

Abstract
The cerebellum is critical for an array of motor functions. During postnatal development, the Purkinje cells (PC) guide afferent topography to establish the final circuit. Perturbing PC morphogenesis or activity during development can result in climbing fiber (CF) multi-innervation or mis-patterning. Structural defects during circuit formation typically have long-term effects on behavior as they contribute to the phenotype of movement disorders such as cerebellar ataxia. The Car8wdl mouse is one model in which early circuit destruction influences movement. However, although the loss of Car8 leads to the mis-wiring of afferent maps and abnormal PC firing, adult PC morphology is largely intact and there is no neurodegeneration. Here, we sought to uncover how defects in afferent connectivity arise in Car8wdl mutants to resolve how functional deficits persist in motor diseases with subtle neuropathology. To address this problem, we analyzed CF development during the first three weeks of life. By immunolabeling CF terminals with VGLUT2, we found evidence of premature CF synapse elimination and delayed translocation from PC somata at postnatal day (P) 10 in Car8wdl mice. Surprisingly, by P15, the wiring normalized, suggesting that CAR8 regulates the early, but not the late stages of CF development. The data support the hypothesis of a defined time window in which cerebellar circuits must establish function.
Keywords: Cerebellum, Development, Climbing Fiber, Circuitry, CAR8, Ataxia
Introduction
An intact cerebellar circuit is crucial for proper motor function. Motor function is altered when the cells and synapses comprising this circuit are incorrectly positioned or improperly connected. For example, when differentiated granule cells (GC) in the external granule layer (EGL) lack a scaffold (i.e. Bergmann glia), they cannot migrate to form the inner granule layer, cells die, and the mice become severely ataxic [1]. Similarly, when Purkinje cells (PC) are displaced because of EGL injury, interneurons degenerate and eventually ataxia and tremor ensue [2,3]. Other ataxias and behavioral deficits involve aberrant mossy fiber (MF) or climbing fiber (CF) topography and connectivity [4,5]. Importantly, circuit dysfunction typically depends on PC development.
How afferent connectivity and functioning become compromised, ranges from poor targeting to poor cell-cell communication. In the case of CFs, several events are coordinated throughout postnatal development for each PC to receive input from one CF: (1) CFs project from the inferior olive to the cerebellum, (2) “winner” CFs translocate from the PC somata to innervate the PC dendrites, and (3) CF terminals are eliminated from the PC somata [6,7,8]. When CFs do not translocate, are not functional, or multi-innervate one PC, motor deficits persist [5, 9,10]. There is evidence that these behavioral deficits can result from a combination of abnormalities in CF and PC development because these two morphogenetic processes are interconnected [11,12,13]. PCs are among the first cerebellar cell types to be born and establish clear-cut patterns of anatomical and molecular expression patterns. The patterned architecture of PCs is thought to control the targeting of different sensory afferent types as well as the positional organization of other cerebellar cortical cells through the expression of molecular cues and synaptic activity [14,15]. PCs also control afferent circuitry at the single cell level. For instance, genetically ablating molecules such as RORα, Nogo-A, and NT-3 results in impaired PC dendritic sprouting, CF elimination, and CF translocation during cerebellar development [5,13,16]. Genetically inhibiting PC GABAergic neurotransmission also impairs circuit formation by mis-patterning PCs and altering the topography of afferent fibers [17]. In many of these examples, the manipulation of cerebellar circuitry results in the development of ataxia [5,17].
