

Abstract
Proteins in the hedgehog family undergo self-catalyzed endoproteolysis involving nucleophilic attack by a molecule of cholesterol. Recently, a conserved aspartate residue (D303, or D46) of hedgehog was identified as the general base that activates cholesterol during this unusual autoprocessing event; mutation of the catalyzing functional group (D303A) reduces activity by >104-fold. Here we report near total rescue of this ostensibly dead general base mutant by a synthetic substrate, 3β-hydroperoxycholestane (3HPC) in which the sterol −OH group is replaced by the hyper nucleophilic −OOH group. Other hedgehog point mutants at D303, also unreactive with cholesterol, accepted 3HPC as a substrate with the rank order: WT > D303A ≈ D303N ≫ D303R, D303E. We attribute the revived activity with 3-HPC to the α-effect, where tandem electronegative atoms exhibit exceptionally high nucleo- philicity despite relatively low basicity.
General base variants of enzymes can present structure− function enigmas: though remarkably similar in structure to the native enzyme, point mutants often show turnover numbers (kcat) diminished by 104- to 108-fold. For example, general base mutation of HIV protease (D25N) suppresses catalytic activity to such a degree that the protein can be cocrystallized in “tight” complex with intact substrate.1 A 108 fold reduction in kcat was reported with the E104A general base mutant of cytidine deaminase; notwithstanding, the protein retained nM affinity for a mechanism-based inhibitor.2 General base mutants of aspartate aminotransferase (K258A) and ketosteroid isomerase (D38G) are among many other examples that display similar characteristics: structurally sound and catalytically inert.3,4 Toney and Kirsch established that mutants like these can be “rescued” to varying degrees by addition in trans of the missing functional group.4 Alternatively, activity may be restored by appending the catalyzing group to substrate itself.5 Here we present a nonclassical approach to chemical rescue, one that sidesteps the general base imperative altogether and instead restores activity through the use of a hyper- nucleophilic substrate.
Hedgehog proteins, extracellular signaling ligands of importance to embryogenesis and cancer,6 undergo a specialized endoproteolytic lipidation called cholesterolysis as part of their complex biosynthesis.7 In cholesterolysis, a precursor form of hedgehog (Hh) separates into N- and C- terminal polypeptides, and the departing N-terminal fragment (HhN) becomes linked covalently to a molecule of cholesterol (Scheme 1).8–10 The reaction begins with an amide to thioester rearrangement at a conserved GCF sequence (step 1); this is followed by transesterification to cholesterol (step 2). Both steps can be brought about in vitro independent of cofactors or accessory proteins by the Hh precursor’s catalytic C-terminal region, HhC (Scheme 1, green)9
Scheme 1.
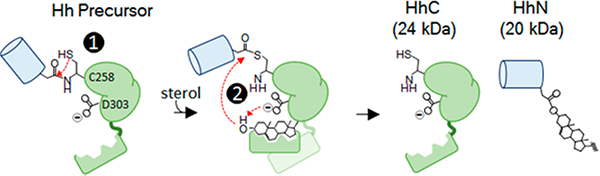
Hedgehog Precursor Autoprocessing
Recently, Xie et al. proposed that an aspartic acid residue (D303, or D46) in the HhC from Drosophila melanogaster functions as a general base to facilitate attack of cholesterol at the thioester intermediate.11 Multiple lines of evidence support this idea: (1) alignment of distantly related HhC sequences reveal strong conservation of the D303 residue; (2) crystal structure of an HhC fragment places D303 in proximity to the thioester forming residue, C258; (3) NMR measurements indicate that the pKa value of the D303 side chain is elevated to 5.6, which would aid in deprotonating the sterol 3-OH group; finally, (4) an alanine point mutant at D303 renders HhC unreactive toward cholesterol.9
In Figure 1A-C and Table 1 (left column), we summarize the diminished cholesterolysis activity of HhC D303A. For quantitative analysis, we used an optical assay that monitors reaction kinetics continuously by loss of Forster resonance energy transfer (FRET) from an engineered Hh precursor (Figure 1A).12 The FRET-active precursor, C−H−Y, is a translational fusion of Cyan fluorescent protein, HhC, and Yellow fluorescent protein. FRET from C−H−Y is lost as autoprocessing liberates C-sterol from H−Y. Results are verified with a gel-based assay, where precursor and products separate by molecular weight allowing reaction progress to be visualized directly (Figure 1B). Plots of initial velocity, determined using the FRET assay, as a function of cholesterol concentration were fit by nonlinear regression to a Michealis− Menten equation to obtain Km values (Figure 1C). First-order rate constants for cholesterolysis (kmax) were calculated at saturating substrate concentration by fitting the entire reaction progress curve to an exponential decay.
