

Abstract
The PD-1 coinhibitory receptor regulates the balance between T cell activation and tolerance. Although the PD-1 ligands, PD-L1 and PD-L2 are expressed on a variety of cell types, the cell type-specific functions of PD-1 ligands in inducing signals through PD-1 are unknown. Here we use PD-L1 conditional knockout mice to investigate cell type specific functions of PD-L1. We demonstrate that PD-L1 expressed on dendritic cells and to a lesser extent, B cells, attenuates the progression of experimental autoimmune encephalomyelitis (EAE) and inhibits both naïve and effector T cells. PD-1 is highly expressed on effector populations including TFH and TFR cells, which reside in germinal centers. We also show that dendritic cell PD-L1 is essential for limiting TFH and TFR cell differentiation. In addition we find that PD-1 suppresses TFH cell differentiation and help for Ig class switching even in the presence of WT TFR cells. Our work points to critical roles for PD-L1 expressed on dendritic cells in mediating PD-1 functions.
Introduction
Programmed death (PD) 1 (CD279) is a coinhibitory receptor expressed mainly on T cells, but also on some B cells (1). Signaling through PD-1 attenuates TCR signals and suppresses T cell expansion, cytokine production and cytolytic function. PD-1 has two ligands, PD-L1 (B7-H1) and PD-L2 (B7-DC)(1), which are highly expressed on B cells and dendritic cells as well as on other types of hematopoietic and non-hematopoietic cells. In addition to PD-1, PD-L1 can also bind to B7-1 (CD80)(2), and PD-L2 can bind to Rgmb(3). Inhibitory signals through the PD-1 pathway regulate T cell activation, T cell tolerance and T cell exhaustion.
PD-1 inhibitory signals control peripheral tolerance in several ways. PD-1:PD-L1 interactions regulate both the induction and maintenance of peripheral T cell tolerance. PD-1:PD-L1 interactions inhibit the initial activation of self-reactive CD4+ and CD8+ T cells, and the responses of self-reactive effector CD4+ and CD8+ T cells. The PD-1 pathway restrains self-reactive T cells in target organs, maintaining tolerance in tissues and protecting them from immunopathology. The important role of the PD-1 pathway in limiting immune-mediated damage caused by potentially pathogenic self reactive T cells is illustrated by the exacerbated disease that develops in mouse models of autoimmunity when PD-1 pathway signals are abrogated. For example, blockade or genetic deletion of PD-L1 or PD-1 can exacerbate experimental autoimmune encephalomyelitis (EAE)(4–7). Anti-PD-1 administration during myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide-induced EAE accelerated EAE onset and severity, greatly increased CD4+ cell infiltration into the CNS and the frequency of IFN-γ producing MOG-reactive T cells, and led to more MOG-specific antibodies in the serum. Our studies with PD-L1−/− mice identified critical inhibitory functions for PD-L1 in tissue tolerance. PD-L1 prevents activation of naïve self-reactive T cells, and inhibits destructive inflammation mediated by pathogenic effector T cells in tissues that are the target of autoimmune attack. Our studies point to critical roles for PD-L1 on hematopoietic and non-hematopoietic cells in regulating immunopathology, but how PD-L1 on specific cell types controls the initiation and progression of EAE is not clear.
PD-1 inhibitory signals also regulate humoral immune responses, which require a delicate balance of positive and negative signals that control antibody production. In the CD4+ T cell compartment, T follicular helper (TFH) cells stimulate, whereas T follicular regulatory (TFR) cells inhibit, B cells to produce antibodies(8). TFH cells provide costimulation to B cells through ICOS and CD40L as well as cytokines such as IL-21(9). TFR cells potently inhibit TFH and/or B cells through poorly understood mechanisms(8, 10). CTLA-4 expressed by TFR cells is essential for their suppressive capacity(11, 12). Moreover, recent data suggest that TFR cells alter B cell responses by inhibiting metabolic pathways in B and TFH cells(13). Despite the opposing functions of these cells, both TFH and TFR cells have similar expression of CXCR5, ICOS and PD-1 and depend on the transcription factor Bcl6 for development. Since PD-1 is more highly expressed on germinal center TFH cells (which have the highest CXCR5 expression) compared to TFH cells outside the GC, it is commonly used as a surrogate marker for this population(14, 15). Despite the use of PD-1 to identify both TFH and TFR cells, the role of PD-1 signaling in these cells is only beginning to be understood. Some studies have found that humoral responses are suppressed(16–18), while others have reported that humoral responses are augmented(19–21) when PD-1 signaling is prevented. Since PD-1 is expressed on B cells as well as T cells, PD-1 may exert its immunoregulatory effects on antibody responses by regulating B cells directly as well as through TFH and TFR cells. We previously demonstrated that PD-1 and/or PD-L1 inhibit both the differentiation and suppressive function of TFR cells(22). Some studies have demonstrated that PD-L2 expressed on B cells can bind to and signal into PD-1 on TFH cells resulting in reduced long-lived plasma cell numbers(16). However, further work is needed to understand how PD-1 on T cells and PD-1 ligands on antigen presenting cells control TFH/TFR cell differentiation and function.
To study the role of PD-L1 on different cell types in regulating T cell responses, we generated PD-L1 conditional knockout mice. Our studies reveal that PD-L1 expressed on dendritic cells is a key mediator of tolerance and regulator of effector cells in germinal centers. PD-L1 on dendritic cells limits self-reactive T cells during the initiation and progression of EAE, and inhibits the differentiation of both TFH and TFR cells. In addition, we examined the role of PD-1 on TFH cells in the absence of effects on TFR cells using cell sorting and adoptive transfer approaches, and demonstrate that PD-1 inhibits TFH cell differentiation and effector function.
Materials and Methods
Mice
WT, CD11cCre, CD19Cre and 2D2 TCR transgenic mice were purchased from The Jackson Laboratories. Pdcd1−/− (22, 23), WT FoxP3 IRES-GFP mice(24) and Pdcd1−/− FoxP3-IRES-GFP mice have been published previously(22). All mice were used according to the Harvard Medical School Standing Committee on Animals and National Institutes of Health Guidelines.
To generate PD-L1 Floxed mice we designed a targeting construct with LoxP sites surrounding exons 2 and 3 of murine Cd274. Linearized DNA was electroporated into Bruce 4 ES cells. After selection with G418, the resulting neo-resistant clones were picked, DNA prepared and analyzed to identify clones that carry the appropriate homologous recombination event. ES cells carrying the appropriate recombination event were microinjected into blastocysts. The resultant chimeras were bred to albino C57BL/6 mice and gave rise to germline transmission of the targeted allele, as demonstrated by progeny carrying the neo allele. We bred these mice with Flpe expressing mice to delete the neo cassette. Mice were then bred to Cre-expressing CD11cCre and CD19Cre mice.