The majority of cerebellar ataxias in humans and animal models result from PC death and obvious mis-wiring defects [18, 19, 20, 21, 22, 23]. However, a number of cerebellar ataxias initiate and even persist in the absence of obvious structural defects, with limited/restricted neurodegeneration, or without widespread cell loss [24, 25, 26], whereas in other cases movement is affected before clear cerebellar pathology is detected [27, 28, 29]. The Car8wdl mouse model is an interesting example where the overall morphology of PCs is preserved, but PCs fire abnormally and the ataxia and other motor deficits worsen over time [30]. Car8wdl mutant mice have a null allele resulting from a spontaneous 19-basepair deletion in exon 8 of the carbonic anhydrase related-protein VIII (Car8) gene [31]. Car8 is expressed predominantly by PCs where it is thought to function as an intracellular Ca2+ regulator. Car8 binds to inositol 1,4,5-triphosphate receptor type 1 (IP3R1) and lowers its affinity for IP3 [32]. Mutations in the human homologues CA8 and ITPR1 cause ataxia and tremor [33, 34, 35, 36]. While loss of Car8 is thought to cause Ca2+ dysregulation, overexpression of Car8 stunts PC dendrite growth [37]. Although this overexpression study suggests a role for CAR8 during PC development, we do not know how the fine circuitry of the afferents is impaired when Car8 is deleted. However, these previous data raise the possibility that the structure and function of the developing cerebellar circuit might not only underlie the onset of PC dysfunction, but perhaps also the motor behavior defects later in life.
Since PCs are morphologically but not functionally normal in Car8wdl adults, we asked whether motor deficits develop because of impaired developmental connectivity. The two major classes of cerebellar afferents—mossy fibers (MF) and CFs—must wire up precisely before they are able to modulate PC firing during motor behavior. We have previously shown that the loss of Car8 delays parasagittal zone formation, alters MF development, and ultimately mis-patterns topographical maps [30]. Loss of Car8 has also been shown to lead to an increase in the total number of CF terminals in the molecular layer and an increase in CF occupancy of more distal areas of the molecular layer in adult mice [38]. There are several possibilities for how Car8 might impact the establishment of precise CF circuitry. Loss of Car8 could lead to ectopic CF innervation potentially due to an early increase in the number of terminals or loss of Car8 could promote early or enhanced CF translocation into the molecular layer. A third possibility could be that loss of Car8 delays PCs wiring so that CFs are forced to translocate onto the PC dendrites beyond the normal developmental time window. We would thus expect to find additional CFs in postnatal mutants. To distinguish between these possibilities, we followed CF development from postnatal day (P) 5 to P20 in Car8wdl mice.
Materials and Methods
Animals
C57BLKS/J (background control strain) and Car8wdl mice were purchased from The Jackson Laboratory (Bar Harbor, ME), crossed, and then maintained in our animal colony at Baylor College of Medicine following approved IACUC and institutional animal care protocols. We studied littermate and non-littermate C57BLKS/J controls and Car8wdl homozygotes since the heterozygotes do not exhibit obvious motor phenotypes [31]. Mutants were distinguished from controls through standard PCR genotyping, using previously published primer sequences [31]. We performed timed pregnant matings and noon on the day a vaginal plug was detected was considered embryonic day 0.5. Mice of either sex were analyzed at ages P5, P10, P15, and P20.
Mouse Perfusion and tissue processing procedures
Control and Car8wdl mice were anesthetized with 2,2,2-tribromoethanol (Avertin), then transcardially perfused with 0.1 M phosphate-buffered saline (PBS; pH 7.2) and 4% paraformaldehyde (PFA). Dissected brains were post-fixed in 4% PFA for at least 24 hours. To obtain sagittal sections, brains were sequentially transferred from ~24 hours in 18% sucrose to ~36 hours in 30% sucrose solutions, then embedded in Optimal Cutting Temperature (OCT) solution. Tissue was cut sagittally into 40 μm sections on a cryostat. Free-floating midline sagittal sections were stained following previously published protocols [4,39,40,41]. A VGLUT2 anti-guinea pig polyclonal antibody (Synaptic Systems, #135404) was used at a 1:500 dilution to label CF terminals. We used either anti-rabbit calbindin polyclonal (Swant #CB38) or anti-mouse calbindin monoclonal antibodies (Swant #300) both at a 1:10,000 dilution to label PCs. The tissue sections were fluorescently labeled using Alexa 488- and 647-conjugated immunoglobins at a 1:1,500 dilution (Invitrogen Molecular Probes Inc., Eugene, OR, USA #A-21202 and #A-31573). Tissues were also counterstained with DAPI upon cover-slipping (Vectashield Antifade Mounting Medium with DAPI #H-1200).
