

Abstract
Freshwater mussels (Bivalvia: Unionida) serve an important role as aquatic ecosystem engineers but are one of the most critically imperilled groups of animals. Here, we used a combination of sequencing strategies to assemble and annotate a draft genome of Venustaconcha ellipsiformis, which will serve as a valuable genomic resource given the ecological value and unique “doubly uniparental inheritance” mode of mitochondrial DNA transmission of freshwater mussels. The genome described here was obtained by combining high-coverage short reads (65× genome coverage of Illumina paired-end and 11× genome coverage of mate-pairs sequences) with low-coverage Pacific Biosciences long reads (0.3× genome coverage). Briefly, the final scaffold assembly accounted for a total size of 1.54 Gb (366,926 scaffolds, N50 = 6.5 kb, with 2.3% of “N” nucleotides), representing 86% of the predicted genome size of 1.80 Gb, while over one third of the genome (37.5%) consisted of repeated elements and >85% of the core eukaryotic genes were recovered. Given the repeated genetic bottlenecks of V. ellipsiformis populations as a result of glaciations events, heterozygosity was also found to be remarkably low (0.6%), in contrast to most other sequenced bivalve species. Finally, we reassembled the full mitochondrial genome and found six polymorphic sites with respect to the previously published reference. This resource opens the way to comparative genomics studies to identify genes related to the unique adaptations of freshwater mussels and their distinctive mitochondrial inheritance mechanism.
Keywords: genome assembly, annotation, high-throughput sequencing, freshwater mussels, Unionida
Introduction
Through their water filtration action, freshwater mussels (Bivalvia: Unionida) serve important roles as aquatic ecosystem engineers (Gutiérrez et al. 2003; Spooner and Vaughn 2006), and can greatly influence species composition (Aldridge et al. 2007). From a biological standpoint, they are also well known for producing obligate parasitic larvae that metamorphose on freshwater fishes (Lopes-Lima et al. 2014), for being slow-growing and long-lived, with several species reaching >30 years old and some species >100 years old (see Haag and Rypel 2011 for a review), and for exhibiting an unusual system of mitochondrial transmission called Doubly Uniparental Inheritance or DUI (see Breton et al. 2007; Passamonti and Ghiselli 2009; Zouros 2013 for reviews). From an economic perspective, freshwater mussels are also exploited to produce cultured pearls (Haag 2012). Regrettably however, habitat loss and degradation, overexploitation, pollution, loss of fish hosts, introduction of nonnative species, and climate change have resulted in massive freshwater mussel decline in the last decades (reviewed in Lopes-Lima et al. 2017, 2018). For example, >70% of the ∼300 North American species are considered endangered at some level (Lopes-Lima et al. 2017).
While efforts are currently underway to sequence and assemble the genome of several marine bivalves such as the mussel Mytilus galloprovincialis (Murgarella et al. 2016), genomic resources for mussels in general are still extremely scarce. In addition to M. galloprovincialis, the genomes of several other mytilid mussel species, such as the deep-sea vent/seep mussel Bathymodiolus platifrons, the shallow-water mussel Modiolus philippinarum and the golden mussel Limnoperna fortunei have recently been published (Sun et al. 2017; Uliano-Silva et al. 2018). In all cases, genomes have proven challenging to assemble due to their large size (∼1.6–2.4 Gb), widespread presence of repeated elements (∼30% of the genome, and up to 62% of the genome for the shallow-water mussel M. philippinarum, Sun et al. 2017) and high heterozygosity (Murgarella et al. 2016; Mun et al. 2017; Uliano-Silva et al. 2018). For example, the M. galloprovincialis genome remains highly fragmented, with only 15% of the gene content estimated to be complete (Murgarella et al. 2016). With respect to freshwater mussels (order Unionida), no nuclear genome draft currently exists. An assembled and annotated genome for freshwater mussels has the potential to be utilized as a valuable resource for many researchers given the biological value and threatened features of these animals. In addition, contrary to most other sequenced bivalve species, heterozygosity of V. ellipsiformis is expected to be relatively low, given its history of genetic bottlenecks due to repeated glaciations events over its current geographical distribution (Zanatta and Harris 2013). Genomic resources are also needed to help identifying genes essential for survival (and/or the genetic mechanisms that led to decline) and ultimately for developing monitoring tools for endangered biodiversity and plan sustainable recoveries (Savolainen et al. 2013; Pavey et al. 2017). Finally, a sequenced genome will help answer more fundamental questions of sex determination (Breton et al. 2011, 2017) and genome evolution through comparative genomics approaches (Sun et al. 2017).
Given the challenges in assembling a reference genome for saltwater mussels (Murgarella et al. 2016; Sun et al. 2017), we used a combination of different sequencing strategies (Illumina paired-end and mate pair libraries, Pacific Biosciences long reads, and a recently assembled reference transcriptome, Capt et al. 2018) to assemble the first genome draft in the family Unionidae. Hybrid sequencing technologies using long-read−low-coverage and short-read−high-coverage offer an affordable strategy with the advantage of assembling repeated regions of the genome (for which short reads are ineffective) and circumventing the relatively higher error rate of long reads (Koren et al. 2012; Miller et al. 2017). Here, we present a de novo assembly and annotation of the genome of the freshwater mussel V. ellipsiformis.
Materials and Methods
To determine the expected sequencing effort to assemble the V. ellipsiformis genome, that is, the necessary software and computing resources required, we first searched for C values from other related mussel species. C values indicate the amount of DNA (in picograms) contained within a haploid nucleus and is roughly equivalent to genome size in megabases. Two closely related freshwater mussel species in the same order (Unionida) as V. ellipsiformis (Elliptio sp., C value = 3; Uniomerus sp., C value = 3.2), in addition to other bivalve species from different orders (e.g., Mytilus spp. [order Mytilida], C value = 1.3–2.1; Dreissena polymorpha [order Venerida], C value = 1.7) were identified on the Animal Genome Size Database (http://www.genomesize.com). As such, we estimated the V. ellipsiformis genome size to be around ∼1.5–3.0 Gb, and this originally served as a coarse guide to determine the sequencing effort required, given that when the sequencing for V. ellipsiformis was originally planned, no mussel genome had yet been published.