Immunizations
For TFH and TFR cell studies mice were immunized with 100μg MOG 35–55 (UCLA biopolymer facility), MOG 1–125 or NP18-OVA (Biosearch Technologies) emulsified 1:1 with H37RA Mtb. CFA (Difco laboratories) in the flanks as previously described(25). 7 days later draining lymph node or blood was harvested for analysis. For studies of TFH and TFR cell differentiation in PD-L1 conditional knockout mice, all mice received 200μg anti-PD-L2 (clone 3.2) on d2 and d4. For EAE induction, similar emulsions were prepared as above but contained an extra 2mg/ml H37RA Mtb. Mice were additionally given 200ng pertussis toxin i.p. on days 0 and 2. Clinical disease was assessed using the following criteria: 1 = limp tail, 2 = weak gait, 3 = hind limb paralysis, 4 = hind+forelimb paralysis, 5 =moribund as described previously(26).
Histopathology
Mice were perfused with PBS, and brains and spinal cords were fixed in 10% formalin as described previously(27). Paraffin-embedded sections were stained with hematoxylin and eosin (H&E) or Luxol fast blue/periodic acid-Schiff for light microscopic analysis by the Dana Farber/Harvard Cancer Center Rodent Histopathology Core. As previously described(28), stained tissue specimens were blindly examined by a neuropathologist and inflammatory foci (>10 clustered inflammatory cells) in meninges and parenchyma were counted.
Antibodies
The following antibodies were used for surface staining: anti-CD4 (RM4-5), anti-ICOS (15F9), anti-CD19 (6D5), anti-CXCR5 biotin (2G8) and GL7 (GL-7). For intracellular staining, samples were fixed with the FoxP3 Fix/Perm buffer set according to manufacturer’s instructions (eBioscience). Samples were then intracellularly stained with IFN-γ (XMG1.2), IL-17A (TC11-18H10.1), anti-IgG1 (A85-1), anti-FoxP3 (FJK-16S), and Ki-67 (B56). Antibodies were from BioLegend, BD Biosciences, or eBioscience. Flow cytometry data were acquired on a FACSCalibur (BD) or LSRII (BD) using FACSDiva software (BD), and analyzed with FlowJo software (FlowJo LLC).
Analysis of mononuclear cells in the lymphoid organs and CNS of mice with EAE
Lymph nodes were mechanically dissociated through nylon cell strainers (70μm) and RBC lysed with ACK lysing buffer (Lonza). Mice were perfused with PBS before CNS tissue collection; brains and spinal cords were mechanically dissociated through a nylon cell strainer, and mononuclear cells isolated by centrifugation through a Percoll gradient (37% and 70%). Cells were resuspended in culture medium (RPMI 1640 [Gibco] with 10% FBS, 10mM HEPES [Gibco], 100U/mL Penicillin/Streptomycin [Gibco], 50μM 2-ME [Sigma]).
2D2 Adoptive Transfer Studies for EAE
CD4+ T cells from 2D2 TCR Tg mice were cultured using Th1-polarizing conditions at 2×106/mL with 1×107/mL irradiated feeder cells (CD4+ T cell-depleted WT splenocytes), IL-12 (10ng/mL, PeproTech), anti-IL-4 (20μg/mL, 11B11, BioXCell), and anti-CD3 (3μg/mL, 2C11, BioXCell), and supplemented with 100U/mL rhIL-2 (R&D Systems) every 2 days. On day 6 live cells were collected using Lymphocyte Separation Media (Mediatech, Inc) and restimulated on anti-CD3/anti-CD28 (37.51, BioXCell)-coated plates (3μg/mL) for 24 hours. 5×106 CD4+ T cells were injected i.p. into recipients that were monitored for clinical EAE.
In vivo analyses of TFR cell differentiation
Mice were immunized with NP-OVA in CFA s.c. or MOG35–55 in CFA and given anti-PD-L2 on d0 and d2 (to eliminate potential compensatory effects of PD-L2 for PD-L1 deficiency). 7 days after immunization TFR cells were analyzed (gated as CD4+ICOS+CXCR5+CD19-FoxP3+) in the blood and draining LN.
TFH in vitro stimulation assays
Stimulation assays were performed as described(25, 29, 30). Briefly, mice were immunized with NP-OVA as described above. 7 days later draining lymph nodes were harvested. CD4+ cells were enriched by magnetic positive selection (Miltenyi Biotec), and TFH cells were sorted as CD4+ICOS+CXCR5+GITR-CD19− or CD4+ICOS+CXCR5+FoxP3−CD19−. 3×105 TFH cells were incubated with 5×105 CD19+ B cells from WT mice in the presence of 2μg/ml anti-CD3 (BioXcell) and 5μg/ml anti-IgM (Jackson Immunoresearch) for 5–6 days. In some cases 20μg/ml anti-PD-L1 blocking antibody (clone 10F.9G2) or isotype control was added to the well.
Naïve 2D2 Cell Transfers for TFH studies
Spleens of WT Foxp3IRES-GFP or Pdcd1−/−Foxp3IRES-GFP unimmunized mice were harvested, CD4+ cells enriched by magnetic positive selection (Miltenyi Biotec), and naïve CD4+FoxP3-CD62L+ cells were sorted by flow cytometry. 1×106 naïve cells were adoptively transferred to WT mice, which were immunized with 100μg of MOG 35–55 in a 1:1 emulsion in CFA subcutaneously in the flanks. 7 days later inguinal lymph nodes were harvested and transferred cells were identified by Vα3.2/Vβ11 staining.
Statistics
Analyses were performed using Prism 6.0 (GraphPad). Statistical significance was calculated utilizing Student’s two-tailed unpaired t test or 1-way ANOVA as specified. EAE clinical scores were analyzed by a Mann–Whitney U test. A p value <0.05 was considered statistically significant.
Results
PD-L1 Expressed on Dendritic Cells Controls EAE Progression
To analyze PD-L1 function on specific cell types in controlling T cell responses we generated a PD-L1 conditional knockout mouse. In this mouse, referred to as PD-L1 F/F, loxP sites were inserted around exons 2 and 3 of the Cd274 gene (Fig 1A) to eliminate the IgV-like and IgC-like domains. We crossed the PD-L1 F/F mouse with CD11c Cre and CD19 Cre mice to conditionally delete PD-L1 on dendritic cells and B cells, respectively. PD-L1 is expressed on DC and B cells in unimmunized wild type mice. To validate the PD-L1 F/F CD11c Cre mice, we first analyzed PD-L1 expression on DC in unimmunized mice. PD-L1 expression was comparably reduced to background levels on DC in unimmunized PD-L1 F/F CD11c Cre mice and PD-L1−/− mice. PD-L1 expression was decreased in CD11c Cre− PD-L1 F/F mice compared to WT unimmunized mice (Supplemental Figure 1A,B). To further validate this strain, we immunized PD-L1 F/F CD11c Cre mice with MOG 35–55 in CFA subcutaneously and found that PD-L1 was selectively deleted on CD11c+IA+ DCs in the dLN (Fig. 1B). The levels of PD-L1 expression on DC and percentages of PD-L1 expressing DC in PD-L1 F/F CD11c Cre mice and PD-L1−/− mice were similarly reduced to background levels. However, expression of PD-L1 was attenuated in CD11c Cre− PD-L1 F/F mice compared to WT immunized mice.
Figure 1.
