C, Dangond F. Benefits of cladribine tablets on magnetic resonance imaging (MRI) outcomes in patients with multiple sclerosis: analysis of pooled double-blind data from the CLARITY and ONWARD studies (Abstract 603; Poster P642). Mult Scler J. 2016;22(3_Suppl):88.68
Abbreviations: DMD, disease-modifying drug; EDSS, Expanded Disability Status Scale; Gd+, gadolinium enhancing; HDA, high disease activity (≥2 relapses in previous year regardless of treatment status, or ≥1 relapse in the previous year while on DMD therapy and ≥1 T1 Gd+ lesion or 9 T2 lesions); OR, odds ratio; RR, relative risk; NE, not estimated.
Cladribine tablets’ effects on markers of neurodegeneration
As discussed, one of the most important global changes in MS is CNS atrophy, arising from focal and diffuse tissue damage, but not necessarily limited to sites of active inflammation and irrespective of the overall inflammatory load.17–19 It is, therefore, interesting to consider the effect that cladribine tablets, with its potent antilymphocytic mechanism of action, have on neurodegenerative atrophy. In exploratory analysis, brain volume measurements were performed on pre-Gd T1 MRI scans from the original CLARITY study.70 Between months 6 and 24 of CLARITY, patients receiving cladribine tablets 3.5 or 5.25 mg/kg experienced a significantly less percentage of brain volume change (PBVC) than patients receiving placebo (−0.77, −0.44, and −0.95, respectively; p=0.02 for both doses vs placebo).70 When annualized, the PBVC/year for months 6–24 was similarly reduced for cladribine tablets 3.5 and 5.25 mg/kg compared to placebo (−0.56, −0.57, and −0.70, respectively; p<0.02 for both doses vs placebo).70 Overall, patients treated with cladribine tablets showed significantly less annualized brain atrophy over 2 years than patients receiving placebo.70 It is not known whether these findings are entirely due to cladribine tablets’ anti-inflammatory action or if there may be another underlying neurodegenerative process that is affected by treatment with cladribine tablets, but the changes in PBVC were associated with differences in the risk of disability progression.70
An interesting sidenote to the PBVC analysis is the reason for analyzing the data only after 6 months. A recognized phenomenon of initiating anti-inflammatory drugs for the treatment of MS is a temporary acceleration of brain volume loss (“pseudoatrophy”).71 Although changes in the water content of the brain has been proposed as a potential mechanism, it may be that changes in inflammatory and glial cell volume are the underlying drivers of “pseudoatrophy.”71–73
Safety and tolerability
During the CLARITY study, the incidence of most reported AEs was comparable across treatment groups, except for lymphopenia (an expected event due to the mechanism of action of cladribine tablets), which was higher in the groups treated with cladribine tablets.74 The safety and tolerability findings for CLARITY Extension were broadly similar to CLARITY. Approximately 90% of patients completed treatment, and most AEs occurred at similar frequencies across treatment groups and most were classed as mild or moderate, with the exception of lymphopenia.48 In CLARITY and CLARITY Extension, incidence rates of the most common infections, including severe infections, were similar between patients treated with placebo and those treated with cladribine tablets, with the exception of herpes zoster, which was more frequently observed in patients treated with cladribine tablets compared with placebo.47,48
For ORACLE-MS, the incidence and severity of AEs (most were mild or moderate) were comparable across treatment groups, except for lymphopenia, which was higher in groups treated with cladribine tablets.62 Given the early termination of ORACLE-MS, the safety data from that trial are not directly comparable with the safety data from CLARITY and CLARITY Extension.
Lymphopenia
As cladribine was specifically designed to be selectively cytotoxic to lymphocytes, a background level of lymphopenia is to be expected.52 However, severe and sustained lymphopenia should be avoided. A retrospective analysis of CLARITY showed that severe lymphopenia could be mitigated using re-treatment guidelines.75 A year-by-year analysis of lymphopenia by grade in patients who were treated using re-treatment guidelines with cladribine tablets 3.5 mg/kg in CLARITY and with cladribine tablets 3.5 mg/kg in CLARITY Extension showed that >85% of patients had recovered to grade 0 or 1 lymphocyte counts and no patient had grade 4 lymphopenia at the end of each treatment year.76
Conclusion
MS is a complex disease that displays aspects of an autoimmune inflammatory and neurodegenerative disorder.4,17,24 In reality, it is probably both of these in a feedback loop, with inflammatory processes driving neurodegeneration, particularly in early disease, but with the possibility that neurodegenerative changes may be triggering additional inflammatory responses. It is true that Ig oligoclonal bands are characteristic in the CSF of patients with MS,14 showing ongoing immune activity, and that many of the known genetic loci associated with MS overlap with other autoimmune disease.7–9 However, it is also true that acute demyelination can occur without inflammatory cells and that demyelination begins at the inner myelin sheaths, with the outer sheaths still intact; a pattern that does not appear to support an antibody-mediated process.4,17 Furthermore, there are reports of neurodegeneration and atrophy occurring in patients with MS that are independent of the underlying inflammation.17,20–23 However, regardless of the balance between inflammation and the underlying neurodegeneration, anti-inflammatory DMTs do have proven efficacy in delaying disease and disability progression in MS.
