

Abstract
Aims
Doxorubicin (DOX) is among the most effective chemotherapies used in paediatric cancer patients. However, the clinical utility of DOX is offset by its well-known cardiotoxicity, which often does not appear until later in life. Since hypertension significantly increases the risk of late-onset heart failure in childhood cancer survivors, we investigated whether juvenile DOX exposure impairs the ability to adapt to angiotensin II (Ang II)–induced hypertension later in life and tested a treatment that could prevent this.
Methods and results
Five-week-old male mice were administered a low dose of DOX (4 mg/kg) or saline once a week for 3 weeks and then allowed to recover for 5 weeks. Following the 5-week recovery period, mice were infused with Ang II or saline for 2 weeks. In another cohort, mice were fed chow containing 0.4% resveratrol 1 week before, during, and 1 week after the DOX administrations. One week after the last DOX administration, p38 mitogen-activated protein kinase (MAPK) was activated in hearts of DOX-treated mice demonstrating molecular signs of cardiac stress; yet, there was no change in cardiac function between groups. However, DOX-treated mice failed to develop compensatory cardiac hypertrophy in response to Ang II–induced hypertension later in life. Of importance, mice receiving DOX with resveratrol co-administration displayed normalization in p38 MAPK activation in the heart and a restored capacity for cardiac hypertrophy in response to Ang II–induced hypertension.
Conclusion
We have developed a juvenile mouse model of DOX-induced cardiotoxicity that displays no immediate overt physiological dysfunction; but, leads to an impaired ability of the heart to adapt to hypertension later in life. We also show that co-administration of resveratrol during DOX treatment was sufficient to normalize molecular markers of cardiotoxicity and restore the ability of the heart to undergo adaptive remodelling in response to hypertension later in life.
Keywords: Cardiovascular disease, Resveratrol, Cardiotoxicity, Chemotherapy, Paediatrics
1. Introduction
As a result of advanced diagnosis and treatment, the 5-year survival rate of children diagnosed with cancer has increased from less than 60% in the 1970s to 83% in 2010.1,2 Although these increased survival rates are promising, survivors are often more susceptible to adverse health effects later in life due to the anticancer treatments that they received as children.3 In fact, cardiovascular-related disease is the leading cause of morbidity and mortality in childhood cancer survivors.4 Of importance, approximately 30% of children diagnosed with cancer are treated with anthracyclines such as doxorubicin (DOX).4 Thus, despite the fact that anthracyclines are known to have cardiotoxic effects that may go undetected but progress to cardiac dysfunction and eventually heart failure later in life,1 anthracyclines continue to be used in cancer therapy.
It is widely accepted that the risk of anthracycline-induced cardiotoxicity increases with a higher anthracycline cumulative dose.5 However, recent studies have discovered that sub-clinical cardiotoxic events occur in children who receive even lower doses of anthracyclines,6,7 suggesting that there is no safe dose of anthracyclines in terms of cardiotoxicity. Since anthracyclines do not cause cardiac dysfunction in every childhood cancer survivor, it is important to study co-morbidities that may increase the odds of experiencing a late-onset cardiac dysfunction in these patients. For instance, since hypertension significantly increases the risk of developing late-onset heart failure in childhood cancer survivors,8 it is possible that anthracycline-treated children are more susceptible to hypertension-induced cardiomyopathy later in life. If this holds true, developing strategies that can prevent anthracycline-induced cardiac injury during treatment may prevent the eventual onset of cardiomyopathy as adults.
To investigate this, we developed an animal model of anthracycline-induced cardiotoxicity whereby young mice were subjected to a once weekly regimen of DOX for a period of 3 weeks and then followed-up later in life. DOX was administered to juvenile mice in doses low enough to produce no overt cardiac dysfunction, yet at a dose sufficient to induce molecular signalling pathways indicative of cardiotoxicity. The effects of this latent cardiotoxicity were then studied in adult mice administered angiotensin II (Ang II) to determine if early DOX-induced cardiac injury increased the risk of hypertension-induced cardiomyopathy later in life. Our study investigated the molecular and physiological mechanisms that may contribute to the sub-clinical anthracycline-induced cardiac injury that leaves paediatric patients susceptible to late-occurring recurrent or progressive cardiac dysfunction9 as well as an intervention strategy that could be used to prevent this.
2. Methods
2.1 Experimental animals
All protocols involving mice were approved by the University of Alberta and the University of Minnesota Institutional Animal Care and Use Committees that conform to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (Eighth edition; revised 2011). The University of Alberta and the University of Minnesota adhere to the principles for biomedical research involving animals developed by the Council for International Organizations of Medical Sciences and comply with the Canadian Council on Animal Care guidelines. All C57BL/6N mice were purchased from Charles River Laboratories (St. Foy, QC).