Image acquisition and data quantification
Images were captured using a Zeiss AxioCam MRm (fluorescence) camera that was mounted on a Zeiss Axio Imager.M2 microscope. We processed the images with Zeiss ZEN software (2012 edition). The number of VGLUT2 immunopositive punctae on each PC soma was counted from at least 2 sagittal sections taken from around the midline of 3 or more control (n=20) and Car8wdl (n=16) cerebella at ages P5, P10, P15, and P20. The molecular layer (ML) lengths and CF distribution were quantified from at least 2 sagittal sections taken from the midline and the immediately adjacent regions of the vermis per animal. We used ImageJ to quantify the data. We analyzed the CF data to calculate the percent CF territory (% CF territory = (CF length/ML length)*100) at each of the postnatal ages as stated above. All analyses were performed on similar anterior-posterior regions located within the primary fissure, which separates lobules IV/V and VI. Between-group differences were analyzed for significance (p<0.05) using the Student’s t-test for all of the ages that were analyzed in this study.
Results
We previously showed that MFs are mis-patterned in developing Car8wdl mice, contributing to an altered spinocerebellar map and motor dysfunction in adults [30]. Because both MF and CF connectivity rely on PC development, it is likely that CFs are also affected by the loss of Car8. We hypothesized that impaired CF wiring could contribute to cerebellar dysfunction in Car8wdl mice. To characterize CF development in postnatal Car8wdl mice, we stained control and mutant cerebella for PCs and CF terminals using calbindin and VGLUT2, respectively (Fig. 1a). We found that there were no significant differences in the number of puncta or CF occupancy of the molecular layer, on average, between PC somata at P5 (Fig. 1b; n=5 mutants, n=4 controls). As expected, we found fewer somatic VGLUT2 puncta, as the mice got older (Fig. 1b). At P10, Car8wdl mice have significantly fewer somatic VGLUT2 puncta (p<0.0001), suggesting that CFs are eliminated prematurely from mutant PCs (Fig. 1b-c; n=4 mutants, 6 controls). Also, at P10, we found a significant difference in CF occupancy of the molecular layer (p=0.0159). Greater than half (55%) of the P10 control tissue analyzed (compared to 13.3% of P10 Car8wdl tissue) have CF terminals that occupy more of the molecular layer than they did, on average, at P5 (Fig. 1b-c; n=4 mutants, 6 controls). At P15 and P20, we found no differences between the number of VGLUT2-positive puncta on PC somata or the percent CF territory in control compared to the mutant mice (Fig. 1b; n=3 mutants, 5 controls at P15; n=4 mutants, 5 controls at P20). Together, these data suggest that (1) CFs are correctly targeted from the inferior olive to the cerebellar cortex, (2) CF elimination may start earlier in developing Car8wdl mice, and (3) CF translocation may be delayed and occur over a longer period of time in developing Car8wdl mice. Because the number of VGLUT2-positive puncta on the PC somata is highly variable in P10 controls (which perhaps is a reflection of CF and PC heterogeneity [42]), in the Car8wdl mutant mice CF elimination may not only be completed sooner, but it may also proceed more uniformly across a given cerebellar lobule, or set of lobules (Fig. 1c; n=4 mutants, 6 controls).
Fig. 1.