Mussel Specimen Sampling, Genomic DNA Extraction, and Library Preparation
Adult specimens of V. ellipsiformis were collected from Straight River (Minnesota; Lat 44.006509, Long −93.290899), and species was identified according to Badra (2007). Specimens were sexed by microscopic examination of gonad smears. Gills were dissected from a single female individual and genomic DNA was extracted using a Qiagen DNeasy Blood and Tissue Kit (QIAGEN Inc., Valencia, CA) using the animal tissue protocol. The quality and quantity of DNA, respectively, were assessed by electrophoresis on 1% agarose gel and with a BioDrop mLITE spectrophotometer (a total of 15 µg of DNA was quantified using the spectrophotometer). For whole genome shotgun sequencing and draft genome assembly, we used two sequencing platforms: Illumina (San Diego, CA) Hiseq2000 and Pacific Biosciences (Menlo Park, CA) PacBio RSII. First, three paired-end libraries with insert size of 300 bp were constructed using Illumina TruSeq DNA Sample Prep Kit. One mate pair library with insert sizes of ∼5 kb was constructed for scaffolding process using Illumina Nextera mate-pair library construction protocol. For high-quality genome assembly, Pacific Biosciences system was employed for final scaffolding process using long reads. Pacific Biosciences long reads (>10 kb) were generated using SMRT bell library preparation protocol (ten SMRT cells were sequenced). Construction of sequencing libraries and sequencing analyses were performed at the Genome Quebec Innovation Centre (McGill University, Qc, Canada).
Preprocessing of Sequencing Reads
We quality trimmed paired-end and mate-pair reads using Trimmomatic 0.32 (Bolger et al. 2014) with the options illuminaclip: TruSeq3-PE.fa: 2: 30: 10 leading: 3 trailing: 3 slidingwindow: 6: 10 minlen: 36. This allowed removal of base pairs below a threshold Phred score of three at the leading and trailing end, in addition to removing base pairs based on a sliding window calculation of quality (mininum Phred score of ten over six base pairs). Finally, if trimmed reads fell below a threshold length (36 bp), both sequencing pairs were removed. We verified visually the quality (including contamination with Illumina paired-end adaptors) before and after trimming using fastqc (Andrews 2010). This allowed us to only keep high-quality reads prior to the assembly steps.
Following quality trimming, we used bfc (Li and Durbin 2009) to perform error correction for the Illumina paired-end sequencing data. bfc suppresses systematic sequencing errors, which helps to improve the base accuracy of the assembly and reduce the complexity of the de Bruijn graph based assembly, described below.
Corrected paired-end reads were subsequently used to identify the optimal K value that provides the most distinct genomic k-mers using KmerGenie v1.7016 (Chikhi and Medvedev 2014). We tested k = 10–100, in incremental steps of 10, and we then refined the interval from 20 to 40, in incremental steps of 2 to get a more precise estimate of K.
Genome Size and Heterozygosity Estimation
We used jellyfish 2.1.4 (Marcais and Kingsford 2011) for counting k-mers of lengths 17, 19, 21, 31, and 41, and obtain their frequency distributions, using the error-corrected, trimmed paired end reads. Based on k-mer frequency distributions, we then used genomescope (Vurture et al. 2017) in R version 3.4.4 (R Core Team 2017) to estimate the overall characteristics of the genome, including genome size, heterozygosity rate, and repeat content. GenomeScope attempts to fit mixture models of four evenly spaced negative binomial distributions to each k-mer profile in order to measure the relative abundances of heterozygous, homozygous, unique, and duplicated sequences.
Genome Assembly Strategy
We used ABySS 2.0 (Jackman et al. 2017), a modern genome assembler specifically built for large genomes and reads acquired by different sequencing strategies. ABySS 2.0 works similarly to ABySS (Simpson et al. 2009), by using a distributed de Bruijn graph representation of the genome, therefore allowing parallel computation of the assembly algorithm across a network of computers. In addition, the software makes use of long-sequencing reads (Illumina mate-pair libraries and Pacific BioSciences long reads) to bridge gaps and scaffold contigs. Yet, as memory requirements and computing time scale up exponentially with genome size, for large genomes (>1 Gb), these rapidly become very large (>100 GB of RAM) and unpractical. Consequently, Jackman et al. (2017) introduced ABySS 2.0, which employs a probabilistic data structure called a Bloom filter (Bloom 1970) to store a de Bruijn graph representation of the genome and, consequently, greatly reduces memory requirements and computing time. The Bloom filter allows removing from memory the majority of nearly identical k-mers likely caused by sequencing errors, as k-mers with an occurrence count below a user-specified threshold are discarded. The caveat is that it can generate false positive extension of contigs, but through optimization, this can be kept well <5%, and in fact, false positives can be corrected later on in the assembly step (Jackman et al. 2017).
In the current study, we combined different types of high-throughput sequencing to aid in assembling the genome (table 1). ABySS 2.0 (Jackman et al. 2017) performs a first genome assembly step without using the paired-end information, by extending unitigs until either they cannot be unambiguously extended or come to an end due to a lack of coverage (uncorrected unitigs). This first de Bruijn graph representation of the genome is further cleaned of vertices and edges created by sequencing errors (unitigs). Paired-end information is then used to resolve ambiguities and merge contigs. Following this, mate-pairs are mapped onto the assembly to create scaffolds, and finally long reads (Pacific Biosciences long reads) and the V. ellipsiformis reference transcriptome from Capt et al. (2018) were also mapped onto the assembly to create long-scaffolds. This reference transcriptome was assembled from a pool of sequences coming from four different male and female individuals and further details are provided in Capt et al. (2018). Although ideally sequencing information would all come from a single individual, the current study design did not allow for this. In addition, given that coding sequences are conserved compared with noncoding regions, it remains highly valuable to use a transcriptome in a de novo genome assembly.
Table 1.