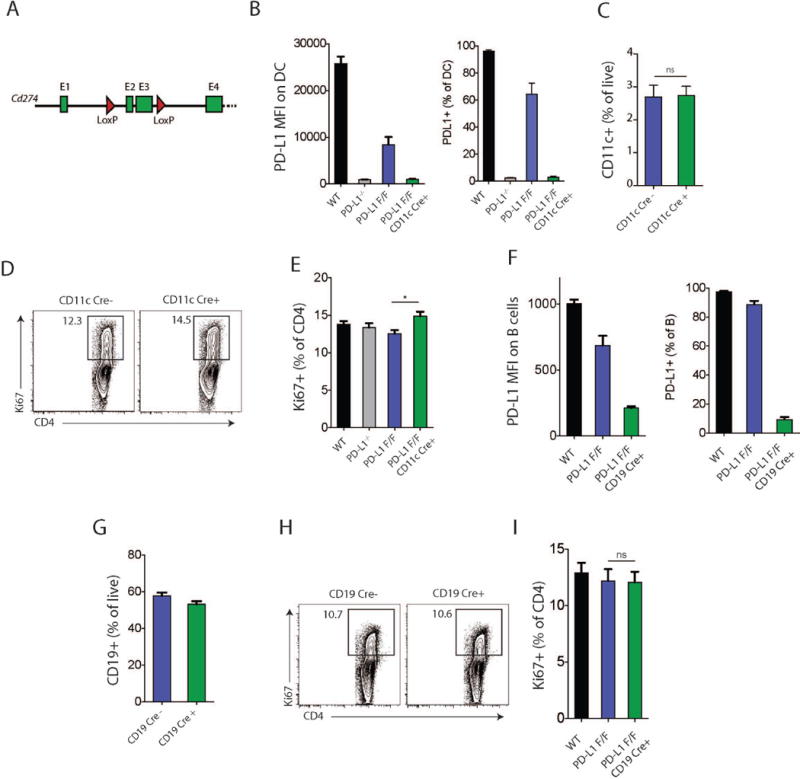
Characterization of PD-L1 conditional knockout mice. (A) Conditional deletion strategy. Cd274 gene exons 1 to 4 are shown as green boxes and loxP sites flanking exons 2 and 3 depicted as red triangles. (B-E) CD11cCre+ PD-L1 floxed (F/F) and indicated control mice were immunized with NP-OVA in CFA s.c. and analyzed 7 days later. (B) PD-L1 expression (MFI and frequency) on dLN DCs in CD11cCre+ PD-L1 floxed (F/F) mice compared to the indicated controls. (C) Percentage of CD11c+ cells in dLN after NP-OVA immunization. (D) Intracellular Ki67 expression in total CD4+ T cells from the dLN of NP-OVA immunized mice. (E) Quantification of Ki67+ cells as in (D). (F-I) CD19Cre+ PD-L1 floxed (F/F) and indicated mice were immunized with NP-OVA in CFA s.c. and analyzed 7 days later. (F) PD-L1 expression (MFI and frequency) on dLN B cells in CD19Cre+ PD-L1 floxed (F/F) mice and indicated controls. (G) Percentage of CD19+ B cells in dLN after NP-OVA/CFA immunization. (H) Intracellular Ki67 expression in total CD4+ T cells from the dLN of NP-OVA immunized mice. (I) Quantification of Ki67+ cells as in (H). Data indicate means +/− standard error of at least 4 mice per group and are representative of 2 independent experiments. *p < 0.05, ns = not significant, Student’s two-tailed unpaired t test.
Deletion of PD-L1 on DCs did not alter the frequency of CD11c+IA+ DCs in the dLN after immunization with MOG 35–55 in CFA (Fig. 1C). To determine if deletion of PD-L1 on DCs altered T cell activation, we assessed intracellular Ki67 staining in total CD4+ T cells on day 7 after immunization. Deletion of PD-L1 on DCs resulted in a small but significant increase in the frequency of Ki67+ CD4+ T cells (Fig. 1D–E).
To validate the CD19 Cre+ PD-L1 F/F strain, we first analyzed PD-L1 expression on B cells in unimmunized mice. PD-L1 expression was comparably reduced in unimmunized PD-L1 F/F CD19Cre mice and PD-L1−/− mice. PD-L1 expression was modestly reduced in CD19Cre− PD-L1 F/F mice compared to WT unimmunized mice (Supplemental Figure 1C,D). To further validate this strain, we immunized PD-L1 CD19 Cre+ PD-L1 F/F mice with MOG35-55 and determined that PD-L1 expression on B cells was markedly reduced in MOG 35-55 immunized CD19 Cre+ PD-L1 F/F mice compared to PD-L1 F/F CD19 Cre− controls (Fig. 1F). PD-L1 expression on B cells was slightly attenuated on CD19Cre− PD-L1 F/F mice compared to WT mice (Fig. 1F). The frequency of B cells in the dLN did not differ in CD19Cre+PD-L1 F/F and control mice, demonstrating that B cell-specific deletion of PD-L1 does not alter B cell development (Fig. 1G). There also were no differences in the frequency of Ki67+ CD4+ T cells in CD19Cre+PD-L1 F/F and control mice at day 7 post immunization (Fig. 1H–I).
We next investigated the relative roles of PD-L1 on DC and B cells during EAE because DCs have key immunoregulatory roles in EAE(31-34), and data support roles for B cells in EAE pathogenesis as APC, cytokine producers (particularly IL-6), antibody producers, and regulatory cells(35-39). We first immunized PD-L1 F/F CD11c Cre+ mice and control mice with MOG35-55 in CFA, and monitored the course of EAE. Mice lacking PD-L1 specifically on DCs had more severe clinical disease compared to control mice (Fig. 2A and Table 1). PD-L1 F/F CD11c Cre+ mice had earlier onset of disease, higher peak clinical disease scores and delayed resolution of EAE compared to controls. Histologic EAE was also increased in the PD-L1 F/F CD11c Cre+ mice with an increase in inflammatory foci in the meninges and parenchyma of the brain (Table 1).
Figure 2.