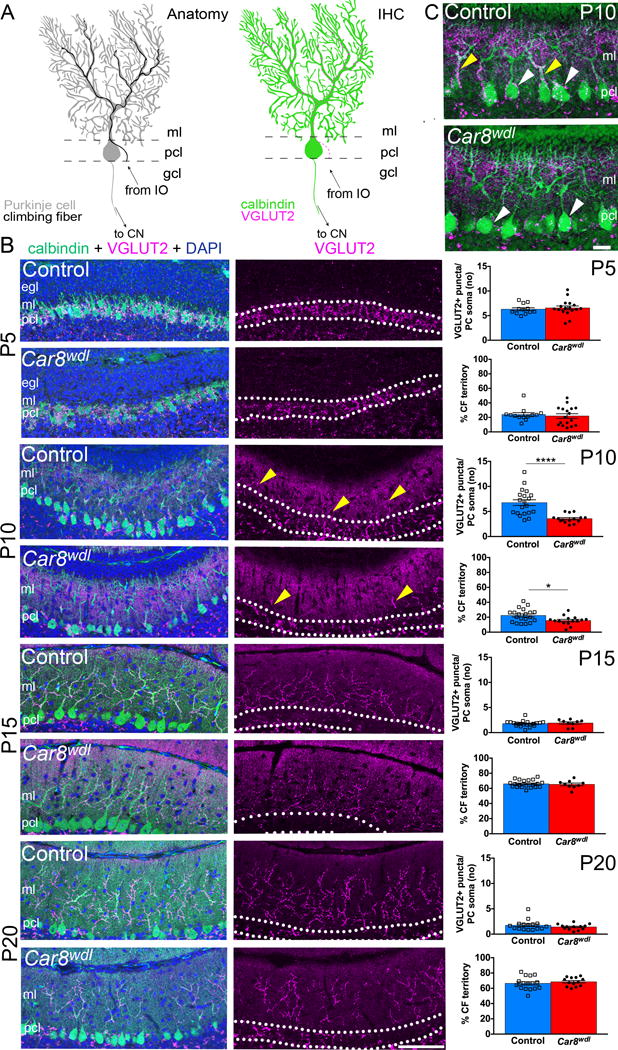
(a) Schematics of the olivo-cerebellar-nuclei circuit (left) and CF terminals on a PC (right). Abbreviations: IHC = immunohistochemistry, ml = molecular layer, pcl = Purkinje cell layer (white dotted lines), gcl = granule cell layer, cn = cerebellar nuclei, IO = inferior olive. (b) VGLUT2 and calbindin double staining reveals premature CF elimination and delayed CF translocation into the molecular layer at P10 (scale bar = 50 μm). The number (no) of VGLUT2-positive puncta on PC somata (p<0.0001) and percent CF territory (p=0.0159) were significantly different between Car8wdl and control mice at P10. In the graphs, * = 0.0159, and **** < 0.0001. Error bars reflect ± the standard of the mean (SEM). Yellow arrowheads point to the CF terminals that have translocated from the PC somata onto the PC dendrites. (c) Higher magnification of VGLUT2-positive CF terminals on PC somata (white arrowheads) and dendrites (yellow arrowheads) at P10 (scale bar = 20 μm). Control mice have more VGLUT2-positive CF terminals on PC somata and dendrites compared to the Car8wdl mutant mice.
Discussion
We have previously shown that loss of Car8 from PCs delays zone formation and alters MF patterning [30]. Here, we show that the loss of Car8 impacts CF development in postnatal mice in two ways: through premature CF elimination from PC somata and delayed CF translocation to PC dendrites. Although we did not find ectopic CF terminals in the most superficial portions of the molecular layer, our data instead suggests that (1) CAR8 may regulate the initiation of CF elimination and translocation, and (2) as discussed below, CAR8 may restrict the CF terminal field to the lower two thirds of the molecular layer as some CFs may expand further into the molecular layer after P20 in Car8wdl. Moreover, without CAR8, CFs are not only eliminated earlier, but within a given region of cerebellar cortex they also appear to be eliminated more uniformly from PC somata (Fig. 1b-c). Because the number of VGLUT2-positive puncta innervating the PC somata and dendrites normalizes between controls and mutants by P15, other molecules expressed by cerebellar cortical neurons may compensate for the loss of Car8. Although by P20 CF territories in control and mutant cerebella are not significantly different, it is possible that CF translocation continues past the normal developmental time window to contribute to the increased area and the increased total number of VGLUT2-positive terminals reported to occupy the molecular layer of Car8wdl adults [38]. Given that PC activity is altered in Car8wdl [30], it could be that specific functional changes in the developing circuit contribute to the phenotype. Interestingly, genetically altering PC excitability causes an increase in supernumerary CFs and an associated increase in complex spikes at P15 [43]. Coupled with the normal dynamics of complex spike activity during postnatal development [44], our Car8wdl CF data could reflect a fascinating intersection of structural and functional plasticity in developing mice.