DNA Sequencing Strategy
| Raw reads |
Trimmed Reads |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Type | Insert Size (bp) | Read Length (bp) | No. Reads (paired) | Total Length (Mb) | No. Reads (paired) | Total Length (Mb) | Total Length (% raw) | Read Length (bp, trimmed) | Coverage |
| Paired-end | 300 | 2×100 | 189,876,842 | 37,975 | 185,721,156 | 36,274 | 95.5 | 97.6 | |
| Paired-end | 300 | 2×100 | 195,394,768 | 39,079 | 191,002,987 | 37,319 | 95.5 | 97.7 | |
| Paired-end | 300 | 2×100 | 178,820,287 | 35,764 | 174,954,230 | 34,224 | 95.6 | 98.9 | |
| Total | 564,091,897 | 112,818 | 551,678,373 | 107,818 | 95.6 | 98.1 | 65× | ||
| Mate pair | 5000 | 2×100 | 97,801,148 | 19,560 | 94,350,168 | 18,717 | 95.7 | 99.3 | 11× |
| Pacific Biosciences Long reads | 4,406.4 (average) | 103,096 | 454 | 0.27× | |||||
| assembled transcriptome | 1,170.9 (average) | 285,260 | 334 | ||||||
| 301–50,048 (min–max) | |||||||||
We ran the ABySS 2.0 assembly stage (abyss-bloom-dbg) with a k-mer size of 41 (ABySS requires an odd number k-mer), a Bloom filter size of 24 GB, 4 hash functions and a threshold of k-mer occurrence set at 3. These parameters were chosen after performing several test assemblies, in order to minimize the false positive rate (<5%), maximize the N50 of the assembly and keep the virtual memory (95 GB) and CPU (24 CPUs) requirements within a reasonable computational limit for our resources. In addition, we adjusted parameters at the mapping stage to create contigs, scaffolds, and long-scaffolds to maximize N50 (overlap required in realignments, distance between mate-pairs, number of reads aligned to support assembly, see pipeline available at https://github.com/seb951/venustaconcha_ellipsiformis_genome).
Genome completeness was assessed using busco 3.0.2 (Benchmarking Universal Single-Copy Orthologs, Simao et al. 2015). Briefly, busco uses curated lists of known core single copy orthologs to produce evolutionarily informed quantitative measures of genome completeness (Simao et al. 2015). Here, we tested both the eukaryotic (303 single copy orthologs) and metazoan (978 single copy orthologs) gene lists to assess the completeness of our genome assembly.
Genome Contamination
Mussels are filter feeders and tissues such as gills can potentially harbour microbial fauna. In addition, freshwater mussels are prone to infection by trematodes (Müller et al. 2015; Capt et al. 2018). As suggested by Takeuchi et al. (2012), we first checked for the presence of double peaks in the distribution of the GC content of the raw reads, which would indicate contamination in the genome. In addition, we checked for potential contaminant sequences in the gene space of our current V. ellipsiformis genome assembly. Accordingly, we created a custom database of all trematodes, nematodes, and bacterial protein sequences available from NCBI (39,617 and 156,174 protein sequences available from refseq database for trematodes and nematodes, respectively, and 337,035 bacterial sequences available from uniprot database). We then compared this custom database of protein sequences to our predicted Open Reading Frames (315,932 V. ellipsiformis ORFs) to identify putative contaminants genes (BLASTp, Altschul et al. 1990; mininum evalue of 1e-20 and minimum 90%/99% Percentage of identical matches).
Characterization of Repetitive Elements
Given that repetitive elements can occupy large proportions of a genome, the characterization of their proportion and composition is an essential step during genome annotation. RepeatModeler open-1.0.10 (Smit and Hubley 2015) was used to create an annotated library of repetitive elements contained in the V. ellipsiformis genome assembly (excluding sequences <1 kb). Then, with RepeatMasker open-4.0.7 (Smit et al. 2015), we extracted libraries of repetitive elements for the taxa “Bivalvia” and “Mollusca” from the RepeatMasker combined database (comprising the databases Dfam_consensus-20170127 and RepBase-20170127) using built-in tools. Sequences classified as “artifact” were removed from the last two libraries before the subsequent steps. The three libraries were used alone and/or in combination (except for the Mollusca+Bivalvia combination) to mask the cut-down assembly again with RepeatMasker, specifying the following options: -nolow (to avoid masking low-complexity sequences, which may enhance subsequent exon annotation), -gccalc (to calculate the overall GC percentage of the input assembly), -excln (to exclude runs of ≥20 Ns in the assembly sequences from the masking percentage calculations). Option -species was used to specify the taxon for the runs with Bivalvia and Mollusca libraries, while option -lib used to specify the V. ellipsiformis library and the combined ones. Results summaries for the latter three runs were refined with the RepeatMasker built-in tools. Linear model fit for genome size and repeats content for all available bivalve genomes were calculated in R, using the highest masking value found for V. ellipsiformis.
Genome Annotation
We used quast (Gurevich et al. 2013) to calculate summary statistics on the genome assembly. In addition quast uses GlimmerHMM (Majoros et al. 2004), a gene predictor that uses Hidden Markov Models to identify putative genes in the final assembly. Following this, we translated Open Reading Frames identified in the annotation files into protein sequences using bedtoolsv2.27.1 (Quinlan and Hall 2010) and the program transeq from embossv6.6.0 (Rice et al. 2000) bioinformatics pipeline. These were then compared against the manually curated UniProt database (556,388 reference proteins, downloaded January 11, 2018, e-value cut-off of 10−5) using BLASTp (Altschul et al. 1990). These steps were done on the long-scaffolds assembly, the masked long-scaffolds assembly (with low-complexity regions replaced with N), in addition to the broken long-scaffolds assembly (scaffolds broken into smaller contigs by quast, based on long stretches of N nucleotides).