Cladribine tablets 3.5 mg/kg have recently been approved for use in patients with highly active RRMS, as defined by clinical or imaging features.49 Cladribine has a well-defined and specifically designed mechanism of action for selectively depleting lymphocytes, an example of SIRT for MS.52 In CLARITY and CLARITY Extension, treatment with cladribine tablets provided durable efficacy in patients with RRMS: 2 years of active treatment in CLARITY produced significant reductions in relapse risk, disability progression, and MRI activity that were maintained for at least 2 further years in patients who received placebo in CLARITY Extension.47,48 ORACLE-MS showed that the risk of conversion from FCDE/CIS to CDMS (by the Poser and McDonald 2005/2010 criteria) was significantly reduced by treatment with cladribine tablets.62,63 The clinical benefits of cladribine tablets in these studies, such as relapse rate and disability progression, are also reflected by profound effects on neurological findings, with large reductions in lesion activity, and protection against brain volume loss.47,48,62,63,68,70 Whether all of these neurological findings are direct results of lymphocyte depletion, or if there are downstream effects on other, unknown, neurodegenerative processes are yet to be determined, but clearly point the way to an interesting area of research.
Cladribine tablets represent an important addition to the treatment options for patients with RRMS. They are a well-tolerated oral form of SIRT that place relatively little treatment burden on patients, and do not increase the monitoring burden for clinicians. The CLARITY and CLARITY Extension studies have demonstrated durable efficacy with cladribine tablets, even in the absence of re-treatment at least after 2 years. The clinical and MRI data for cladribine tablets show that there is rapid treatment effect on both markers of inflammation and on signs, such as brain volume loss, of neurodegeneration.
Acknowledgments
Medical writing assistance was provided by Ash Dunne of inScience Communications, Chester, UK, and supported by a Medical Writing Grant from Merck KGaA, Darmstadt, Germany.
Footnotes
Disclosure
Boyko AN received honoraria as member of working groups, advisory boards, and participated in clinical trials supported by Biogen, Schering, Merck, Teva, Novartis, Sanofi-Genzyme, Actelion, Biocad, and Generium. Boyko OV participated in clinical trials supported by Biogen, Merck, Teva, Novartis, Sanofi-Genzyme, Biocad, and Generium.
References
- 1.Browne P, Chandraratna D, Angood C, et al. Atlas of multiple sclerosis 2013: a growing global problem with widespread inequity. Neurology. 2014;83(11):1022–1024. doi: 10.1212/WNL.0000000000000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Multiple Sclerosis International Federation Atlas of MS 2013. Mapping multiple sclerosis around the world. 2013. Available from: https://www.msif.org/about-us/who-we-are-and-what-we-do/advocacy/atlas/.