The abnormalities we observed in CF innervation of PC soma and translocation in Car8wdl mutant mice likely contribute to the circuit dysfunction in these mice [30]. Previous electrophysiological analyses showed that Car8wdl PCs have fewer complex spikes and receive less excitatory input despite increased VGLUT2 expression in the molecular layer and normal paired pulse ratios at their parallel fiber (PF) synapses [30,38]. Furthermore, through electron microscopy, Hirasawa et al. (2007) found abnormalities in synaptogenesis. PC spines were either devoid of input or multiple PC spines shared one input, suggesting that fewer functional synapses form during development [38]. Although the CF impairments we found were either transient or delayed in Car8wdl mice, these could contribute to diminished PC excitation in the adult because several of the morphogenetic events that occur during cerebellar development—granule cell (GC) proliferation, PC dendritogenesis, and CF translocation—are interdependent and crucial for setting up a functional circuit. Impairments or delays to one of these events can impact the timing of others, thereby leading to permanent wiring defects and compromised motor function. For instance, transiently prolonging the cell cycle of GC precursors temporarily delays GC migration, as observed in Matrix metalloproteinase-2 (MMP-2) deficient mice [45]. Despite these transient developmental alterations at P3 and P9-P15, PC size is permanently affected, PC spine density and lengths are altered, and mild deficits in balance and motor coordination persist in the adult [46,47]. In hypothyroid mice, delayed GC migration and differentiation prolongs afferent innervation of PC somata, manifesting in vestibular problems and circling behavior [47,48]. Thus, the spatial and temporal features of cerebellar wiring are finely controlled and the behavioral consequences can be severe even when specific developmental processes are only temporarily disturbed.
The cerebellar circuit supports motor function in developing and adult animals. While we know that disrupting circuit development severely impairs motor behavior, we lack a comprehensive view of how short delays and transient defects contribute to long-lasting deficits that cause motor disease. Further elucidating how transiently impairing the cerebellar circuit affects movement in Car8wdl mice will advance efforts towards understanding the function of CAR8 in postnatal development. This in turn will help uncover how different forms of ataxia and tremor arise and how they might be treated.
Acknowledgments
This work was supported by funds from Baylor College of Medicine (BCM) and Texas Children’s Hospital, BCM IDDRC Grant U54HD083092 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (The IDDRC Neuropathology Sub-Core contributed to the tissue staining experiments), and by National Institutes of Neurological Disorders and Stroke (NINDS) R01NS089664.