Mitochondrial Genome
Given the atypical mode of mitochondrial inheritance of freshwater mussels and therefore its evolutionary importance, we first aimed to check if the mitochondrial female genome had been properly assembled. Using BLASTn (Altschul et al. 1990) with high stringency (e-value <1e-50), we identified a fragmented mitochondrial genome. We then created a mt specific data set containing 1,396,004 sequence reads by aligning paired-end reads to the reference mt genome of Breton (2009, GenBank Acc. No. FJ809753) using samtoolsv1.3.1 and bedtoolsv2.27.1 (Li et al. 2009; Quinlan and Hall 2010). We then rebuilt the mt genome de novo using abyss 2.0, testing different k-mers (17–45). In addition, we aligned reads to the reference transcriptome using bwav0.7.12-r1039 (Li and Durbin 2009) and identified Single Nucleotide Polymorphisms (SNPs) with respect to the reference mt genome using samtools and bcftools v1.3.1 (Li et al. 2009).
Results and Discussion
We generated 564 million (M) paired-end reads (2×100 bp) representing an average 65× coverage of the genome (table 1). This was complemented by 98 M mate-pairs (5 kb insert, 11× average genome coverage) and 103,000 Pacific Biosciences long reads (0.3× average genome coverage), and a recently published reference transcriptome comprised of 285,000 contigs (Capt et al. 2018). Filtering and trimming the raw paired-end and mate-pair sequences removed ∼5% of the total base pairs from further analyses, indicating that the quality of the raw sequences was high (table 1). K-mer analysis indicated that the number of unique k-mers peaked at 42. In addition, model fitting predicted a genome assembly size of 1.80 Gb (see fig. 1 for k = 21, but note that similar values were found at other k-mer values analyzed), which is smaller than the predicted genome size according to C value for other freshwater mussel species in the order Unionida (Elliptio sp., C value = 3; Uniomerus sp., C value = 3.2), but in general agreement with the recent draft genome of other sequenced bivalves (0.55–3.2 Gb, see table 2).
Fig. 1.
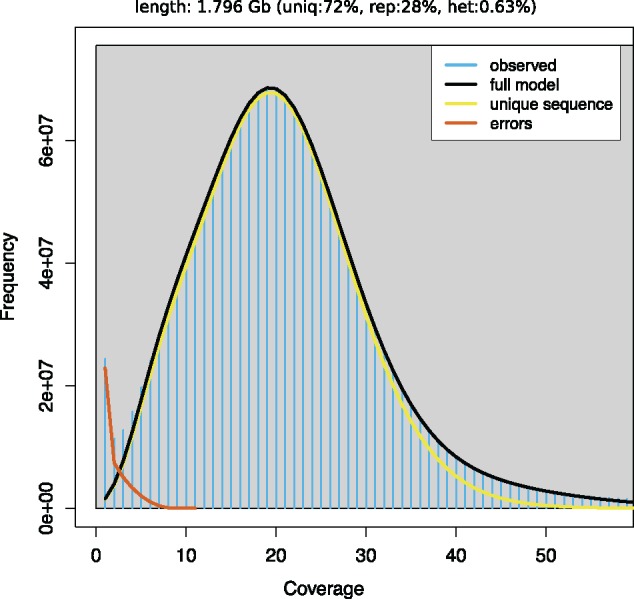
—k-mer distribution (k = 21) as calculated by genomescope (Vurture et al. 2017). Blue bars represent the observed k-mer distribution; black line represents the modelled distribution without the error k-mers (red line) and up to a maximum k-mer coverage specified in the model (yellow line). Length, estimated genome length; Uniq, unique portion of the genome (nonrepetitive elements); Rep, repetitive portion of the genome; Het, genome heterozygosity.
Table 2.
Genome Size, Heterozygosity, and Repeat Elements
| Subclass | Order | Family | Species | Estimated Genome Size (Gb) | Heterozygosity (%) | % of Repeated Elements |
|---|---|---|---|---|---|---|
| Palaeoheterodonta | Unionida | Unionidae | Venustaconcha ellipsiformis | 1.80 | 0.63 | 37.81 |
| Heterodonta | Veneroida | Veneridae | Ruditapes philippinarum | 1.37 | high* | 26.38 |
| Pteriomorphia | Mytiloida | Mytilidae | Bathymodiolus platifrons | 1.64 | 1.24 | 47.90 |
| Modiolus philippinarum | 2.38 | 2.02 | 62.00 | |||
| Mytilus galloprovincialis | 1.60 | high* | 36.13 | |||
| Limnoperna fortune | 1.67 | 2.3 | 33.00 | |||
| Ostreoida | Ostreidae | Crassostrea gigas | 0.55 | 1.95 | 36.00 | |
| Pectinidae | Chlamys farreri | 0.95 | 0.8 | 32.10 | ||
| Patinopecten yessoensis | 1.43 | 0.45 | 38.87 | |||
| Pterioida | Pteriidae | Pinctada fucata | 1.15 | high* | 37.00 | |
| Pteriomorphia mean (SD) | 1.39 (0.58) | 1.29 (0.70) | 41.43 (10.29) | |||
| Mytiloida mean (SD) | 1.87 (0.44) | 1.52 (0.68) | 48.68 (12.95) | |||
| Ostreoida mean (SD) | 0.98 (0.44) | 1.07 (0.78) | 35.66 (3.40) | |||
| Pectinidae mean (SD) | 1.19 (0.34) | 0.63 (0.25) | 35.49 (4.79) | |||
| All subclasses mean (SD) | 1.41 (0.51) | 1.20 (0.69) | 39.35 (10.23) |
Note.—Estimates of genome size, heterozygosity, and percentage of repeated elements in the currently available bivalve nuclear genomes. Data for each single species were retrieved from the literature: P. yessoensis (highly inbreed individual, Wang et al. 2017), V. ellipsiformis (wild, recurrent population bottlenecks, this study), C. farreri (selective breeding in aquaculture, Li et al. 2017), B. platifrons (recurrent population bottlenecks in the wild, Sun et al. 2017), C. gigas (highly inbreed individual, Zhang et al. 2012), M. philippinarum (large wild population, Sun et al. 2017), L. fortunei (invasive worlwide, Uliano-Silva et al. 2018), R. philippinarum (selective breeding in aquaculture, Mun et al. 2017), P. fucata (selective breeding in aquaculture, Takeuchi et al. 2012), M. galloprovincialis (large wild population, Murgarella et al. 2016). The genome size for V. ellipsiformis was based on k-mer analysis (see Materials and Methods). Mean and standard deviation (SD) values are also shown for the taxa comprising more than one species and for all subclasses, that is, the class Bivalvia. Note that all species are marine, except for V. ellipsiformis and L. fortunei (freshwater).
no rate calculated, but “high” heterozygosity documented.