2.2 DOX treatment
Following 1 week of acclimatization, male C57BL/6N mice at 5 weeks of age were randomly assigned to Saline-injected (Control) or DOX-injected groups. The mice were administered an intraperitoneal injection of DOX (4 mg/kg body weight) or an equal volume of normal saline once per week for 3 weeks. A cohort of mice was euthanized 1 week after the last DOX injection to determine the acute effects of DOX by an intraperitoneal injection of sodium pentobarbital, and another cohort was left to recover for 5 weeks to determine the late-occurring effects of DOX. Following the 5-week recovery period at the age of 12 weeks (the age of young adult mice), the mice were infused with a low (1.4 mg/kg/day) or high (2.8 mg/kg/day) dose of Ang II or saline for 14 days through subcutaneously implanted ALZET osmotic mini-pumps (Durect Corp, Cupertino, CA) to induce hypertension as previously reported.10,11 Briefly, mice were anesthetized using 1.0–1.5% isoflurane with an intramuscular injection of 5 mg/kg meloxicam to obtain the surgical plane of anaesthesia. Thereafter, mice were placed in the prone position and a midscapular incision made. Osmotic mini-pumps were implanted subcutaneously and skin was closed with surgical staplers. To investigate the preventative effect of resveratrol on DOX-induced cardiotoxicity, juvenile mice were fed a control chow diet or a diet containing 0.4% resveratrol (as we have performed previously10,12) for 1 week prior to initiation of saline or DOX injections. Following the 1 week resveratrol pre-treatment, mice received 3 weekly intraperitoneal injections of either saline or DOX. Resveratrol administration was discontinued at 1 week after the last DOX injection. Body weight and food consumption were measured weekly. Body composition was assessed by EchoMRI (Echo Medical System, Houston, TX). Mice had ad libitum access to food and water.
2.3 Locomotor activity
Home-cage activity was recorded in individually housed animals during several days under 12:12 h light:dark cycle. Locomotor activity was determined by means of an automated system that used small passive infrared sensors positioned on the top of each cage (ActiMeter, TechnoSmart, Rome, Italy). This system allowed continuous monitoring of undisturbed mouse locomotor activity.13 Activity was quantified as average values for either the dark (active) or light (rest) phase of the cycle.
2.4 Echocardiography
Mice were anaesthetized using 0.75%–1.5% isoflurane (induction with 3% isoflurane), and transthoracic echocardiography was performed in a blinded manner to assess cardiac function and remodelling using a Vivo 770 High-Resolution Imaging System equipped with a 30-MHz transducer (Visual Sonics, Toronto, ON, Canada) as previously described.12
2.5 Immunoblot analysis
Mice were euthanized with an intraperitoneal injection of sodium pentobarbital (120 mg/kg body weight). Hearts were snap-frozen in liquid nitrogen. Frozen heart tissue was homogenized according to previously reported methods14 and protein concentration was measured using the bicinchoninic acid protein assay kit (number 23255; Pierce, Thermo Fisher Scientific, Rockford, IL). About 25 μg of protein was resolved by SDS-PAGE and transferred to a nitrocellulose membrane. The following antibodies were used: anti-p53 catalog # 2524, anti-phospho extracellular signal–regulated kinases (ERK)1/2 catalog # 9101, anti-ERK1/2 catalog # 9102, anti-phospho c-Jun N-terminal kinase (JNK) catalog # 4668, anti-JNK catalog # 9252, anti-phospho p38 catalog # 9211, anti-p38 catalog # 9212, alpha-Tubulin catalog # 2144, Cell Signaling Technology, Danvers, MA; anti-VEGF catalog # sc-152 and the secondary antibody goat anti-rabbit IgG-HRP catalog # sc-2054, Santa Cruz Biotechnology, Dallas, TX, USA. Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and corrected against alpha-Tubulin protein levels as a loading control.
2.6 Quantitative real time-PCR
Total RNA from the frozen tissues was isolated using TRIzol® reagent (Life Technologies, Carlsbad, CA). First-strand cDNA was synthesized by using the High-Capacity cDNA reverse transcription kit (Life Technologies). Quantitative analysis of specific mRNA expression was performed by real time-PCR, by subjecting the resulting cDNA to PCR amplification using 384-well optical reaction plates in the ABI 7900HT instrument (Applied Biosystems, Foster City, CA). Gene expression of a cardiac stress marker (atrial natriuretic peptide: ANP, forward primer; 5′-GGA GCC TAC GAA GAT CCA GC-3′, reverse; 5′-TCC AAT CCT GTC AAT CCT ACC C-3′) were analysed as previously described.15 Data are presented as the fold change in gene expression normalized to the endogenous reference gene (beta-actin, forward primer; 5′-TAT TGG CAA CGA GCG GTT CC-3′, reverse; 5′-GGC ATA GAG GTC TTT ACG GAT GTC-3′).