Figure 1.
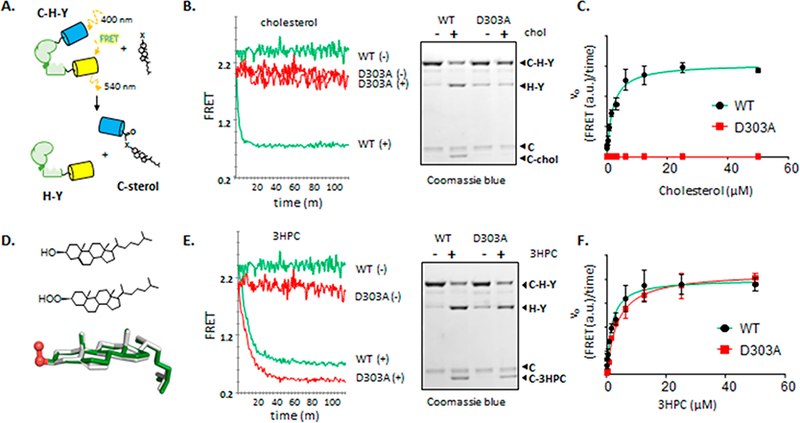
Activation of general base mutant D303A with an α-effect substrate. (A) Continuous assay to measure kinetics of Hh activity by using a FRET-active precursor (C−H−Y). (B) Kinetics of precursor processing with wild-type and D303A constructs (1 × 10−7 M) ± cholesterol (5 × 10−5 M), monitored by FRET (left) and by SDS-PAGE (right). (C) Initial velocity of wild-type and D303A HhC constructs plotted as a function of increasing cholesterol concentration. Trend line for wild-type was drawn using the fitted KM value of 1 μΜ. (D) Bond line formulas and structural overlay of cholesterol and 3-β hydroperoxycholestane (3HPC). (E) Kinetics of precursor processing with wild-type and D303A HhC constructs ±3HPC (50 μΜ), monitored by FRET (left) and by SDS-PAGE (right). (F) Initial velocity of wild-type and D303A constructs plotted as a function of increasing 3HPC. Trend lines were drawn using the fitted Km values of 1.5 × 10−6 M for wild-type, and 3.5 × 10−6 M for D303A.
Table 1.
Substrate Activity of Cholesterol and 3HPC with Wild-Type and D303 Mutant HhCa
| substrate |
||
|---|---|---|
| cholesterol | 3HPC | |
| wild-type HhC | ||
| kmax(s−1) | 4.70 × 10−3 | 1.21 × 10−3 |
| KM(μM) | 1.87 | 1.54 |
| kmax/KM(M−1s−1) | 2.53 × 103 | 785 |
| D303A HhC | ||
| kmax(s−1) | nd | 1.03 × 10−3 |
| Kmax(μM) | nd | 3.47 |
| kmax/KM(M−1s−1) | <0.27 | 296 |
Standard errors were less than or equal to ±14% for KM values and ±20% for kmax values. n ≥ 3.
In contrast to HhC wild-type (HhC WT), the D303A mutant exhibits an autoprocessing rate with cholesterol that is reduced by a factor of >300-fold (Table 1) with cholesterol added at concentrations 50-fold above the KM value of HhC WT. A limiting value of kmax/KM for the D303A is thereby reduced by ≥104 for cholesterol, equivalent to 5.4 kcal/mol in free energy. The very large deleterious effect of removing the D303 carboxyl group is within range of general base mutations mentioned earlier. Despite inactivity toward cholesterol, the D303A variant continues to generate internal thioester in amounts comparable to wild type HhC, and similar behavior was observed with D303E, R, and N variants.11 Thus, general base mutants of HhC can bring about step 1 (Scheme 1) but autoprocessing advances no further with substrate cholesterol.