References
- 1.Rakic P, Sidman RL. Organization of cerebellar cortex secondary to deficit of granule cells in weaver mutant mice. J Comp Neurol. 1973;152(2):133–161. doi: 10.1002/cne.901520203. [DOI] [PubMed] [Google Scholar]
- 2.Woodward DJ, Bickett D, Chanda R. Purkinje cell dendritic alterations after transient developmental injury of the external granular layer. Brain Res. 1975;97(2):195–214. doi: 10.1016/0006-8993(75)90445-x. [DOI] [PubMed] [Google Scholar]
- 3.Haddad RK, Rabe A, Dumas R. Comparison of effects of methylazoxymethanol acetate on brain development in different species. Fed Proc. 1972;31:1520–1523. [PubMed] [Google Scholar]
- 4.Sillitoe RV, Benson MA, Blake DJ, Hawkes R. Abnormal dysbindin expression in cerebellar mossy fibers synapses in the mdx mouse model of Duchenne Muscular Dystrophy. J Neurosci. 2003;23(16):6576–6585. doi: 10.1523/JNEUROSCI.23-16-06576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crepel F, Delhaye-Bouchaud N, Guastavino JM, Sampaio I. Multiple innervation of cerebellar Purkinje cells by climbing fibres in staggerer mutant mouse. Nature. 1980;283:483–484. doi: 10.1038/283483a0. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe M, Kano M. Climbing fiber synapse elimination in cerebellar Purkinje cells. Eur J Neurosci. 2011;34(10):1697–1710. doi: 10.1111/j.1460-9568.2011.07894.x. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto K, Kano M. Synapse elimination in the developing cerebellum. Cell Mol Life Sci. 2013;70(24):4667–4680. doi: 10.1007/s00018-013-1405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White JJ, Sillitoe RV. Development of the cerebellum: From gene expression patterns to circuit maps. WIREs Dev Bio. 2013;2(1):149–164. doi: 10.1002/wdev.65. [DOI] [PubMed] [Google Scholar]
- 9.Rossi F, Jankovski A, Sotelo C. Target neuron controls the integrity of afferent axon phenotype: A study on the Purkinje cell-climbing fiber system in cerebellar mutant mice. J Neurosci. 1995;15(3):2040–2056. doi: 10.1523/JNEUROSCI.15-03-02040.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JJ, Sillitoe RV. Genetic silencing of olivocerebellar synapses causes dystonia-like behavior in mice. Nat Commun. 2017;8:14912. doi: 10.1038/ncomms14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason CA, Christakos S, Catalano SM. Early climbing fiber interactions with Purkinje cells in the postnatal mouse cerebellum. J Comp Neurol. 1990;297(1):77–90. doi: 10.1002/cne.902970106. [DOI] [PubMed] [Google Scholar]
- 12.Bradley P, Berry M. The effects of reduced climbing and parallel fibre input on Purkinje cell dendritic growth. Brain Res. 1976;109(1):133–151. doi: 10.1016/0006-8993(76)90384-x. [DOI] [PubMed] [Google Scholar]
- 13.Sherrard RM, Bower AJ. Climbing fiber development: do neurotrophins have a part to play? Cerebellum. 2002;1(4):265–275. doi: 10.1080/147342202320883579. [DOI] [PubMed] [Google Scholar]
- 14.Apps R, Hawkes R. Cerebellar cortical organization: A one-map hypothesis. Nature Rev Neurosci. 2009;10(9):670–681. doi: 10.1038/nrn2698. [DOI] [PubMed] [Google Scholar]
- 15.Dahmane N, Ruiz I, Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 16.Petrinovic MM, Hourez R, Aloy EM, Dewarrat G, Gall D, Weinmann O, Gaudias J, Bachmann LC, Schiffmann SN, Vogt KE, Schwab ME. Neuronal Nogo-A negatively regulates dendritic morphology and synaptic transmission in the cerebellum. Proc Natl Acad Sci. 2013;110(3):1083–1088. doi: 10.1073/pnas.1214255110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White JJ, Arancillo M, Stay TL, George-Jones NA, Levy SL, Heck DH, Sillitoe RV. Cerebellar zonal patterning relies on Purkinje cell neurotransmission. J Neurosci. 