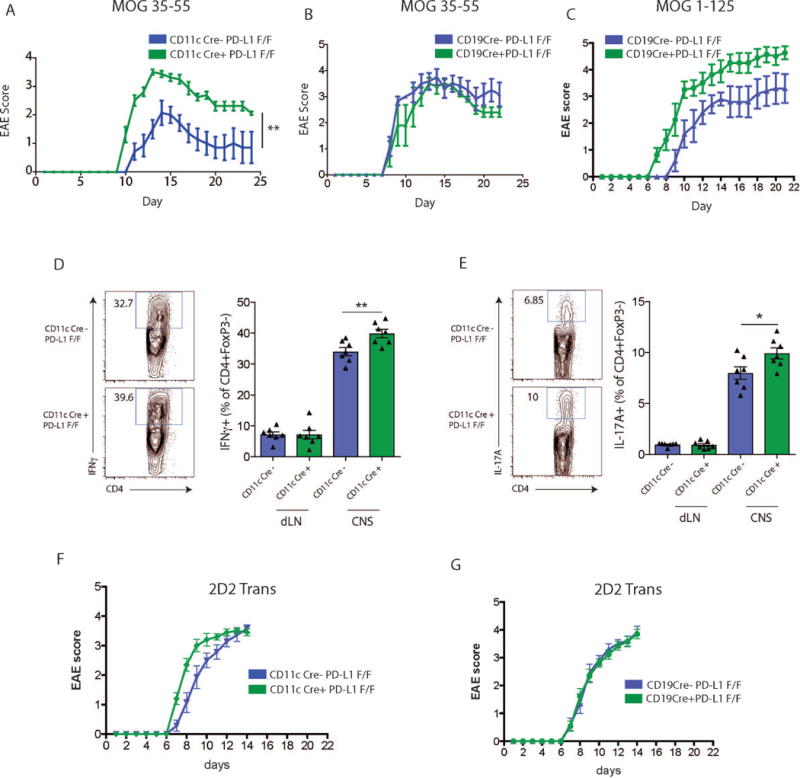
PD-L1 deficiency only on dendritic cells exacerbates experimental autoimmune encephalomyelitis. (A) PD-L1 F/F or CD11cCre+ PD-L1 F/F mice were immunized with MOG 35-55 and monitored for clinical EAE according to materials and methods. (B) PD-L1 F/F or CD19Cre+ PD-L1 F/F mice were immunized with MOG 35-55 and monitored for clinical EAE. (C) PD-L1 F/F or CD19Cre+ PD-L1 F/F mice were immunized with MOG 1-125 and monitored for development of clinical EAE. (D) Expression of IFNγ in CD4+ FoxP3− dLN or CNS cells of CD11cCre+PD-L1F/F or PD-L1F/F mice immunized with MOG 35-55 and harvested at the peak of EAE disease (~day 13). Representative flow cytometry plots (left) and quantification (right). (E) Expression of IL-17A in CD4+ FoxP3− dLN or CNS cells of CD11cCre+PD-L1F/F or PD-L1F/F mice immunized with MOG 35-55 and harvested at the peak of EAE disease (~day 13). Representative flow cytometry plots (left) and quantification (right). (F) Effects of PD-L1 deficiency on DC during the effector phase of EAE. Activated WT Th1 polarized 2D2 cells were adoptively transferred to CD11cCre+PD-L1F/F or PD-L1F/F mice, and recipients monitored for development of clinical EAE. (G) Effects of PD-L1 deficiency on CD19+ B cells during the effector phase of EAE. Activated WT Th1 polarized 2D2 cells were adoptively transferred to CD19cCre+PD-L1F/F or PD-L1F/F mice, and recipients monitored for clinical EAE. Data indicate means +/− standard error of 5-10 mice per group and are representative of at least 2 independent experiments. EAE clinical scores were analyzed by a Mann–Whitney U test (A-C, F+G). Significance of flow cytometry data (D+E) was calculated using Student’s two-tailed unpaired t test. A p value <0.05 was considered statistically significant. *p < 0.05, **p < 0.01.
Table 1.
EAE in PD-L1 conditional knockout mice.
| Clinical EAE | ||||
|---|---|---|---|---|
| Parameters | Method | Incidence | Day of onset (Mean +/− SE) |
Mean maximal score (Mean/− SE) |
| CD11c-Cre− PD-L1fl/fl | MOG 35–55 | 7/7 (100%) | 12.1 +/− 0.6 | 2.3 +/− 0.3 |
| CD11c-Cre+ PD−L1fl/fl | MOG 35–55 | 8/8 (100%) | 10.1 +/− 0.1** | 3.7 +/− 0.3** |
| CD19-Cre− PD-L1fl/fl | MOG 1–125 | 5/5 (100%) | 10.0 +/− 0.5 | 3.6 +/− 0.4 |
| CD19-Cre+ PD-L1fl/fl | MOG 1–125 | 8/8 (100%) | 7.9 +/− 0.5* | 4.6 +/− 0.2* |
| Histological EAE | Brain | Spinal Cord | ||||||
|---|---|---|---|---|---|---|---|---|
| Parameters | Method | Incidence | Meningeal foci (Mean +/− SE) |
Parenchyma foci (Mean/− SE) |
Total foci (Mean/− SE) |
Meningeal foci (Mean +/− SE) |
Parenchyma foci (Mean/− SE) |
Total foci (Mean/− SE) |
| CD11c-Cre− PD-L1fl/fl | MOG 35–55 | 10/10 (100%) | 29.5 +/− 3.766 | 40 +/− 6.673 | 69.5 +/− 10.15 | 85.5 +/− 6.937 | 82.2 +/− 12.62 | 167.7 +/− 18.04 |
| CD11c-Cre+ PD−L1fl/fl | MOG 35–55 | 9/9 (100%) | 44.33 +/− 7.225 | 51.33 +/− 9.739 | 95.67 +/− 13.6 | 91.67 +/− 7.927 | 86.89 +/− 3.889 | 178.6 +/− 10.35 |
| CD19-Cre− PD-L1fl/fl | MOG 1–125 | 8/8 (100%) | 16.75 +/− 4.148 | 11.13 +/− 3.705 | 27.88 +/− 7.453 | 62.5 +/− 6.118 | 50.63 +/− 4.488 | 113.1 +/− 10.04 |
| CD19-Cre+ PD-L1fl/fl | MOG 1-125 | 9/9 (100%) | 12.67 +/− 2.723 | 10.11 +/− 4.158 | 22.78 +/− 6.839 | 74.22 +/− 9.857 | 65.56 +/− 8.569 | 140.9 +/− 17.91 |
Mice were immunized with MOG35-55 or MOG1-125 and pertussis toxin to induce EAE as described in materials and methods, and clinical disease was monitored. For histologic analysis, mice were sacrificed on day 15, and brain and spinal cord were collected and processed according to materials and methods. All p-values are compared with Cre− control.
p < 0.05,
p < 0.01.
We next immunized PD-L1 F/F CD19 Cre+ and control mice with MOG35-55 in CFA to induce EAE. In contrast to PD-L1 F/F CD11c Cre+ mice, mice lacking PD-L1 specifically on B cells showed similar onset and severity of clinical EAE as control mice (Fig. 2B). These data indicate that PD-L1 expression on DCs, but not B cells, is essential for controlling MOG 35-55-induced EAE. Since B cells have been implicated in modulating EAE disease progression when whole MOG protein is used as the immunizing agent(40, 41), we also compared the effects of immunization with MOG 35-55 peptide and MOG 1-125 protein as the immunogen. When immunized with MOG 1-125, mice lacking PD-L1 specifically on B cells showed earlier onset and slightly higher severity of EAE compared to control mice (Fig. 2C). There was an increase in inflammatory foci, particularly in the spinal cord of MOG 1-125 immunized PD-L1 F/F CD19 Cre+ mice compared to controls (Table 1).
To determine if the increased severity of disease in mice lacking PD-L1 from dendritic cells may be due to increased infiltration of cytokine producing cells, we analyzed CD4+ T cells from the CNS. CD4+FoxP3− T cells in the CNS of the PD-L1 F/F CD11c Cre+ produced more IFNγ as well as IL-17A (Fig. 2D–E). These studies indicate an important role for PD-L1 on DC in EAE. Thus, PD-L1 expressed on dendritic cells and/or B cells can reduce the severity of the immune response in EAE.
