

Abstract
Langerhans cell histiocytosis (LCH) is an inflammatory myeloid neoplasm characterized by constitutive activation of extracellular signal-regulated kinase (ERK). Genomic characterization has identified activating point mutations including mutually exclusive BRAFV600E and activating MAP2K1 mutations to be responsible for ERK activation in a majority of pediatric LCH patients. Here, we report the discovery of a novel BRAF kinase fusion, PACSIN2-BRAF, in a child with multisystem LCH. This is the second reported case of an activating BRAF kinase fusion and indicates a recurrent pathologic mechanism. Genomic evaluation for activating kinase fusions should be strongly considered in pediatric LCH patients lacking more common mutations.
Keywords: BRAF, kinase fusion, Langerhans cell histiocytosis, LCH, myeloid neoplasia, PACSIN2
1 |. INTRODUCTION
Langerhans cell histiocytosis (LCH) is an inflammatory myeloid neoplasm characterized by clonal proliferation of CD1a+/CD207+ cells.1 At a molecular level, pathologic constitutive activation of extra cellular signal-regulated kinase (ERK) is universally implicated in LCH and thought to drive the disease.2 Genomic characterization of pediatric LCH has identified a number of activating mutations responsible for ERK activation including mutually exclusive BRAFV600E2–7 and MAP2K1 mutations in 40–70% and 10–20% of patients, respectively.4,8,9 There are also case reports of mutations in ARAF10 and ERBB3,4 a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases, in pediatric LCH. A recent report identified a single case of an in-frame BRAF kinase fusion (FAM73A-BRAF) in a child with LCH.11 Due to the limited mRNA sequencing (RNA-seq) of LCH to date, however, it is unclear if fusions in BRAF or other kinases are recurrent in pediatric LCH. Here, we report the discovery of a novel BRAF kinase fusion, PACSIN2-BRAF, produced by chromosomal translocation t(22;7)(q13.2;q34) in a child with refractory cutaneous LCH. This is the first report of this fusion in human disease and represents the second reported kinase fusion in a child with LCH. This finding suggests that in-frame activating BRAF kinase fusions are a recurrent phenomenon in pediatric LCH and a recurrent genomic mechanism for ERK activation in patients without BRAFV600E activating mutations.
2 |. METHODS
Parent-informed consent and patient-informed assent were obtained for photographic documentation of the patient’s skin lesions for clinical use and in adeidentified manner to report this case. Assent and consent were obtained for enrollment on specimen acquisition and molecular profiling protocols approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center. Histopathologic and molecular data were obtained as part of clinical care. No patient tissue was used for experimental purposes. Sequencing platforms and experimental methods are described in Supplementary Methods S1.
2.1 |. Case description and experimental results
At disease presentation, our patient was a healthy 6-year-old male child with no significant personal or family medical history. He developed a physically detectable right temporal bone lesion, further visualized by computed tomography (CT) scan (Fig. 1A). He also presented with several 5–8 mm erythematous papular skin lesions on his trunk (Fig. 1B). Upon resection, the temporal bone lesion consisted of CD1a+/CD207+ cells, consistent with a diagnosis of LCH (Fig. 1C). The cutaneous lesions were positive for CD1a and phospho-ERK1/2 (pERK1/2) and negative for BRAFV600E by immunohistochemistry (Fig. 1D). This child’s LCH was classified as multisystem (cutaneous, temporal bone), risk-organ negative (no bone marrow, liver, or spleen involvement) with a central nervous system (CNS)-risk lesion (temporal bone; CNS-risk lesions are a specific group of craniofacial bones, including the temporal bone, whose involvement is associated with increased risk of developing diabetes insipidus [DI]).12 The patient was treated according to the LCH-III protocol13 with routine extent of disease evaluations including brain magnetic resonance imagings and dermatology assessments.
FIGURE 1.