2014;34(24):8231–8245. doi: 10.1523/JNEUROSCI.0122-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker EBE, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EMC, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. PNAS. 2009;106(16):6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim J, Hao T, Shaw C, Patel AJ, Szabó G, Rual JF, Fisk CJ, Li N, Hill DE, Barabási AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125(4):801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 20.He Y, Zu T, Benzow KA, Orr HT, Clark HB, Koob MD. Targeted deletion of a single Sca8 ataxia locus allele in mice causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits. J Neurosci. 2006;26(39):9975–9982. doi: 10.1523/JNEUROSCI.2595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJA, Dutia MB, Jackson M. Loss of β-III Spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of Spinocerebellar Ataxia Type 5 in humans. J Neurosci. 2010;30(14):4857–4867. doi: 10.1523/JNEUROSCI.6065-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim J, Jafa-Nejad P, di Ronza A, Kang H, Sayegh LS, Cooper TA, Orr HT, Sillitoe RV, Zoghbi HY. Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type ataxin1 levels. Cell. 2015;160(6):1087–1098. doi: 10.1016/j.cell.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramani B, Harris GM, Huang R, Seki T, Murphy GG, Costa Mdo C, Fischer S, Saunders TL, Xia G, McEachin RC, Paulson HL. A knockin mouse model of spinocerebellar ataxia type 3 exhibits prominent aggregate pathology and aberrant splicing of the disease gene transcript. Hum Mol Genet. 2015;24(5):1211–1224. doi: 10.1093/hmg/ddu532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65(10):1296–1303. doi: 10.1001/archneur.65.10.1296. [DOI] [PubMed] [Google Scholar]
- 25.Novak MJU, Sweeney MG, Li A, Treacy C, Chandrashekar HS, Giunti P, Goold RG, Davis MB, Houlden H, Tabrizi SJ. An ITPR1 gene deletion causes Spinocerebellar Ataxia 15/16: A genetic, clinical and radiological description. Mov Disord. 2010;25(13):2176–2182. doi: 10.1002/mds.23223. [DOI] [PubMed] [Google Scholar]
- 26.Hisatsune C, Miyamoto H, Hirono M, Yamaguchi N, Sugawara T, Ogawa N, Ebisui E, Ohshima T, Yamada M, Hensch TK, Hattori M, Mikoshiba K. IP3R1 deficiency in the cerebellum/brainstem causes basal ganglia-independent dystonia by triggering tonic Purkinje cell firings in mice. Front Neural Circuits. 2013;7:156. doi: 10.3389/fncir.2013.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alviña K, Khodakhah K. The therapeutic mode of action of 4-Aminopyridine in cerebellar ataxia. J Neurosci. 2010;30(21):7258–7268. doi: 10.1523/JNEUROSCI.3582-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease Spinocerebellar Ataxia Type 3. J Neurosci. 2011;31(36):13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22(2):271–283. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White JJ, Arancillo M, King A, Lin T, Miterko LN, Gebre SA, Sillitoe RV. Pathogenesis of severe ataxia and tremor without the typical signs of neurodegeneration. Neurobiol Dis. 2016;86(2016):86–98. doi: 10.1016/j.nbd.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiao Y, Yan J, Zhao Y, Donahue LR, Beamer WG, Li X, Roe BA, LeDoux MS, Gu W. Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics. 2005;171(3):1239–1246. doi: 10.1534/genetics.105.044487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirota J, Ando H, Hamada K, Mikoshiba K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J. 2003;372:435–441. doi: 10.1042/BJ20030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaya N, Aldhalaan H, Al-Younes B, Colak D, Shuaib T, Al-Mohaileb, Al-Suigair A, Nester M, Al-Yamani S, Al-Bakheet A, Al-Hashmi N, Al-Sayed M, Meyer B, Jungbluth H, Al-Owain M. Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am J Med Genet. 2011;156(7):826–834. doi: 10.1002/ajmg.b.31227. [DOI] [PubMed] [Google Scholar]