Because we and others have published that the PD-1:PD-L1 pathway can inhibit effector T cell responses, we also investigated the role of PD-L1 on DCs (or B cells) in regulating the function of MOG-specific effector T cells using an adoptive transfer approach. We cultured naïve 2D2 CD4+ T cells (which recognize MOG35-55) in vitro under Th1-polarizing conditions to generate effector T cells and transferred the activated 2D2 CD4+ T cells to mice lacking PD-L1 on DCs or B cells. Transfer of 2D2 effector cells to PD-L1F/F CD11c Cre+ mice resulted in earlier onset of EAE and but similar EAE severity compared to transfer to control mice (Fig. 2F). Transfer of 2D2 effector cells into CD19Cre+PD-L1 F/F did not alter the course of EAE compared to control mice (Fig. 2G). The modest changes in EAE after adoptive transfer into recipients lacking PD-L1 on DCs or B cells suggest that PD-L1 on DC or B cell alone has a minor role in inhibiting effector T cell responses, and that PD-L1 on DC has a predominant role in controlling the initial activation of naïve CD4+ T cells.
PD-L1 Expressed on Dendritic Cells Inhibits TFR Cell Differentiation
Because we have previously shown that PD-L1-deficient mice have increased percentages of TFR cells, we next assessed the role of PD-L1 on the DC during TFR cell differentiation/expansion. We immunized CD11cCre+PD-L1F/F mice or PD-L1F/F controls with NP-OVA in CFA s.c. and administered anti-PD-L2 on d0 and d2 (to eliminate potential compensatory effects of PD-L2 for PD-L1 deficiency) and harvested the draining lymph nodes 7 days after immunization. We found a small but significant increase in the frequency of CD4+FoxP3+ Treg cells in dLN of mice that had PD-L1 deleted on DCs (Fig. 3A). There was a significant increase in the percentage of TFR cells (gated as CD4+ICOS+CXCR5+CD19-FoxP3+) in the dLN and blood of PD-L1F/F CD11c Cre+ mice (Fig. 3B–C). TFR cells in the dLN of DC specific PD-L1 deleted mice had a minor increase in intracellular Ki67 compared to controls (Fig. 3D). Similarly, when we immunized WT, PD-L1−/−, CD11cCre+PD-L1F/F and PD-L1F/F mice with MOG 35-55 in CFA and analyzed TFR cell percentages 7 days later, we found that PD-L1 deficiency on DCs resulted in increased TFR cell percentages in the dLN and blood compared to control mice (Fig. 3E).
Figure 3.
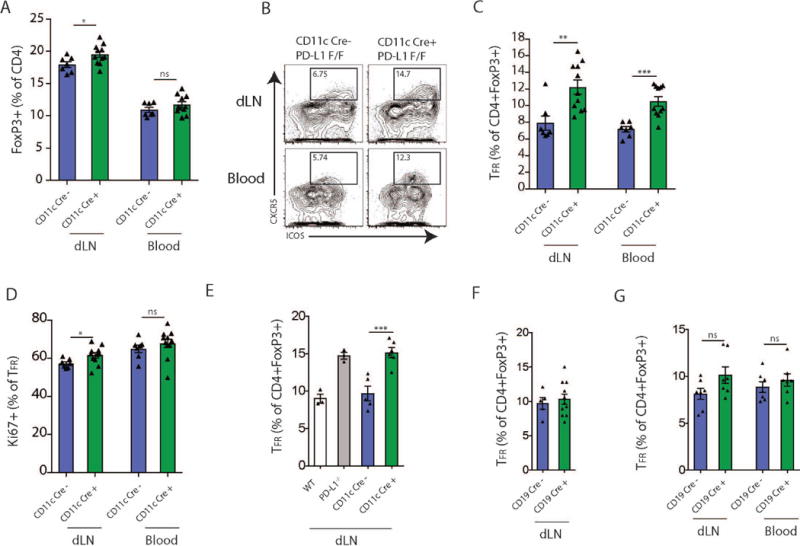
PD-L1 on dendritic cells regulates TFR cell differentiation. (A) Quantification of FoxP3+ Tregs in dLN of CD11cCre− PD-L1 F/F or CD11cCre+ PD-L1 F/F mice that were immunized with NP-OVA s.c. and given anti-PDL2 according to Materials and Methods. (B) TFR cell differentiation in conditional knockout mice from (A). Representative gating (plots pregated on CD4+FoxP3+) are shown. (C) Quantification of TFR cells from conditional mice as in (A) in dLN and blood after immunization. (D) Ki67 expression in TFR cells from mice as in (A). (E) TFR cell differentiation in dLN of CD11cCre− PD-L1 F/F or CD11cCre+ PD-L1 F/F mice that were immunized with MOG s.c. and given anti-PDL2 according to Materials and Methods. PD-L1−/− mice are included for comparison. (F) TFR cell differentiation in dLN of CD19Cre− PD-L1 F/F or CD19Cre+ PD-L1 F/F mice that were immunized with NP-OVA s.c. and given anti-PDL2 according to Materials and Methods. (G) TFR cell differentiation in dLN of CD19Cre− PD-L1 F/F or CD19Cre+ PD-L1 F/F mice that were immunized with MOG s.c. and given anti-PDL2 according to Materials and Methods. Data indicate means +/− standard error of at least 4 mice per group and are representative of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant, Student’s two-tailed unpaired t test.
Next we determined whether deletion of PD-L1 only in B cells affected TFR cell differentiation. We immunized CD19Cre+PD-L1F/F or PD-L1F/F mice with NP-OVA subcutaneously (and administered anti-PD-L2 blocking antibody) and assessed the draining lymph nodes for TFR cells 7 days later. There were no differences in the percentages of TFR cells in mice that lacked PD-L1 specifically on B cells and controls (Fig. 3F). Similarly, when we immunized CD19Cre+PD-L1F/F and PD-L1F/F mice with MOG 35-55 and analyzed TFR cell percentages 7 days later, there was not any statistically significant difference in TFR cell percentages (Fig. 3G). Taken together, these data demonstrate that PD-L1 expressed on DCs, but not B cells, is important for inhibiting TFR cell generation/maintenance.
PD-L1 Expressed on Dendritic Cells Inhibits Circulating TFH Cells
Next we determined if PD-L1 expression on B cells or DCs regulates TFH cell differentiation. We have previously demonstrated that PD-L1−/− mice have increased percentages of TFH cells in the blood, but not the lymph node of immunized mice. However the PD-L1 expressing cell type responsible for controlling TFH cells in the blood is not known. To determine if PD-L1 expression on DCs reduces TFH cell numbers in the blood, we immunized CD11cCre+PD-L1F/F mice and Cre− PD-L1 F/F control mice with NP-OVA. We injected all groups of mice with a PD-L2 blocking antibody at d0 and d2 to ensure that PD-L2 does not have a compensatory effect, and analyzed the dLN and blood of CD11cCre+ and Cre− PD-L1 F/F mice 7 days later. We identified TFH cells as CD4+ICOS+CXCR5+FoxP3-CD19− cells. Consistent with the data in PD-L1−/− mice, we did not measure any significant increases in TFH cells in the draining lymph node of CD11cCre+ PD-L1F/F mice after immunization compared to controls. However, there were increased TFH cell percentages in the blood of CD11cCre+ PD-L1F/F mice compared to controls (Fig. 4A). These data indicate that deletion of PD-L1 specifically on dendritic cells leads to enhanced differentiation of TFH cells destined for the circulation. The TFH cells from PD-L1F/F CD11cCre+ and Cre− mice had similar expression of Ki67 in both dLN and blood, suggesting that the TFH cells in the circulation of CD11cCre+ mice and Cre− control mice are comparably activated (Fig. 4B). Likewise, subcutaneous immunization with another antigen, MOG 35-55, in CFA (with PD-L2 blockade) also resulted in heightened percentages of TFH cells in the blood of CD11cCre+ mice compared to control mice (Fig. 4C). In contrast, immunization of CD19Cre+ PD-L1F/F mice with MOG 35-55 did not alter TFH cell percentages in the blood compared to control mice (Fig. 4D). These data suggest that PD-L1 expressed on dendritic cells, but not B cells, suppresses TFH cell differentiation and/or migration into the circulation.