- 3.Cohen JA, Rae-Grant A. In: Handbook of Multiple Sclerosis. 2nd ed. Salazar T, editor. London: Springer Healthcare; 2012. pp. 1–6. [Google Scholar]
- 4.Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 5.Herranz E, Gianni C, Louapre C, et al. Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol. 2016;80(5):776–790. doi: 10.1002/ana.24791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koudriavtseva T, Mainero C. Neuroinflammation, neurodegeneration and regeneration in multiple sclerosis: intercorrelated manifestations of the immune response. Neural Regen Res. 2016;11(11):1727–1730. doi: 10.4103/1673-5374.194804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beecham AH, Patsopoulos NA, Xifara DK, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45(11):1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bashinskaya VV, Kulakova OG, Boyko AN, Favorov AV, Favorova OO. A review of genome-wide association studies for multiple sclerosis: classical and hypothesis-driven approaches. Hum Genet. 2015;134:11–12. 1143–1162. doi: 10.1007/s00439-015-1601-2. [DOI] [PubMed] [Google Scholar]
- 9.Baranzini SE, Oksenberg JR. The genetics of multiple sclerosis: from 0 to 200 in 50 years. Trends Genet. 2017;33(12):960–970. doi: 10.1016/j.tig.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015;14(3):263–273. doi: 10.1016/S1474-4422(14)70267-4. [DOI] [PubMed] [Google Scholar]
- 11.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–558. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- 12.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 13.Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. 2012;8(11):613–623. doi: 10.1038/nrneurol.2012.203. [DOI] [PubMed] [Google Scholar]
- 14.Meinl E, Krumbholz M, Hohlfeld R. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol. 2006;59(6):880–892. doi: 10.1002/ana.20890. [DOI] [PubMed] [Google Scholar]
- 15.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 16.Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(Pt 9):2755–2771. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhuri A. Multiple sclerosis is primarily a neurodegenerative disease. J Neural Transm (Vienna) 2013;120(10):1463–1466. doi: 10.1007/s00702-013-1080-3. [DOI] [PubMed] [Google Scholar]
- 18.Filippi M, Rocca MA, Barkhof F, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2012;11(4):349–360. doi: 10.1016/S1474-4422(12)70003-0. [DOI] [PubMed] [Google Scholar]
- 19.Matthews PM, Roncaroli F, Waldman A, et al. A practical review of the neuropathology and neuroimaging of multiple sclerosis. Pract Neurol. 2016;16(4):279–287. doi: 10.1136/practneurol-2016-001381. [DOI] [PubMed] [Google Scholar]
- 20.Anderson VM, Fox NC, Miller DH. Magnetic resonance imaging measures of brain atrophy in multiple sclerosis. J Magn Reson Imaging. 2006;23(5):605–618. doi: 10.1002/jmri.20550. [DOI] [PubMed] [Google Scholar]
- 21.Bø L, Vedeler CA, Nyland H, Trapp BD, Mork SJ. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9(4):323–331. doi: 10.1191/1352458503ms917oa. [DOI] [PubMed] [Google Scholar]
- 22.DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM. The contribution of demyelination to axonal loss in multiple sclerosis. Brain. 2006;129(Pt 6):1507–1516. doi: 10.1093/brain/awl074. [DOI] [PubMed] [Google Scholar]
- 23.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50(3):389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 24.Fischer MT, Wimmer I, Hoftberger R, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(Pt 6):1799–1815. doi: 10.1093/brain/awt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14(3):271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 26.Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 2010;17(7):1083–1091. doi: 10.1038/cdd.2009.179. [DOI] [PubMed] [Google Scholar]
- 27.Stadelmann C, Wegner C, Bruck W. Inflammation, demyelination, and degeneration – recent insights from MS pathology. Biochim Biophys Acta. 2011;1812(2):275–282. doi: 10.1016/j.bbadis.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Hecker M, Hartmann C, Kandulski O, et al. Interferon-beta therapy in multiple sclerosis: the short-term and long-term effects on the patients’ individual gene expression in peripheral blood. Mol Neurobiol. 2013;48(3):737–756. doi: 10.1007/s12035-013-8463-1. [DOI] [PubMed] [Google Scholar]
- 29.Duddy M, Haghikia A, Cocco E, et al. Managing MS in a changing treatment landscape. J Neurol. 2011;258(5):728–739. doi: 10.1007/s00415-011-6009-x. [DOI] [PubMed] [Google Scholar]
- 30.Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- 31.Multiple Sclerosis IFNB, Group Study. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multi-center, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43(4):655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 32.Hartung HP, Montalban X, Sorensen PS, Vermersch P, Olsson T. Principles of a new treatment algorithm in multiple sclerosis. Expert Rev Neurother. 2011;11(3):351–362. doi: 10.1586/ern.11.15. [DOI] [PubMed] [Google Scholar]