Clinical and histopathological presentation of PACSIN2-BRAF expressing multisystem Langerhans cell histiocytosis (LCH). (A) Computed tomography (CT) scan showing a 4.0 × 2.6 cm right retroauricular mass centered on the posterosuperior right temporal bone, with focal dehiscence of the bone, scalloped margins, and a small intracranial epidural component. (B) Dermatology exam demonstrating scattered erythematous papules at low and high magnification. (C) Histopathological examination of resected right temporal scalp lesion showing dermal infiltration of histiocytes, multinucleated giant cells, and eosinophils (hematoxylin and eosin (H&E), 400× magnification) with immunoreactivity for Langerin (CD207) and CD1a (200× magnification). (D) Histopathological examination of diagnostic skin lesions with perifollicular histiocytic infiltration within the dermis (H&E, 200× magnification) that is negative for BRAF V600E (not shown) and immunoreactive for CD1a (400× magnification) and phospho-ERK1/2 (200× magnification). (E) Histopathological examination of LCH skin lesions (H&E, 200× magnification) at disease progression after 12 weeks of prednisone and vinblastine therapy with residual histiocytic infiltrate and scattered eosinophils in the dermis that retain scattered immunoreactivity for CD1a (100× magnification) and phospho-ERK1/2 (400× magnification)
After 12 weeks of therapy with prednisone and vinblastine (LCH-III, initial course 1, 213), the patient had persistent cutaneous lesions. To assess for refractory disease, a biopsy from two representative lesions was performed. Pathologic assessment confirmed active LCH with residual immunoreactivity for CD1a and pERK1/2 (Fig. 1E). In order to identify a potential genetic alteration responsible for ERK activation, genomic DNA from these lesions underwent targeted mutational analysis using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a hybrid capture-based DNA sequencing assay of 410 cancer genes (Supplementary Methods S1).14 The lesional DNA was compared to genomic DNA from the patient’s blood as a paired normal control. However, this failed to reveal mutations in any regions of BRAF (exon 11 [439–478], exon 15 [581–620], or introns 7–10), MAP2K1, ERK, ARAF, or ERBB3, which have previously been described as mutated in LCH. The patient completed 52 weeks of prednisone and vinblastine according to previously established standard of care.13 At the end of treatment, he had several stable cutaneous lesions with no other sites of disease. During routine surveillance 4 months later, however, his cutaneous lesions had progressed. These asymptomatic lesions were < 1 cm without secondary changes.
Due to the recent description of BRAF kinase fusions in histiocytic disorders identified by RNA seq,11,15 we utilized a next-generation sequencing (NGS), RNA-based fusion gene detection assay known as the Archer FusionPlex Custom Solid Panel (Supplementary Methods S1).16 This assay evaluates for fusions across 35 cancer-related genes involved in chromosomal rearrangements. The BRAF-specific primers provide coverage for exons 7, 9, and 11. Using this method, we identified a novel BRAF fusion, PACSIN2-BRAF (Fig. 2A), generated through an in-frame fusion between genes PACSIN2 exon 7 (NM_001184970) on chromosome 22q13.2 and BRAF exon 9 (NM_004333) on chromo some 7q34. Although low tumor purity in the skin biopsy sample precluded detection of the fusion by genomic DNA mutational analysis, high expression of the fusion RNA transcript allowed for its detection by Archer FusionPlex.
FIGURE 2.
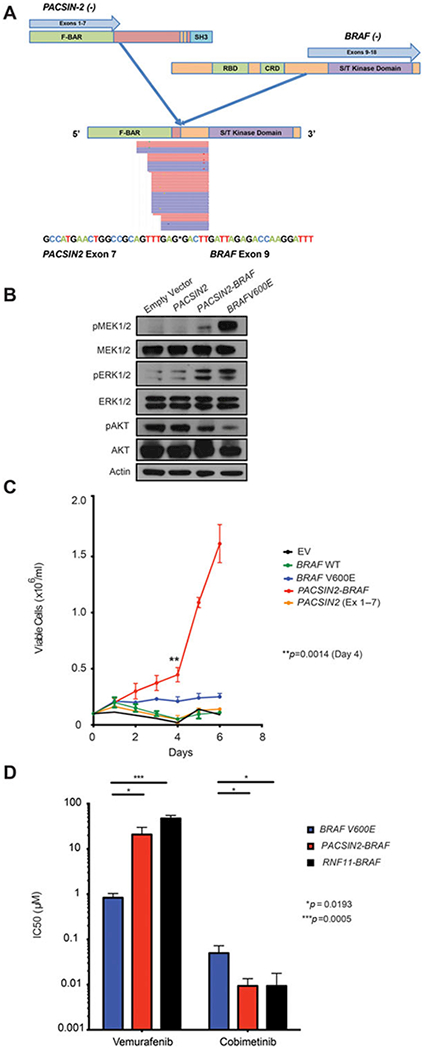
Characterization of the PACSIN2-BRAF fusion. (A) Schematic illustration of protein structure and transcript sequence of the PACSIN2 (NM_007229)-BRAF (NM_004333) in-frame fusion product with exons 1–7 of PACSIN2 fused to exons 9–18 of BRAF. Red and blue rectangles represent bidirectional RNA sequencing reads supporting the fusion breakpoint. (B) Effect of stable expression of an empty vector, PACSIN2 exons 1–7, PACSIN2-BRAF, and BRAFV600E in GP2–293 cells on MAP kinase and AKT signaling and (C) cytokine independent growth of Ba/F3 cells. Mean viable cell number post-IL-3 withdrawal from a triplicate experiment is shown. Error bars indicate standard deviation of mean. The P value was calculated using the Friedman test. (D) CellTiter-Glo luminescent viability IC50 results from three independent triplicate experiments of Ba/F3 cells from (C), as well as RNF11-BRAF-expressing Ba/F3 cells, exposed to the MEK inhibitor cobimetinib and the RAF inhibitor vemurafenib. Log10 IC50 values are shown on the y-axis. Error bars indicate standard deviation of mean. The P value was calculated using the unpaired t-test. F-BAR domain, Fes/CIP4homology-Bin-amphiphysin-Rvs + coiled-coil domain; RBD, RAS binding domain; CRD, cysteine-rich domain; SH3, SRC homology 3 domain; S/T kinase domain, serine/threonine kinase; RNF11-BRAF, ring finger protein 11-B-raf protooncogene