Figure 4.
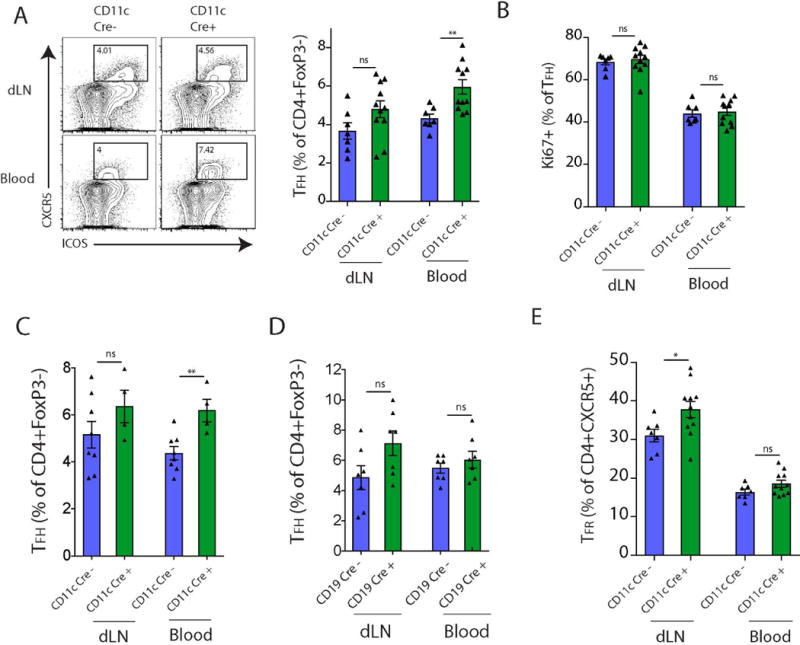
PD-L1 on dendritic cells regulates TFH cell differentiation. (A) CD11cCre− PD-L1 F/F or CD11cCre+ PD-L1 F/F mice were immunized with NP-OVA s.c. and given anti-PDL2 according to Materials and Methods, and TFH cells analyzed 7 days later. Representative gating of TFH cells in the dLN and blood (left; pregated on CD4+FoxP3− cells) and quantification (right) are shown. (B) Intracellular Ki67 staining on TFH cells as in (A). (C) CD11cCre− PD-L1 F/F or CD11cCre+ PD-L1 F/F mice were immunized with MOG s.c. and given anti-PDL2. TFH cells were analyzed as in A (D) CD19Cre− PD-L1 F/F or CD19Cre+ PD-L1 F/F mice were immunized with MOG s.c. and given anti-PDL2. TFH cells were analyzed as in A. (E) Quantification of TFR cells expressed as a percentage of total CD4+CXCR5+ cells. Data indicate means +/− standard error of at least 4 mice per group and are representative of 3 independent experiments. **p < 0.01, ns = not significant, Student’s two-tailed unpaired t test.
Since we found increased numbers of TFR cells in both dLN and blood (Fig 3) but only increased TFH cells in blood (Fig 4) with loss of PD-L1 on DCs, we next measured the frequencies of TFR cells of the total CXCR5+ CD4+ T cell population in draining LN and blood. Loss of PD-L1 on DCs resulted in a much higher relative frequency of TFR cells in the CD4+ CXCR5+ population compared to controls in the draining LN, but not in the circulation (Fig. 4E). Therefore, selective PD-L1 deficiency on DCs resulted in an increased ratio of TFR to TFH cells in the draining LN but similar proportional increases in TFH and TFR cells in the blood.
PD-1 suppresses TFH cell differentiation and function in the absence of PD-1 deficient TFR cells
To further investigate the role of the PD-1 pathway in regulating TFH cells, we immunized WT or Pdcd1−/− mice with NP-OVA subcutaneously and 7 days later evaluated TFH cells in the draining lymph node. The percentage of TFH cells of total CD4+FoxP3− cells was significantly decreased (Fig. 5A). Therefore, in PD-1 deficient mice there are slightly less TFH cells after immunization. These studies suggest that PD-1 deficiency has modest effects on TFH cell differentiation when PD-1 is deficient in all cell types.
Figure 5.

PD-1 deficient mice show modest cell extrinsic alteration in TFH cells differentiation. (A) WT or Pdcd1−/− mice were immunized with NP-OVA in CFA s.c. and 7 days later draining lymph nodes were analyzed for TFH development. (left) Representative plots pregated on CD4+CD19-FoxP3− and (right) quantification. (B-C) Enhanced TFH cell differentiation from PD-1 deficient naïve CD4+ T cells. WT or Pdcd1−/− 2D2+FoxP3−CD62L+ cells were adoptively transferred to WT mice which were immunized with MOG/CFA s.c., 7 days later dLN were analyzed for transferred cells and CXCR5+ICOS+ TFH cells were quantified. (left) Gating strategy (pregated on CD4+). (right) Quantification (pregated on CD4+Vα3.2+Vβ11+). (D) Cell death analysis of populations as in (A) using the fluorescent caspase reagent VAD-FMK. Total transferred CD4+ T cells (CD4+Vα3.2+Vβ11+) were included as controls. End. = endogenous. (E) TFH stimulation of B cells in the presence of anti-PD-L1 blocking antibody. WT mice were immunized with NP-OVA and 7 days later TFH cells were sorted and plated with B cells along with anti-CD3/IgM, plus isotype control or anti-PD-L1 antibody. Cultures were harvested 6 days later and B cells analyzed for IgG1 class switching. (left) Representative gates (pregated on CD19+IA+). (right) Quantification. (F) TFH cells were cultured with WT or PD-L1 deficient B cells similar to (E). (Left) Representative histograms. (Right) Quantification. Data indicate means +/− standard error of 5 replicate mice (A-B) or 3-4 replicate wells (C-D) and are representative of 3 experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant, Student’s two-tailed unpaired t test.