One of the main reason for the highly fragmentation of many bivalve genomes is thought to be high heterozygosity and repetitive elements. In fact, heterozygosity rates of most sequenced bivalve species appear to be high, even for highly inbred individuals under strong artificial selection for many generations (see table 2). In the current V. ellipsiformis genome assembly, we did not observe the typical double peak patterns in the k-mer distribution (fig. 1) previously reported in most other bivalve genomes (Murgarella et al. 2016; Mun et al. 2017; Uliano-Silva et al. 2018). In fact, heterozygosity appears remarkably low (0.63%, table 2) and more in line with previous reports for the deep-sea vent/seep mussel (Sun et al. 2017), where recurrent population bottlenecks as a result of population extinctions and recolonizations of hydrothermal vents are common (Faure et al. 2015; Sun et al. 2017). Similarly, V. ellipsiformis populations have experienced severe genetic bottlenecks due to glaciation events (Zanatta and Harris 2013). The last glaciation in North America ended ∼12,000 BP, after which individuals from glacial refuges were able to recolonize previously uninhabitable regions. As a consequence, effective population size and heterozygosity for V. ellipsiformis is assumed to be fairly low, which was confirmed with the present data set, and in contrast to most published bivalve genomes so far (table 2).
Running the ABySS 2.0 assembly stage (abyss-bloom-dbg) led to a low False Positive Rate (<0.05%). The N50 for the contig assembly was 3.2 kb with 551,875 contigs (discarding contigs <1 kb, given that small contigs likely represent artifacts and provide little information for the overall genome assembly; Murgarella et al. 2016; Pavey et al. 2017; see table 3). Once these were corrected and paired-end, mate-pairs and long read information were added, the scaffolds N50 increased to 5.5 kb, with 2.3% of nucleotides represented as “N” (see table 3 for the summary statistics and table 4 for overall genome assembly statistics acquired from quast analysis). Adding the Pacific Biosciences long reads only slightly improved the scaffolds N50 (from 5.5 to 5.7 kb, table 3) and slightly decreased the number of long-scaffolds >1 kb (from 423,853 to 410,237), likely because our long read coverage was quite low (0.3×, table 1). In addition, it is also possible that the more error prone Pacific Biosciences sequences, compared with Illumina paired-end reads, reduced their usability (Miller et al. 2017). Once the reference transcriptome was added, it improved the N50 to 6.5 kb, and substantially decreased the number of long-scaffolds to 366,926. This final long-scaffold assembly accounted for a total size of 1.54 Gb (with 2.3% of “N” nucleotides) and represented 86% of the predicted genome size of 1.80 Gb. Yet, it remained highly fragmented (366,926 scaffolds, table 3). Genome annotation statistics can also be viewed in html format and downloaded here: https://github.com/seb951/venustaconcha_ellipsiformis_genome/tree/master/annotation_quast_v3; last accessed June 12, 2018
Table 3.
Assembly Statistics (ABySS2.0)
| Assembly | n ( ×10e6) | n: 1000 | L50 | Min | N80 | N50 | N20 | Max | Sum (Mb) |
|---|---|---|---|---|---|---|---|---|---|
| Raw unitigs | 39.8 | 347,879 | 101,624 | 1,000 | 1,361 | 2,181 | 3,891 | 25,883 | 707 |
| Unitigs | 18.5 | 444,734 | 127,617 | 1,000 | 1,485 | 2,452 | 4,273 | 25,944 | 984 |
| Contigs | 14.0 | 551,875 | 141,012 | 1,000 | 1,704 | 3,117 | 5,817 | 39,408 | 1,449 |
| Scaffolds | 13.7 | 423,853 | 92,607 | 1,000 | 2,303 | 5,477 | 9,099 | 45,260 | 1,539 |
| Long scaffolds (PacBio) | 13.7 | 410,237 | 86,661 | 1,000 | 2,391 | 5,708 | 9,893 | 47,610 | 1,548 |
| Long scaffolds (PacBio+transcriptome) | 13.6 | 366,926 | 58,906 | 1,000 | 2,534 | 6,523 | 16,660 | 298,135 | 1,549 |
Assembly (raw unitigs=raw assembly, not taking into account paired-end information, unitigs=filtering, merging, and popping bubbles in de Bruijn graph, contigs=unitigs with paired-end information mapped, scaffolds=contigs with mate-pairs information mapped, long scaffolds=scaffolds with PacBio/transcriptome information integrated), n=number of contigs, n: 1,000 = number of contigs of mininum length of 1,000, L50 = minimum number of sequences required to represent 50% of the entire assembly, min=mininum length of sequences analyzed, N80, N50, N20 = weighted median statistic such that 80/50/20% of the entire assembly is contained in contigs equal to or larger than this value in bp, max=maximum size of contig in bp, sum=sum of all contigs of size>min.
Table 4.