- 33.Winkelmann A, Loebermann M, Reisinger EC, Hartung HP, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. 2016;12(4):217–233. doi: 10.1038/nrneurol.2016.21. [DOI] [PubMed] [Google Scholar]
- 34.Clanet MC, Wolinsky JS, Ashton RJ, Hartung HP, Reingold SC. Risk evaluation and monitoring in multiple sclerosis therapeutics. Mult Scler. 2014;20(10):1306–1311. doi: 10.1177/1352458513513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tur C, Tintore M, Vidal-Jordana A, et al. Risk acceptance in multiple sclerosis patients on natalizumab treatment. PloS One. 2013;8(12):e82796. doi: 10.1371/journal.pone.0082796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehr SR, Zimmerman MP. Reviewing the unmet needs of patients with multiple sclerosis. Am Health Drug Benefits. 2015;8(8):426–431. [PMC free article] [PubMed] [Google Scholar]
- 37.Evans C, Marrie RA, Zhu F, et al. Adherence and persistence to drug therapies for multiple sclerosis: a population-based study. Mult Scler Relat Disord. 2016;8:78–85. doi: 10.1016/j.msard.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Menzin J, Caon C, Nichols C, White LA, Friedman M, Pill MW. Narrative review of the literature on adherence to disease-modifying therapies among patients with multiple sclerosis. J Manag Care Pharm. 2013;19(1 Suppl A):S24–S40. doi: 10.18553/jmcp.2013.19.s1.S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novakova L, Axelsson M, Khademi M, et al. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J Neurochem. 2017;141(2):296–304. doi: 10.1111/jnc.13881. [DOI] [PubMed] [Google Scholar]
- 40.Tomioka R, Matsui M. Biomarkers for multiple sclerosis. Intern Med. 2014;53(5):361–365. doi: 10.2169/internalmedicine.53.1246. [DOI] [PubMed] [Google Scholar]
- 41.Brureau A, Blanchard-Bregeon V, Pech C, et al. NF-L in cerebrospinal fluid and serum is a biomarker of neuronal damage in an inducible mouse model of neurodegeneration. Neurobiol Dis. 2017;104:73–84. doi: 10.1016/j.nbd.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Suarez I, Rodriguez de Antonio L, Orviz A, et al. Catastrophic outcome of patients with a rebound after natalizumab treatment discontinuation. Brain Behav. 2017;7(4):e00671. doi: 10.1002/brb3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giovannoni G. Cladribine to treat relapsing forms of multiple sclerosis. Neurotherapeutics. 2017;14(4):874–887. doi: 10.1007/s13311-017-0573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. 2017;13(7):391. doi: 10.1038/nrneurol.2017.81. [DOI] [PubMed] [Google Scholar]
- 45.Genzyme Therapeutics LEMTRADA®. EMA summary of product characteristics. 2016. [Accessed November 28, 2017]. Available from: http://www.medicines.org.uk/emc/medicine/28917.
- 46.Ocrevus™ [FDA prescribing information] South San Francisco, CA: Genentech Inc; 2017. [Accessed November 28, 2017]. Available https://www.accessdata.fda.gov/drug-satfda_docs/label/2017/761053lbl.pdf. [Google Scholar]
- 47.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 48.Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler. 2017 Aug 1; doi: 10.1177/1352458517727603. Epub. [DOI] [PubMed] [Google Scholar]
- 49.Merck Serono Ltd. MAVENCLAD®. EMA summary of product characteristics. 2017. [Accessed November 28, 2017]. Available from: http://www.medicines.org.uk/emc/medicine/34044.
- 50.PubChem Compound Database [database on the Internet] Bethesda, MD: National Center for Biotechnology Information; [Accessed November 28, 2017]. CID=20279. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/20279. [Google Scholar]
- 51.Leist TP, Weissert R. Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol. 2011;34(1):28–35. doi: 10.1097/WNF.0b013e318204cd90. [DOI] [PubMed] [Google Scholar]
- 52.Carson DA, Wasson DB, Kaye J, et al. Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo. Proc Natl Acad Sci U S A. 1980;77(11):6865–6869. doi: 10.1073/pnas.77.11.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carson DA, Kaye J, Seegmiller JE. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s) Proc Natl Acad Sci U S A. 1977;74(12):5677–5681. doi: 10.1073/pnas.74.12.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beutler E. Cladribine (2-chlorodeoxyadenosine) Lancet. 1992;340(8825):952–956. doi: 10.1016/0140-6736(92)92826-2. [DOI] [PubMed] [Google Scholar]
- 55.Leist TP, Vermersch P. The potential role for cladribine in the treatment of multiple sclerosis: clinical experience and development of an oral tablet formulation. Curr Med Res Opin. 2007;23(11):2667–2676. doi: 10.1185/030079907x233142. [DOI] [PubMed] [Google Scholar]