As with other previously described BRAF fusions, this fusion produces a protein lacking the N-terminal regulatory RAS-binding domain of BRAF, thereby placing the BRAF kinase domain under aberrant regulation of the PACSIN2 promoter. PACSIN2 is an F-BAR protein involved in regulating EGFR signaling by mediating intracellular receptor trafficking.17 Of particular importance here, deletion of the C-terminal SRC homology 3 (SH3) domain of PACSIN2 by itself has also been suggested to lead to ERK activation.17 To evaluate the functional effects of PACSIN2-BRAF and the potential contribution of truncated PACSIN2 to ERK activation, we generated cDNA for both the PACSIN2-BRAF fusion and PACSIN2 cDNA truncated at exon 7 to mimic the portion of PACSIN2 incorporated in the fusion (Supplementary Methods S1). Stable expression of PACSIN2-BRAF, along with an empty vector, truncated PACSIN2 (exons 1–7), or BRAFV600E in GP2–293 cells revealed activation of MEK1/2 and ERK1/2 phosphorylation by both PACSIN2-BRAF and BRAFV600E (Fig. 2B) concomitant with reduced phosphorylation of AKT1/2. Consistent with this, stable expression of PACSIN2-BRAF or BRAFV600E resulted in cytokine-independent growth of Ba/F3 cells (Fig. 2C), indicating that the PACSIN2-BRAF fusion is an activating event with pathway activity similar to the BRAFV600E mutation. PACSIN2-BRAF expression also sensitized Ba/F3 cells to MEK inhibition by cobimetinib but not to RAF inhibition by vemurafenib in vitro (Fig. 2D; Supplementary Methods S1).
Given that our patient’s cutaneous lesions remain asymptomatic, and he has no evidence of DI or multisystem disease, we are monitoring his disease with careful surveillance rather than second-line or investigational therapy. As with previously reported BRAF fusions and based on our in vitro kinase inhibitor evaluation, his activating fusion is not expected to be sensitive to RAF inhibitors vemurafenib or dabrafenib but may be sensitive to MEK inhibitors trametinib or cobimetinib.11,15,18,19
3 |. DISCUSSION
Chromosomal rearrangements have not been frequently reported in LCH. In fact, prior to the wider implementation of NGS, a study of 31 patient samples found no detectable translocations.20 The discovery of this BRAF kinase fusion highlights fusions of BRAF as a recurrent alternative mechanism for pathologic ERK activation in LCH. Given these data and the discovery of recurrent fusions in anaplastic lymphoma kinase (ALK) and neurotrophic receptor tyrosine kinase (NTRK) members in non-LCH,15 we suspect activating kinase fusions may have a higher prevalence in pediatric LCH. Genomic evaluation for activating kinase fusions should be strongly considered in pediatric LCH patients who do not have activating point mutations in BRAF, MAP2K1, or other genes known to be affected by coding nucleotide mutations in order to identify potential candidates for targeted therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. S.Z. is supported by CURE Childhood Cancer. B.H.D. is supported by the American Society of Hematology Senior Research Training Award for Fellows and the New York State Council on Graduate Medical Education Empire Clinical Research Investigator Program Fellowship. E.L.D. and O.A.W. are supported by grants from the Erdheim Chester Disease Global Alliance and the Histiocytosis Association. O.A.W. is supported by grants from the Edward P. Evans Foundation, the Department of Defense Bone Marrow Failure Research Program (BM150092 and W81XWH-12–1-0041), NIH/NHLBI (R01 HL128239), an NIH K08 Clinical Investigator Award (1K08CA160647–01), the Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award, an award from the Starr Foundation (I8-A8–075), the Leukemia and Lymphoma Society, and the Pershing Square Sohn Cancer Research Alliance.