Assembly and Annotation Statistics for the Long Scaffold Assembly
| quast Assembly Statistics | Long_scaffolds | Long_scaffolds (>1 kb scaffolds broken based on N streches) | Long_scaffolds (>1 kb scaffolds, masked assembly) |
|---|---|---|---|
| Number of scaffolds (≥0 bp) | 13,635,758 | 821,266 | 374,245 |
| Number of scaffolds (≥1kb) | 371,706 | 549,364 | 374,245 |
| Number of scaffolds (≥5kb) | 94,238 | 50,209 | 95,019 |
| Number of scaffolds (≥10kb) | 26,952 | 5,151 | 27,030 |
| Number of scaffolds (≥25kb) | 5,073 | 23 | 4,976 |
| Number of scaffolds (≥50kb) | 1,456 | 0 | 1,427 |
| Total length (≥0 bp) | 2,638,723,663 | 1,554,026,338 | 1,596,234,060 |
| Total length (≥1kb) | 1,590,292,198 | 1,425,294,273 | 1,596,234,060 |
| Total length (≥5kb) | 1,000,983,904 | 360,423,103 | 1,003,000,325 |
| Total length (≥10kb) | 541,545,133 | 64,766,821 | 538,648,016 |
| Total length (≥25kb) | 231,252,884 | 687,249 | 226,147,564 |
| Total length (≥50kb) | 107,178,666 | 0 | 104,739,660 |
| Number of scaffolds | 371,706 | 821,266 | 374,245 |
| Largest scaffolds | 313,274 | 44,597 | 313,274 |
| Total length | 1,590,292,198 | 1,554,026,338 | 1,596,234,060 |
| Estimated reference length | 1,800,000,000 | 1,800,000,000 | 1,800,000,000 |
| GC (%) | 34.19 | 34.19 | 33.49 |
| N50 | 6,656 | 2,812 | 6,627 |
| Number of N’s per 100kb | 2,293.33 | 13.17 | 39,200.22 |
| Number of predicted genes (unique) | 201,068 | 277,765 | 123,457 |
| Number of predicted genes (≥300 bp) | 74,820 | 82,359 | 41,697 |
| Number of predicted genes (≥1.5kb) | 18,539 | 14,338 | 11,897 |
| Number of predicted genes (≥3kb) | 6,511 | 3,289 | 4,375 |
| Number of annotated ORF (uniprot) | 29,031 | 14,198 | 25,544 |
Note.—All statistics are based on scaffolds of size ≥1 kb, unless otherwise noted (e.g., “No scaffolds [≥ = 0 bp]” and “Total length [≥ = 0 bp]” include all scaffolds).
While assembly numbers (N50 and number of scaffolds) are not directly comparable with other recently published genomes given the diversity of sequencing approaches (Illumina, 454, Sanger, PacBio), library types, sequencing depth, and unique nature of the genome themselves, they can give a broad perspective of the inherent difficulties of assembling large genomes. The best comparison is probably with the saltwater mussel, M. galloprovincialis, giving their similar genome size (1.6 Gb for Mytilus vs. 1.80 Gb for V. ellipsiformis) and Illumina paired-end sequencing approaches (32× for Mytilus vs. 65× for V. ellipsiformis). While the M. galloprovincialis genome project (Murgarella et al. 2016) did not utilize mate-pair libraries or Pacific Bioscience long reads, they did make use of sequencing libraries with varying insert sizes (180, 500, and 800 bp). As such, they obtained a genome assembly quality relatively similar to ours and consisting of 393,000 scaffolds (>1 kb), with however a substantially lower N50 (2.6 kb compared with 6.5 kb for V. ellipsiformis). The recently reported genome for the deep-sea vent/seep mussel B. platifrons (1.64 Gb) made use of nine Illumina sequencing libraries with varying insert sizes (180–16 kb) and an overall coverage of >300× (Sun et al. 2017). With this very thorough sequencing approach and a heterozygosity closer to V. ellipsiformis than other Mytiloida, the scaffold N50 obtained was substantially higher (343.4 kb), but again the genome remained highly fragmented, into >65,000 scaffolds. As exemplified here, high-coverage sequencing libraries with varying insert sizes have become a broadly used approach for large and complex genomes. In fact, it is implemented by default in many genome assembly platforms (e.g., allpaths-lg, Gnerre et al. 2011; SOAPdenovo2, Luo et al. 2012). In the future, these libraries will likely be useful to further assemble the V. ellipsiformis genome, at least until these approaches are superseded by affordable, error free, single molecule long read sequencing (Gordon et al. 2016; Badouin 2017), or mapping approaches that allow reaching chromosome level assemblies such as optical mapping (e.g., Bionano Genomics, San Diego, CA).
Results of the busco (Simao et al. 2015) analyses showed that 664 (68%) of the 978 core metazoan genes (CEGs) were considered complete in our assembly. When the busco analysis was extended to include also fragmented matches, 871 (89%) proteins aligned. Results were similar when compared against the 303 core eukaryotic genes (61% complete, 86% complete or fragmented, table 5). When compared with the previously published reference transcriptome for V. ellipsiformis (Capt et al. 2018), we found fewer complete genes, but also fewer duplicated genes (97.5% complete, and 24% duplicated in the reference transcriptome, compared with 68.1% complete and 1% duplicated here). This likely reflects the fact that the reference transcriptome is nearly complete, while the current reference genome is still fragmented. However, the reference transcriptome also likely contains multiple isoforms of the same genes, in addition to possible nematode contaminating sequences, despite the authors’ best efforts to minimize these problems. Previously analyzed molluscan genomes of similar size (Murgarella et al. 2016; Sun et al. 2017) have found that 16% (M. galloprovincialis, 1.6 Gb), 25% (pearl oyster Pinctada fucata, 1.15 Gb), 36% (California sea hare Aplysia californica, 1.8 Gb) of the core eukaryotic genes were complete. For their part Sun and collaborators (2017), identified 96% of the core metazoan genes to be partial or complete in the deep-sea vent/seep mussel B. platifrons (1.6 Gb), again reflecting that the depth and type of sequencing, in addition to the idiosyncrasies of each genome, can have considerable influence on the end results.
Table 5.
Analysis of Genome Completeness Using busco 3.0.2 (Benchmarking Universal Single-Copy Orthologs, Simao et al. 2015)
| Metazoa | Eukaryota | |
|---|---|---|
| Complete orthologs (C) | 664 (68%) | 185 (61%) |
| Complete and single-copy orthologs (S) | 652 (67%) | 181 (60%) |
| Complete and duplicated orthologs (D) | 12 (1%) | 4 (1%) |
| Fragmented orthologs (F) | 207 (21%) | 76 (25%) |
| Missing orthologs (M) | 107 (11%) | 42 (14%) |
| Total ortholog groups searched | 978 | 303 |
We confirmed the presence of a single GC content peak (supplementary fig. 1, Supplementary Material online), thus supporting a lack of sequence contamination in the raw paired sequencing reads. In addition, we identified a very small percentage of Open Reading Frames matching to our custom database of nematodes–trematodes–bacteria proteins. Out of 315,932 Open Reading Frames identified in V. ellipsiformis, we identify 299 and 29 proteins with >90% and 99% sequence identity (between 0.09% and 0.0009% of all ORF, respectively, supplementary table 1, Supplementary Material online). This confirms that in the current genome assembly, the gene space is effectively free of the most common contaminants of freshwater mussels.