- 56.Rice GP, Filippi M, Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Cladribine MRI Study Group. Neurology. 2000;54(5):1145–1155. doi: 10.1212/wnl.54.5.1145. [DOI] [PubMed] [Google Scholar]
- 57.Beutler E, Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci U S A. 1996;93(4):1716–1720. doi: 10.1073/pnas.93.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sigal DS, Miller HJ, Schram ED, Saven A. Beyond hairy cell: the activity of cladribine in other hematologic malignancies. Blood. 2010;116(16):2884–2896. doi: 10.1182/blood-2010-02-246140. [DOI] [PubMed] [Google Scholar]
- 59.Soelberg Sorensen P, Rieckmann P, Comi G, et al. Effects of cladribine tablets on circulating lymphocyte subsets in the 96-week CLARITY study in relapsing-remitting multiple sclerosis. Neurology. 2010;(73):330. [Google Scholar]
- 60.Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet. 1997;32(2):120–131. doi: 10.2165/00003088-199732020-00003. [DOI] [PubMed] [Google Scholar]
- 61.Romine JS, Sipe JC, Koziol JA, Zyroff J, Beutler E. A double-blind, placebo-controlled, randomized trial of cladribine in relapsing-remitting multiple sclerosis. Proc Assoc Am Physicians. 1999;111(1):35–44. doi: 10.1046/j.1525-1381.1999.09115.x. [DOI] [PubMed] [Google Scholar]
- 62.Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257–267. doi: 10.1016/S1474-4422(14)70005-5. [DOI] [PubMed] [Google Scholar]
- 63.Freedman MS, Leist TP, Comi G, et al. The efficacy of cladribine tablets in CIS patients retrospectively assigned the diagnosis of MS using modern criteria: results from the ORACLE-MS study. Mult Scler J Exp Transl Clin. 2017;3(4):2055217317732802. doi: 10.1177/2055217317732802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montalban X, Cohen B, Leist T, Moses H, Hicking C, Dangond F. Efficacy of cladribine tablets as add-on to IFN-beta therapy in patients with active relapsing MS: final results from the phase II ONWARD study (P03.029) Neurology. 2016;86(16 Supplement) [Google Scholar]
- 65.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Comi G, Cook SD, Giovannoni G, et al. MRI outcomes with cladribine tablets for multiple sclerosis in the CLARITY study. J Neurol. 2013;260(4):1136–1146. doi: 10.1007/s00415-012-6775-0. [DOI] [PubMed] [Google Scholar]
- 67.Kappos L, Polman C, Pozzilli C, Thompson A, Beckmann K, Dahlke F. Final analysis of the European multicenter trial on IFNbeta-1b in secondary-progressive MS. Neurology. 2001;57(11):1969–1975. doi: 10.1212/wnl.57.11.1969. [DOI] [PubMed] [Google Scholar]
- 68.Giovannoni G, Comi G, Montalban X, Hicking C, Dangond F. Benefits of cladribine tablets on magnetic resonance imaging (MRI) outcomes in patients with multiple sclerosis: analysis of pooled double-blind data from the CLARITY and ONWARD studies (Abstract 603; Poster P642) Mult Scler J. 2016;22(3_Suppl):88. [Google Scholar]
- 69.Giovannoni G, Montalban X, Hicking C, Dangond F. Benefits of cladribine tablets on relapse rates and disability progression in patients with multiple sclerosis: analysis of pooled double-blind data from the CLARITY and ONWARD studies. Mult Scler J. 2016;22(3_Suppl):88–399. [Google Scholar]
- 70.De Stefano N, Giorgio A, Battaglini M, et al. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult Scler. 2018;24(2):222–226. doi: 10.1177/1352458517690269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Stefano N, Arnold DL. Towards a better understanding of pseudoatrophy in the brain of multiple sclerosis patients. Mult Scler. 2015;21(6):675–676. doi: 10.1177/1352458514564494. [DOI] [PubMed] [Google Scholar]
- 72.Zivadinov R, Reder AT, Filippi M, et al. Mechanisms of action of disease-modifying agents and brain volume changes in multiple sclerosis. Neurology. 2008;71(2):136–144. doi: 10.1212/01.wnl.0000316810.01120.05. [DOI] [PubMed] [Google Scholar]
- 73.Chen JT, Collins DL, Atkins HL, Freedman MS, Galal A, Arnold DL. Brain atrophy after immunoablation and stem cell transplantation in multiple sclerosis. Neurology. 2006;66(12):1935–1937. doi: 10.1212/01.wnl.0000219816.44094.f8. [DOI] [PubMed] [Google Scholar]
- 74.Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler. 2011;17(5):578–593. doi: 10.1177/1352458510391344. [DOI] [PubMed] [Google Scholar]
- 75.Giovannoni G, Soelberg Sorensen P, Butzkueven G, et al. Mitigating severe lymphopenia: post hoc analysis of data from the 96-week CLARITY study. Mult Scler J. 2011;17(Suppl 1):S454–S455. [Google Scholar]
- 76.Cook S, Comi G, Giovannoni G, et al. Year-by-year lymphopenia rates in patients with relapsing multiple sclerosis (RMS) treated with cladribine tablets 3.5 mg/kg in CLARITY and re-treated in CLARITY Extension (Abstract EP1142) Eur J Neurol. 2017;(24):192. [Google Scholar]