The increased numbers and potency(22) of TFR cells in PD-1 deficient mice may obscure the effects of PD-1 deficiency on TFH cell generation and function. To circumvent this potential problem and determine the role of PD-1 signaling on TFH cell development, we used an adoptive transfer approach. We isolated CD4+CD62L+FoxP3GFP− naïve cells from WT or PD-1 deficient 2D2 TCR transgenic mice crossed to a FoxP3GFP reporter, adoptively transferred these naïve 2D2 cells into WT mice, and immunized the recipient mice with MOG 35-55 in CFA subcutaneously. Importantly, PD-1 deficient TFR cells will not be generated by this approach because PD-1 deficient Treg cells are not being transferred. The WT recipients of WT and PD-1 deficient 2D2 T cells will have wild type TFR cells, enabling us to selectively investigate the effects of PD-1 deficiency on TFH cell generation. We analyzed the adoptive transfer recipients on day 7 after immunization with MOG35-55 and found increased percentages of 2D2 PD-1 deficient CXCR5+ICOS+ TFH cells compared to WT 2D2 TFH cells in the adoptive transfer recipients (Fig. 5B, C). To determine if loss of PD-1 on TFH cells resulted in a change in cell death, we measured active caspase by staining with fluorescent VAD-FMK reagent. PD-1 deficient TFH cells did not show a change in cell death (Fig. 5D). However, there was a slight increase in active caspase staining in total PD-1−/− compared to WT CD4+ 2D2 T cells. Therefore, PD-1 deficiency on naïve CD4+ T cells results in increased TFH cell differentiation/expansion in vivo.
Next we investigated whether blockade of PD-1 signals alters the effector function of TFH cells. To do this, we sorted CD4+ICOS+CXCR5+GITR−CD19− TFH cells from dLN of WT mice 7 days after immunization with MOG35-55 and cultured these cells with WT B cells (from the same lymph nodes) and either isotype control or anti-PD-L1 for 6 days in the presence of anti-CD3 and anti-IgM. Blockade of PD-L1 enhanced class switch recombination of B cells significantly (Fig. 5E). We performed complementary experiments with B cells isolated from dLN of MOG 35-55 immunized WT or PD-L1 deficient mice. In these experiments, TFH cells were sorted from dLNs of FoxP3 reporter mice 7 days after MOG 35-55 immunization. These cells were then cultured for 6 days with WT or PD-L1 deficient B cells from mice immunized with MOG 35-55 7 days previously along with anti-CD3/IgM. WT TFH cells stimulated increased class switch recombination in PD-L1 deficient B cells compared to WT B cells, demonstrating that loss of PD-1 signaling into TFH cells during the effector phase results in increased B cell help (Fig. 5F). Taken together, these data indicate PD-1 signaling can limit both the differentiation and effector function of TFH cells in the presence of WT TFR cells. Moreover, PD-L1 on B cells can inhibit the capacity of TFH cells to promote class switch recombination in B cells.
Discussion
Here we used PD-L1 conditional knockout mice to determine the role of PD-L1 on specific types of APCs in regulating T cell function and identified a key immunoregulatory role for PD-L1 on dendritic cells in tolerance and humoral immunity. Loss of PD-L1 only in dendritic cells resulted in exacerbated EAE, demonstrating the key role for PD-L1 on DC in limiting self-reactive CD4+ T cell responses. In addition, PD-L1 expressed on dendritic cells, but not B cells, inhibits the differentiation of TFH and TFR cells. These studies provide mechanistic insights into the relative roles of PD-L1 on DC and B cells in tolerance and humoral immunity.
Our EAE studies identified a dominant role for PD-L1 expressed on CD11c+ dendritic cells in inhibiting the initial activation of naïve MOG-specific CD4+ T cells. These observations are consistent with earlier studies showing that DCs play a key role in the propagation of CNS inflammation during EAE(42-44). We probed the role of PD-L1 on DC during the induction and effector phases of EAE by immunizing our CD11cCre+ PD-L1F/F mice with MOG 35-55 and using them as recipients of differentiated MOG35-55-specific 2D2 effector T cells. MOG35-55 immunized PD-L1 F/F CD11c Cre+ mice exhibited earlier onset of EAE, higher peak EAE scores and delayed resolution of disease compared to controls. PD-L1F/F CD11c Cre+ recipients of 2D2 effector T cells had earlier onset of disease but similar EAE severity compared to controls. Thus, somewhat surprisingly, PD-L1 on DC has a more significant role in limiting the initial activation of naïve MOG35-55 specific T cells than controlling MOG35-55 effector T cells.
In contrast, selective deletion of PD-L1 on B cells did not alter the onset or course of MOG 35-55-induced EAE. These findings are consistent with studies showing that B cells do not alter disease progression in MOG 35-55-induced EAE. Recent studies have shown that PD-L1 expressing “Breg” cells can suppress EAE when adoptively transferred(45). It is possible that Bregs do not have a strong role in our immunization settings or that Bregs need to be adoptively transferred to confer suppressive capacity. Because B cells are critical to induction of experimental allergic encephalomyelitis by MOG protein but not by a encephalitogenic MOG peptide(40, 45), we also immunized our mice lacking PD-L1 on B cells with MOG 1-125. We found a modest increase in disease severity. These data indicate that PD-L1 on DCs and B cells can play a role in controlling myelin-reactive T cell activation and differentiation.
Our studies further demonstrate that PD-1/PD-L1 interactions can inhibit TFH cell differentiation and function. Loss of PD-L1 specifically on dendritic cells resulted in greater percentages of TFH cells in the blood, similar to germline PD-L1 deficient mice(22). These findings indicate that PD-1 expressed on TFH cells mainly suppresses TFH cell differentiation/maintenance in the T cell zone since dendritic cells reside in this location and are largely excluded from the B cell follicle. Loss of PD-L1 on B cells did not alter TFH cell maintenance or Ki67 expression. Thus, DC PD-L1 has a key role in controlling TFH cell differentiation and/or maintenance.
Similarly, PD-L1 on dendritic cells (but not B cells) inhibits TFR cell differentiation and/or maintenance. It is still unclear which dendritic cell subset expressing PD-L1 is responsible for inhibition of TFR cell responses. We hypothesize that many different dendritic cells can utilize PD-L1 to suppress TFR cell responses, and the specific dendritic cell responsible depends on the immunization. In support of this, it has recently been shown that optimal TFH cell development depends on both migratory and lymph node resident dendritic cells for differentiation(46), but TFR cells were not analyzed. Further experiments are needed to determine which specific DC subset is responsible for enhanced TFR cell development.
We also found that PD-1 can inhibit TFH cell differentiation and function when we examined the effects of PD-1 on TFH cell generation and function in the presence of WT TFR cells. Therefore, during a humoral immune response PD-1/PD-L1 interactions can inhibit both TFR and TFH cell differentiation. Since these two cell types have opposing roles, the effects of PD-1 on humoral immune responses are likely context dependent, with the effects on antibody production dependent on whether TFR or TFH cell predominate. This may help to explain conflicting data on the role of PD-1 in vivo. Under some circumstances such as subcutaneous immunization, PD-1 deficiency has a dominant effect in TFR cells, which then suppress the stimulated TFH cells. In other circumstances, the nature of the antigenic challenge may preferentially stimulate TFH cells resulting in a strong TFH phenotype. In support of this concept, loss of PD-L1 results in heightened TFH cell differentiation in helminth infection and arthritis(17, 19). Additionally, mixed bone marrow chimeras in which PD-L1 is lost predominantly on B cells results in increased TFH cell percentages(45). However, it is important to note that TFR cells were not gated in these studies. We speculate that in settings of strong inflammation (such as viral infection) that increase the TFH: TFR ratio(10), PD-1 pathway deficiency will result in a predominant phenotype on TFH cells. Since PD-L1 blockade increased TFH cell function, there also may be temporal effects of PD-1 pathway modulation on humoral immune responses.