The custom V. ellipsiformis repeat library created de novo with RepeatModeler contained 2,068 families, the majority of them (1,498, 72.44% of the total) classified as “unknown.” Repeat content values reported below are slightly higher than the ones calculated based on k-mer analyses (28%, fig. 1), but should be considered more accurate given that they are based on the assembled sequences, rather than raw reads. The genome masking performed with the Bivalvia and Mollusca libraries had scarce performances (masking 2.38% and 2.59%, respectively; details in supplementary table 2, Supplementary Material online), possibly because of the phylogenetic distance between V. ellipsiformis, which belongs to the early branching bivalve lineage of Palaeoheterodonta, and the other bivalve and mollusk species represented in the database as well as their relative number of sequences. The custom V. ellipsiformis library masked 37.17% of the genome, while the combined V. ellipsiformis +Bivalvia masked 37.69% of the genome and the V. ellipsiformis + Mollusca reached 37.81%, the highest masking percentage (supplementary table 3, Supplementary Material online). After refining, these raw values slightly decreased to, respectively, 36.29%, 36.80%, and 36.91% (supplementary table 4, Supplementary Material online). All these latter values of repeat content fall in the 32–39% range (the median for all species is 37%) where six out of the nine sequenced bivalve species lie, irrespective of their genome size (M. philippinarum and R. philippinarum are the furthest from this interval) (table 2 and supplementary fig. 2, Supplementary Material online). Although the number of species sequenced up to now is still low, this observation indicates that repetitive elements may contribute differently to the total genome size among the different bivalve taxa: indeed, the correlation between genome size and repeats content is weak (supplementary fig. 2, Supplementary Material online). In both, the ab initio masking with the V. ellipsiformis library and the two combined ones, most of the identified repeats are categorized as “unknown” (22.8% of the assembly), followed by retroelements (LINEs 2.9%, LTR elements 2.3–2.4%, and SINEs 1.7%, for a total of 6.9% of the assembly) and DNA elements (5.4–5.6% of the assembly) (supplementary table 4, Supplementary Material online). Direct comparisons of these values with other species should be performed with caution, as the usually large “unclassified” portion of repeats might contain species-specific variants of known elements (Murgarella et al. 2016) that may therefore change the relative weight of each category on the total.
quast was used to calculate summary statistics and identify putative genes in the final assembly using a hidden markov model (table 4). Following this, 29,031; 14,195; and 25,544 Open Reading Frames were annotated using BLASTp against UniProt database in the long-scaffolds, broken, and masked long-scaffolds assemblies, respectively.
Freshwater mussels, marine mussels, as well as marine clams are the only known exception in the animal kingdom with respect to the maternal inheritance of mitochondrial DNA (see Breton et al. 2007 for a review). Their unique system, characterized by the presence of two gender-associated mitochondrial DNA lineages, has therefore attracted studies to better understand mitochondrial inheritance and the evolution of mtDNA in general. Using blastn, we recovered 53 contigs matching to the 15,975-bp female reference mt genome from Breton (2009), indicating that the mt genome was highly fragmented and likely improperly assembled with our current approach, much like what was found in the M. galloprovincialis genome draft of Murgarella et al. (2016). As such, we created a data set of mt specific sequences that could be aligned to the mt genome (1,396,004 reads). This mt specific data set was then reassembled de novo, using different k-mers (17–45). Using a k-mer similar or larger to the one used in the overall assembly (k ≥ 41) resulted in a failed assembly (no contigs created, data not shown), while using a k-mer <21 generated a highly fragmented mt genome (data not shown). Using a k-mer between 21 and 39 generated one large contig of 16,024 bp comprising the entire mitogenome, with a 42-bp insertion in the 16S ribosomal RNA. Given the different rate of evolution of mtDNAs, it is likely that assembly parameters we used for the whole genome were not appropriate for the V. ellipsiformis female mt genome. Finally, we also realigned the mt specific data set to the original mt genome of Breton (2009) and found high coverage (mean = 7,256×, SD = 682) for most positions, while for three regions coverage dropped <300× (fig. 2). Six SNPs with respect to the reference were also identified, indicating possible polymorphism, or sequencing error in the original mt reference genome (fig. 2).
Fig. 2.

—Mitochondrial coverage based on sequence alignment and annotation (from NCBI). Six nucleotide positions were identified in the legend as fixed for an alternative allele compared with the reference of Breton (2009).
Conclusion
High-throughput sequencing has the power to produce draft genomes that were only reserved to model systems 10 years ago. Here, we report the first de novo draft assembly of the V. ellipsiformis genome, a freshwater mussel from the bivalve order Unionida. Our assembly covers over 86% of the genome and contains nearly 90% of the core eukaryotic orthologs, indicating that it is nearly complete. In addition, we calculated relatively low-heterozygosity rates, uncommon in bivalves, but likely explained by the recent evolutionary history of V. ellipsiformis. Finally, as for other mussel genomes recently published, our genome remains fragmented, showing the limits of high-throughput sequencing and the necessity to combine different sequencing approaches to augment the scaffolding and overall genome quality, especially when a large fraction of the genome is comprised of repetitive elements. In the future, the V. ellipsiformis genome will benefit from a larger number of long read sequences, varying library size for paired-end sequencing, and the use of genetic, physical, or optimal maps to subsequently order scaffolded contigs into pseudomolecules or chromosomes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This research was supported by Natural Sciences and Engineering Research Council Discovery Grants awarded to S.B. (RGPIN/435656-2013) and D.T.S. (RGPIN/217175-2013), and by ‘Canziani Bequest’ and ‘Fondazione del Monte’ funding (M.P.) Computations were made on the supercomputer briaree from Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), the ministère de l’Économie, de la science et de l’innovation du Québec (MESI), and the Fonds de recherche du Québec—Nature et technologies (FRQ-NT).