α-Effect nucleophiles, where the attacking atom is adjacent (i.e., alpha) to a second electronegative atom, are known for reacting at exceptionally fast rates.13–15 Large positive deviations from linear free energy relationships are observed with α-effect nucleophiles like hydrazine, hydroxylamine and hydroperoxides.1–17 For example, methyl peroxide anion (CH3OO−) cleaves an aryl ester at a rate 4× faster than methoxide (CH3O−) despite having a pKa value (11.5) 4 units lower than methanol (15.6).18 Said another way, methyl peroxide reacts as if it were ~10 000× more basic than its measured pKa value.
With the potential for augmented acidity and nucleophilicity, we prepared 3β-hydroperoxy cholestane (3HPC) as a potential substrate for the Hh D303A mutant. Structures of 3HPC and cholesterol are similar except for a saturated stanol ring system and the −OOH substituent in place of the sterol −OH (Figure 1D). The analog 3HPC was obtained in 3 steps starting from cholestanone and separated from its C-3 epimer by preparative TLC and HPLC. Stereochemistry of the hydroperoxy group was confirmed by 1H NMR using the chemical shift and splitting pattern of the −CH−OOH (ref 19 and Supporting Information).
We observed robust autoprocessing of HhC WT and D303A with 3HPC in a set of in vitro experiments, summarized in Figure 1E,F and Table 1 (right column). Assays using 3HPC were carried out as before using the optical system for determining kinetic parameters and gel-based assay for orthogonal readout. Results of SDS-PAGE show that the product polypeptide derived from reaction with 3HPC migrates with an Rf value similar to product from the cholesterol reaction; both appear larger than the Rf of the hydrolysis product (Figure 1E, gel, compare C and C-3HPC), which is consistent with steroylation.10,12,20 We note that selected carboxylic acid hydrolases accept hydrogen peroxide as an alternative substrate in a so-called perhydrolysis side reaction.21 Comparison of the specificity constants (kmax/KM) derived from the FRET assay indicate a modest 3-fold preference for cholesterol over 3HPC by HhC WT, diminished only by the kmax value (Table 1). Substrate activity of 3HPC remained strong using the general base D303A mutant (Figure 1E, red traces): the kmax value is within 10% of wild-type, and the apparent KM value is elevated by only 2-fold. In comparison with cholesterol, the t1/2 value for autoprocessing by D303A is accelerated from >19 h to 10 min, equivalent to a rate enhancement of >1000×.
To explore the specificity using 3HPC, we surveyed three other seemingly inert D303 point mutants: charged reversed, D303R; isosteric, charge neutralized, D303N; and charge conserved, D303E.11 In the absence of steric or electrostatic clashes, it seemed reasonable to expect that 3HPC would rescue in a manner similar to the D303A HhC mutant. In fact, comparable activity was observed for D303N, with an apparent kmax value within 2-fold of D303A. Rate constants with D303R and D303E were slowed by ~ 10-fold compared with D303A. We speculate based on these results that at the transition state for transesterification (Scheme 1, step 2) the attacking atom of the sterol and the side chain of residue 303 are narrowly separated, consistent with general base catalysis. Larger side- chains introduced by D303R and E substitution could sterically inhibit approach of the attacking −OOH group of 3HPC, leading to slower reaction rates. Full explanation of the variation awaits complete structural analysis of HhC.
In summary, a synthetic Hh substrate bearing an α-effect functional group, noteworthy for reacting at rates disproportionately faster than expected from its pKa value, supports near wild-type activity of a HhC general base mutant (D303A). Thus, a functioning general base appears critical for HhC to activate the −OH group of cholesterol, but superfluous with the α-effect −OOH group of 3HPC. These results are consistent with a nonclassical mode of chemical rescue, distinct from convention where the missing catalytic group is returned to the mutant enzyme.22 The present study does evoke rescue-type experiments on glucosidases and ribozymes where general acid mutants were revived by substrates bearing hyper-labile leaving groups.23,24 To the extent that any rescue approach succeeds, the restored activity seems to point toward a key structural feature of enzyme and enzyme-like active sites. Though active sites almost certainly provide a template for the transition state,25 that template is to some degree elastic even improvisatory.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge generous support from the National Cancer Institute (Grant R01 CA206592) and Department of Defense (Grant W81XWH-14–1-0155). We also acknowledge the upgrade of the 600 MHz NMR spectrometer at SUNY-ESF under NSF grant CHE-1048516 and NIH grant S10 OD012254.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b05161.