In summary, these studies identify key immunoregulatory roles for PD-L1 expressed on dendritic cells in tolerance and humoral immunity. DC PD-L1 has a predominant role in limiting the initial activation of myelin-reactive CD4+ T cells during EAE. PD-L1 on DC also inhibits the differentiation of TFH and TFR cells. In addition, we demonstrate that during a humoral immune response PD-1 interactions can inhibit TFH cell differentiation and function. Since PD-1 can inhibit both TFH and TFR cell differentiation and function, and TFH and TFR cell have opposing roles in controlling humoral immunity, the PD-1 pathway likely exerts context-dependent effects on humoral immune responses.
Supplementary Material
Acknowledgments
This work was supported by US National Institute of Health through grants 5T32HL007627 (P.T.S.), R37AI38310 (A.H.S.), R01AI46414 (A.H.S.), P01AI56299 (A.H.S., G.J.F, and V.K.K) and by the National Multiple Sclerosis Society (F.A.S.). F.A.S. was supported by the International Society for Advancement of Cytometry (ISAC) through an ISAC Marylou Ingram Scholar award.
References
- 1.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological reviews. 2009;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, Hua P, Duke-Cohan JS, Umetsu DT, Sharpe AH, DeKruyff RH, Freeman GJ. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211:943–959. doi: 10.1084/jem.20130790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Li Y, Proctor TM, Vandenbark AA, Offner H. Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. Journal of neuroscience research. 2010;88:7–15. doi: 10.1002/jnr.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, Milarski KL, Groves C, Brown T, Carito BA, Percival K, Carreno BM, Collins M, Marusic S. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2007;182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends in immunology. 2015;36:410–418. doi: 10.1016/j.it.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crotty S. Follicular helper CD4 T cells (TFH) Annual review of immunology. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 10.Sage PT, Sharpe AH. T follicular regulatory cells. Immunological reviews. 2016;271:246–259. doi: 10.1111/imr.12411. [DOI] [PubMed] [Google Scholar]
- 11.Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor ctla-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–1039. doi: 10.1016/j.immuni.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of TFH cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41:1013–1025. doi: 10.1016/j.immuni.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Sage PT, Ron-Harel N, Juneja VR, Sen DR, Maleri S, Sungnak W, Kuchroo VK, Haining WN, Chevrier N, Haigis M, Sharpe AH. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol. 2016;17:1436–1446. doi: 10.1038/ni.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 15.Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, Barnett B, Crotty S. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150) J Immunol. 2010;185:190–202. doi: 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamel KM, Cao Y, Wang Y, Rodeghero R, Kobezda T, Chen L, Finnegan A. B7-H1 expression on non-B and non-T cells promotes distinct effects on T- and B-cell responses in autoimmune arthritis. European journal of immunology. 2010;40:3117–3127. doi: 10.1002/eji.201040690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, Fagarasan S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science (New York, NY. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 19.Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, Fallon PG. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186:5648–5655. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 20.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13:188–195. doi: 10.1038/ni.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–161. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keir ME, Freeman GJ, Sharpe A. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. Journal of Immunology. 2007;179:5064–5070. doi: 10.4049/jimmunol.179.8.5064. [DOI] [PubMed] [Google Scholar]
- 24.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 25.Sage PT, Sharpe AH. In Vitro Assay to Sensitively Measure TFR Suppressive Capacity and TFH Stimulation of B Cell Responses. Methods in molecular biology. 2015;1291:151–160. doi: 10.1007/978-1-4939-2498-1_13. [DOI] [PubMed] [Google Scholar]
- 26.Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Carcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, Freeman GJ, Sharpe AH. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med. 2015;212:1603–1621. doi: 10.1084/jem.20141030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, Jenne DE, Wekerle H, Sobel RA, Bernard CC, Shlomchik MJ, Zamvil SS. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. The Journal of experimental medicine. 2013;210:2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobel RA, V, Tuohy K, Lu ZJ, Laursen RA, Lees MB. Acute experimental allergic encephalomyelitis in SJL/J mice induced by a synthetic peptide of myelin proteolipid protein. J Neuropathol Exp Neurol. 1990;49:468–479. doi: 10.1097/00005072-199009000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory-like properties. The Journal of clinical investigation. 2014;124:5191–5204. doi: 10.1172/JCI76861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sage P, Ron-Harel N, Juneja VR, Sen D, Maleri S, Sungnak W, Kuchroo V, Haining WN, Chevrier N, Haigis M, Sharpe A. Suppression by TFR cells leads to durable and selective inhibition of B cell effector function. Nat Immunol. 2016 doi: 10.1038/ni.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dittel BN, Visintin I, Merchant RM, Janeway CA., Jr Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. Journal of immunology. 1999;163:32–39. [PubMed] [Google Scholar]
- 32.Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FB, Coyle AJ, Hoogsteden HC, Lambrecht BN. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. European journal of immunology. 2006;36:2472–2482. doi: 10.1002/eji.200635978. [DOI] [PubMed] [Google Scholar]
- 33.Schreiner B, Bailey SL, Shin T, Chen L, Miller SD. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. European journal of immunology. 2008;38:2706–2717. doi: 10.1002/eji.200838137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zozulya AL, Clarkson BD, Ortler S, Fabry Z, Wiendl H. The role of dendritic cells in CNS autoimmunity. Journal of molecular medicine. 2010;88:535–544. doi: 10.1007/s00109-010-0607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, O’Connor RA, Anderton SM, Bar-Or A, Fillatreau S, Gray D. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. The Journal of experimental medicine. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nature immunology. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 37.Mann MK, Ray A, Basu S, Karp CL, Dittel BN. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity. 2012;45:388–399. doi: 10.3109/08916934.2012.665523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, Wekerle H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. The Journal of experimental medicine. 2009;206:1303–1316. doi: 10.1084/jem.20090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. Journal of immunology. 2012;188:3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. European journal of immunology. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. European journal of immunology. 2002;32:1905–1913. doi: 10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 42.Schreiner B, Bailey SL, Shin T, Chen L, Miller SD. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. European journal of immunology. 2008;38:2706–2717. doi: 10.1002/eji.200838137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sosa RA, Forsthuber TG. The critical role of antigen-presentation-induced cytokine crosstalk in the central nervous system in multiple sclerosis and experimental autoimmune encephalomyelitis. J Interferon Cytokine Res. 2011;31:753–768. doi: 10.1089/jir.2011.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zozulya AL, Clarkson BD, Ortler S, Fabry Z, Wiendl H. The role of dendritic cells in CNS autoimmunity. J Mol Med (Berl) 2010;88:535–544. doi: 10.1007/s00109-010-0607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nature communications. 2015;6:5997. doi: 10.1038/ncomms6997. [DOI] [PubMed] [Google Scholar]
- 46.Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity. 2015;42:172–185. doi: 10.1016/j.immuni.2014.12.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.