Literature Cited
- Aldridge DC, Fayle TM, Jackson N.. 2007. Freshwater mussel abundance predicts biodiversity in UK lowland rivers. Aquat Conserv Mar Freshw Ecosyst. 176:554–564. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 2153:p403–p410. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Cambridge, UK: Babraham Institute. Available from: httpswww.bioinformatics.babraham.ac.ukprojectsfastqc Last Accessed May 1, 2018.
- Badouin H. 2017. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 5467656:148. [DOI] [PubMed] [Google Scholar]
- Badra PJ. 2007. Special animal abstract for Venustaconcha ellipsiformis (Ellipse). Lansing (MI: ): Michigan Natural Features Inventory; p. 4. [Google Scholar]
- Bloom BH. 1970. Space/time trade-offs in hash coding with allowable errors. Commun ACM 137:422–426. [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 3015:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S. 2009. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics 1834:1575–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Beaupré HD, Stewart DT, Hoeh WR, Blier PU.. 2007. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 239:465–474. [DOI] [PubMed] [Google Scholar]
- Breton S, Capt C, Guerra D, Stewart D.. 2017. Sex determining mechanisms in bivalves. Preprints 2017060127. [Google Scholar]
- Breton S, et al. , . 2011. Novel protein genes in animal mtDNA: a new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol Biol Evol. 285:1645–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capt C, et al. , . 2018. Deciphering the link between doubly uniparental inheritance of mtDNA and sex determination in bivalves: clues from comparative transcriptomics. Genome Biol Evol. 102:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R, Medvedev P.. 2014. Informed and automated k-mer size selection for genome assembly. Bioinformatics 301:31–37. [DOI] [PubMed] [Google Scholar]
- Faure B, Schaeffer SW, Fisher CR.. 2015. Species distribution and population connectivity of deep-sea mussels at hydrocarbon seeps in the Gulf of Mexico. PLoS One 104:e0118460.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, et al. , . 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 1084:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, et al. , . 2016. Long-read sequence assembly of the gorilla genome. Science 3526281:aae0344–aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G.. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 298:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez JL, Jones CG, Strayer DL, Iribarne OO.. 2003. Mollusks as ecosystem engineers: the role of shell production in aquatic habitats. Oikos 1011:79–90. [Google Scholar]
- Haag WR. 2012. North American freshwater mussels: natural history, ecology, and conservation. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Haag WR, Rypel AL.. 2011. Growth and longevity in freshwater mussels: evolutionary and conservation implications. Biol Rev. 861:225–247. [DOI] [PubMed] [Google Scholar]
- Jackman SD, et al. , . 2017. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 275:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. , . 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 307:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2514:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. , . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2516:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. , . 2017. Scallop genome reveals molecular adaptations to semi-sessile life and neurotoxins. Nat Commun. 81:1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Lima M, et al. , . 2014. Biology and conservation of freshwater bivalves: past, present and future perspectives. Hydrobiologia 7351:1–13. [Google Scholar]
- Lopes-Lima M, et al. , . 2017. Conservation status of freshwater mussels in Europe: state of the art and future challenges. Biol Rev. 921:572–607. [DOI] [PubMed] [Google Scholar]
- Lopes-Lima M, et al. , . 2018. Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia 8101:1–14. [Google Scholar]
- Luo R, et al. , . 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL.. 2004. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2016:2878–2879. [DOI] [PubMed] [Google Scholar]
- Marcais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 276:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, et al. , . 2017. Hybrid assembly with long and short reads improves discovery of gene family expansions. 181:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, et al. , . 2015. Factors affecting trematode infection rates in freshwater mussels. Hydrobiologia 7421:59–70. [Google Scholar]
- Mun S, et al. , . 2017. The whole-genome and transcriptome of the manila clam (Ruditapes philippinarum). Genome Biol Evol. 96:1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgarella M, et al. , . 2016. A first insight into the genome of the filter-feeder mussel Mytilus galloprovincialis. PLoS One 113:e0151561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti M, Ghiselli F.. 2009. Doubly uniparental inheritance: two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 282:79–89. [DOI] [PubMed] [Google Scholar]
- Pavey SA, et al. , . 2017. Draft genome of the American Eel (Anguilla rostrata). Mol Ecol Resourc. 174:806–811. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 266:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a Language and Environment for Statistical Computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 166:276–277. [DOI] [PubMed] [Google Scholar]
- Savolainen O, Lascoux M, Merilä J.. 2013. Ecological genomics of local adaptation. Nat Rev Genet. 1411:807–820. [DOI] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 3119:3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson JT, et al. , . 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 196:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R.. 2015. RepeatModeler Open-1.0 (2008–2015). Seattle, USA:Institute for Systems Biology. Available from: httpwww.repeatmasker.org, Last Accessed May 1, 2018.
- Smit A, Hubley R, Green P.. 2015. RepeatMasker Open-4 (2013–2015), Seattle, USA: Institute for Systems Biology. Last Accessed May 1, 2018.
- Spooner DE, Vaughn CC.. 2006. Context-dependent effects of freshwater mussels on stream benthic communities. Freshwater Biol. 516:1016–1024. [Google Scholar]
- Sun J, et al. , . 2017. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat Ecol Evol. 15:0121–0127. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, et al. , . 2012. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 192:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliano-Silva M, et al. , . 2018. A hybrid-hierarchical genome assembly strategy to sequence the invasive golden mussel, Limnoperna fortunei. GigaScience 72:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vurture GW, et al. , . 2017. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 3314:2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, et al. , . 2017. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat Ecol Evol. 15:0120–0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanatta DT, Harris AT.. 2013. Phylogeography and genetic variability of the freshwater mussels (Bivalvia: Unionidae) ellipse, Venustaconcha ellipsiformis (Conrad 1836), and bleeding tooth. Am Malacolog Bull. 312:267–279. [Google Scholar]
- Zhang G, et al. , . 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 4907418:49–54. [DOI] [PubMed] [Google Scholar]
- Zouros E. 2013. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol Biol. 401:1–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.