- 34.Van de Leemput J, Chandran J, Kinght MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung H, Lin X, Hernandez D, Simon-Sanchez J, Wood NW, Giunti P, Rafferty I, Hardy J, Storey E, Gardner RJM, Forrest SM, Fisher EMC, Russell JT, Cai H, Singleton AB. Deletion at ITPR1 underlies ataxia in mice and Spinocerebellar Ataxia 15 in humans. PLoS Genet. 2007;3(6):e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Synofzik M, Beetz C, Bauer C, Bonin M, Sanchez-Ferrero E, Schmitz-Hubsch T, Wullner U, Nagele T, Riess O, Schols L, Bauer P. Spinocerebellar Ataxia Type 15: Diagnostic assessment, frequency, and phenotypic features. J Med Genet. 2011;48(6):407–412. doi: 10.1136/jmg.2010.087023. [DOI] [PubMed] [Google Scholar]
- 36.Storey E, Gardner RJ. Spinocerebellar Ataxia Type 15. Handb Clin Neurol. 2012;103:561–565. doi: 10.1016/B978-0-444-51892-7.00037-1. [DOI] [PubMed] [Google Scholar]
- 37.Shimobayashi E, Wagner W, Kapfhammer JP. Carbonic anhydrase 8 expression in purkinje cells is controlled by PKCγ activity and regulates purkinje cell dendritic growth. Mol Neurobiol. 2015;53(8):5149–5160. doi: 10.1007/s12035-015-9444-3. [DOI] [PubMed] [Google Scholar]
- 38.Hirasawa M, Xu X, Trask RB, Maddatu TP, Johnson BA, Naggert JK, Nishina PM, Ikeda A. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol Cell Neurosci. 2007;35(1):161–170. doi: 10.1016/j.mcn.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sillitoe RV, Vogel MW, Joyner AL. Engrailed homeobox genes regulate establishment of the cerebellar afferent circuit map. J Neurosci. 2010;30(30):10015–10024. doi: 10.1523/JNEUROSCI.0653-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reeber SL, Sillitoe RV. Patterned expression of a cocaine- and amphetamine-regulated transcript peptide reveals complex circuit topography in the rodent cerebellar cortex. J Comp Neurol. 2011;519(9):1781–1796. doi: 10.1002/cne.22601. [DOI] [PubMed] [Google Scholar]
- 41.White JJ, Sillitoe RV. Postnatal development of cerebellar zones revealed by neurofilament heavy chain protein expression. Front Neuroanat. 2013;7(9):1–10. doi: 10.3389/fnana.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerminara NL, Lang EJ, Sillitoe RV, Apps R. Redefining the cerebellar cortex as an assembly of non-uniform Purkinje cell microcircuits. Nat Rev Neurosci. 2015;16(2):79–93. doi: 10.1038/nrn3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorenzetto E, Caselli L, Feng G, Yuan W, Nerbonne JM, Sanes JR, Buffelli M. Genetic perturbation of postsynaptic activity regulates synapse elimination in developing cerebellum. Proc Natl Acad Sci USA. 2009;106(38):16475–16480. doi: 10.1073/pnas.0907298106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arancillo M, White JJ, Lin T, Stay TL, Sillitoe RV. In vivo analysis of Purkinje cell firing properties during postnatal mouse development. J Neurophysiol. 2015;113(2):578–591. doi: 10.1152/jn.00586.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verselgers M, Van Hove I, Buyens T, Dekeyster E, Knevels E, Moons L. Identification of MMP-2 as a novel enhancer of cerebellar granule cell proliferation. Mol Cell Neurosci. 2013;57(2013):63–72. doi: 10.1016/j.mcn.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Verslegers M, Van Hove I, Dekeyster E, Gantois I, Hu T, D’Hooge R, Arckens L, Moons L. MMP-2 mediates Purkinje cell morphogenesis and spine development in the mouse cerebellum. Brain Struct Funct. 2015;220(3):1601–1617. doi: 10.1007/s00429-014-0747-3. [DOI] [PubMed] [Google Scholar]
- 47.Neveu I, Arena E. Neurotrophins promote survival and development of neurons in the cerebellum of hypothyroid rats in vivo. J Cell Biol. 1996;133(3):631–646. doi: 10.1083/jcb.133.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kincaid AE. Spontaneous circling behavior and dopamine neuron loss in a genetically hypothyroid mouse. Neuroscience. 2001;105(4):891–898. doi: 10.1016/s0306-4522(01)00229-9. [DOI] [PubMed] [Google Scholar]