Protein preparation, activity assay, chemical synthesis, 1-D NMR spectra and additional experimental information (PDF)
AUTHOR INFORMATION
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Prabu-Jeyabalan M; Nalivaika E; Schiffer CA J. Mol. Biol 2000, 301, 1207. [DOI] [PubMed] [Google Scholar]
- (2).Carlow DC; Smith AA; Yang CC; Short SA; Wolfenden R Biochemistry 1995, 34, 4220. [DOI] [PubMed] [Google Scholar]
- (3).Lamba V; Yabukarski F; Pinney M; Herschlag DJ Am. Chem. Soc 2016, 138, 9902. [DOI] [PubMed] [Google Scholar]
- (4).Toney MD; Kirsch JF Science 1989, 243, 1485. [DOI] [PubMed] [Google Scholar]
- (5).Carter P; Wells JA Science 1987, 237, 394. [DOI] [PubMed] [Google Scholar]
- (6).Briscoe J; Therond PP Nat. Rev. Mol. Cell Biol 2013, 14, 418. [DOI] [PubMed] [Google Scholar]
- (7).Mann RK; Beachy PA Annu. Rev. Biochem 2004, 73, 891. [DOI] [PubMed] [Google Scholar]
- (8).Porter JA; Ekker SC; Park WJ; von Kessler DP; Young KE; Chen CH; Ma Y; Woods AS; Cotter RJ; Koonin EV; Beachy PA Cell 1996, 86, 21. [DOI] [PubMed] [Google Scholar]
- (9).Hall TM; Porter JA; Young KE; Koonin EV; Beachy PA; Leahy DJ Cell 1997, 91, 85. [DOI] [PubMed] [Google Scholar]
- (10).Porter JA; Young KE; Beachy PA Science 1996, 274, 255. [DOI] [PubMed] [Google Scholar]
- (11).Xie J; Owen T; Xia K; Callahan B; Wang CJ Am. Chem. Soc 2016, 138, 10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Owen TS; Ngoje G; Lageman TJ; Bordeau BM; Belfort M; Callahan BP Anal. Biochem 2015, 488, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Edwards JO; Pearson RG J. Am. Chem. Soc 1962, 84, 16. [Google Scholar]
- (14).Jencks WP; Carriuolo JJ Am. Chem. Soc 1960, 82, 1778. [Google Scholar]
- (15).Jencks WP; Carriuolo JJ Am. Chem. Soc 1960, 82, 675. [Google Scholar]
- (16).Herschlag D; Jencks WP J. Am. Chem. Soc 1990, 112, 1951. [Google Scholar]
- (17).Kalia J; Raines RT ChemBioChem 2006, 7, 1375. [DOI] [PubMed] [Google Scholar]
- (18).Jencks WP; Gilchrist MJ Am. Chem. Soc 1968, 90, 2622. [Google Scholar]
- (19).Caglion L; Gasparrini F; Misiti D; Palmieri G Tetrahedron 1978, 34, 135. [Google Scholar]
- (20). An anonymous reviewer suggested two alternative mechanisms to explain the observed cleavage of C−H−Y with added 3HPC: first, 3HPC could react transiently with the internal thioester followed by rapid hydrolysis; and second, the −OOH group of 3HPC could activate an adventitious water molecule for thioester cleavage. A third possibility involves oxidative cleavage of the internal thioester. We could not identify by mass spectrometry covalent peroxy esterified protein (C-3HPC), therefore these pathways remain viable alternatives.
- (21).Kirk O; Conrad LS Angew. Chem., Int. Ed 1999, 38, 977. [DOI] [PubMed] [Google Scholar]
- (22).Peracchi A Curr. Chem. Biol 2008, 2, 32. [Google Scholar]
- (23).Das SR; Piccirilli JA Nat. Chem. Biol 2005, 1, 45. [DOI] [PubMed] [Google Scholar]
- (24).Rakic B; Withers SG Aust. J. Chem 2009, 62, 510. [Google Scholar]
- (25).Wolfenden R Annu. Rev. Biophys. Bioeng 1976, 5, 271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.