Grant sponsor: NIH/NCI Cancer Center Support Grant; Grant number: P30 CA008748; Grant sponsor: CURE Childhood Cancer; Grant sponsor: American Society of Hematology Senior Research Training Award for Fellows; Grant sponsor: New York State Council on Graduate Medical Education Empire Clinical Research Investigator Program Fellowship; Grant sponsor: Erdheim Chester Disease Global Alliance and the Histiocytosis Association; Grant sponsor: Edward P. Evans Foundation; Grant sponsor: Department of Defense Bone Marrow Failure Research Program; Grant numbers: BM150092 and W81XWH-12–1-0041; Grant sponsor: NIH/NHLBI; Grant sponsor: R01HL128239; Grant sponsor: NIH K08 Clinical Investigator Award; Grant sponsor: 1K08CA160647–01; Grant sponsor: Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award; Grant sponsor: Starr Foundation; Grant sponsor: I8-A8–075; Grant sponsor: Leukemia and Lymphoma Society; Grant sponsor: Pershing Square Sohn Cancer Research Alliance.
Abbreviations:
- CNS
central nervous system
- CT
computed tomography
- DI
diabetes insipidus
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- LCH
Langerhans cell histiocytosis
- NGS
next-generation sequencing
- pERK1/2
phospho-ERK 1/2
Footnotes
CONFLICT OF INTEREST
I J.D. is a consultant for Bayer HealthCare Pharmaceuticals, Bristol Myers Squibb, Ipsen, Eisai, and Pfizer. M.E.L. has been a consultant with honoraria from Roche-Genentech, Astra Zeneca, and Novartis.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
AUTHORS CONTRIBUTION
B.H.D., S.Z., and O.A.W. designed and interpreted the experiments and BHD executed the experiments. S.Z., B.H.D., P.K., M.E.L., E.L., D.C.L., and I.J.D. collected patient material and interpreted the clinical data. S.Z., B.H.D., R.B., N.N.S., E.L.D., and O.A.W. interpreted the genomic data. S.Z., B.H.D., N.N.S., M.E.L., I.J.D., and O.A.W. wrote and revised the manuscript.
REFERENCES
- 1.Monsereenusorn C, Rodriguez-Galindo C. Clinical characteristics and treatment of langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):853–873. [DOI] [PubMed] [Google Scholar]
- 2.Rollins BJ. Genomic alterations in langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):839–851. [DOI] [PubMed] [Google Scholar]
- 3.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124(19):3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durham BH, Diamond EL, Abdel-Wahab O. Histiocytic neoplasms in the era of personalized genomic medicine. Curr Opin Hematol. 2016;23(4):416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahm F, Capper D, Preusser M, et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012;120(12):e28–e34. [DOI] [PubMed] [Google Scholar]
- 7.Zeng K, Ohshima K, Liu Y, et al. BRAFV600E and MAP2K1 mutations in Langerhans cell histiocytosis occur predominantly in children. Hematol Oncol. 2016. 10.1002/hon.2344. [DOI] [PubMed] [Google Scholar]
- 8.Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E negative Langerhans cell histiocytosis. Blood. 2014;124(10):1655–1658. [DOI] [PubMed] [Google Scholar]
- 9.Nelson DS, van Halteren A, Quispel WT, et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes, Chromosomes & Cancer. 2015;54(6):361–368. [DOI] [PubMed] [Google Scholar]
- 10.Nelson DS, Quispel W, Badalian-Very G, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. 2014;123(20):3152–3155. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty R, Burke TM, Hampton OA, et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood. 2016;128(21):2533–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grois N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatric Blood & Cancer. 2006;46(2):228–233. [DOI] [PubMed] [Google Scholar]
- 13.Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006–5014. [DOI] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, et al. Memorial sloan kettering integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. The Journal of Molecular Diagnostics: JMD. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discovery. 2016;6(2):154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20(12):1479–1484. [DOI] [PubMed] [Google Scholar]
- 17.de Kreuk BJ, Anthony EC, Geerts D, Hordijk PL. The F-BAR protein PACSIN2 regulates epidermal growth factor receptor internalization. J Biol Chem. 2012;287(52):43438–43453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchinson KE, Lipson D, Stephens PJ, et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(24):6696–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105(8):3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes, Chromosomes & Cancer. 2009;48(3):239–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.