2.7 Blood pressure measurement
Blood pressure was measured using the non-invasive tail cuff method with an iiTC blood pressure analysis system (IITC Life Science, Woodland Hills, CA) as previously described.10,11 Blood pressure measurements via tail cuff were corroborated by a telemetric blood pressure recording device, TA11PA-C10 (Data Science International, St. Paul, MN), analysed using PowerLab 4/30 Acquisition and LabChart 7 software (ADInstruments, Colorado Springs, CO). Blood pressure was recorded at 1 week after the last DOX injection, as well as immediately before and 2 weeks after Ang II infusion.
2.8 Histology
Haematoxylin and eosin staining of paraffin-embedded left ventricular (LV) heart sections taken mid-papillary were visualized using a Leica DMLA microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Retiga 1300i FAST 1394 CCD camera (OImaging, Surrey, BC, Canada) as described previously.14 Representative images were taken from each sample. Cardiomyocyte cross-sectional area was assessed using ImageJ software.
2.9 RNA sequencing
Total RNA extracted from heart tissues was subsequently used to generate the cDNA library using standard TruSeq library stranded construction protocol and the samples were sequenced on Illumina NextSeq500. Quality of the sequencing reads was assessed using FastQC v0.11.4 software. Raw reads were trimmed using cutadapt v1.14. Reads shorter than 60 bp after quality trimming were discarded. Trimmed reads were mapped to the mouse Ensembl GRCm38 genome. The mapping was performed using Tophat2 (v2.1) software based on Bowtie (v1.2). Only uniquely mapped reads were kept for the differential expression. Reads were counted on GRCm38 annotated genes using FeatureCounts program (v1.5.0-p2). The differential expression gene were identified by P < 0.05 and an absolute fold change >1.5 using all biological replicates for each sample with DESeq2 R package (1.16) via SARtools (v1.4). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis16 was performed using the Database for Annotation, Visualization and Integrated Discovery. Pathways with P < 0.05 and fold enrichment >1.5 were considered significant.
2.10 Statistical analysis
Data are shown as mean ± standard error of the mean (SEM). Comparisons between groups were performed using the unpaired Student’s two-tailed t-test or analysis of variance (ANOVA) with a Bonferroni post hoc test where appropriate. A probability value of <0.05 was considered significant.
3. Results
3.1 Juvenile mouse model of sub-clinical DOX-induced cardiotoxicity
To represent a clinical scenario of childhood cancer chemotherapy, we administered once weekly DOX (4 mg/kg) to 5-week-old mice for a period of 3 weeks (Figure 1A). The DOX dosage regimen resulted in no change in survival rate compared to controls (saline) through 7 weeks following the first DOX administration (Figure 1B). Although this DOX regimen caused an initial decline in the weight gain of the mice (Figure 1C), the mice resumed their normal growth rate after the last DOX injection (Figure 1C). To determine whether the weight loss was due to DOX-induced change in activity and/or food consumption, we measured the locomotor activity and daily food intake in control and DOX-treated mice. There was no significant difference in activity (Figure 1D) nor in food consumption (Figure 1E) between control and DOX-treated mice. To determine if the weight loss was due to skeletal muscle cachexia or loss of adipose mass, mice were subjected to echoMRI analysis at 5 weeks after the last DOX injection. Although juvenile DOX treatment did not alter the percentage of lean mass later in life (Figure 1F), DOX reduced the percentage of fat mass compared to controls (Figure 1G).
Figure 1.

Low-dose DOX administration does not induce overt morbidity or mortality. (A) Scheme of study design for investigating the effects of low dose DOX treatment. (B) Survival rate (n = 22–23) and (C) weekly body weight change during and after DOX treatment (n = 9). (D) Locomotor activity in the light and dark cycles in control and DOX-treated mice, measured 1 week after the last DOX injection (n = 6). (E) Food consumption during DOX treatment (n = 6). Body composition measured by echo MRI after a 5 weeks of recovery (n = 7): (F) percentage of lean mass and (G) percentage of fat mass. *P < 0.05 vs control. Values are presented as mean ± SEM and analysed by the unpaired Student’s two-tailed t-test.
To investigate whether our protocol of DOX treatment caused overt cardiac dysfunction in mice, we performed echocardiography to assess cardiac function and morphology after a 5-week recovery period. We show that the dose of DOX administered in this study resulted in no changes in systolic or diastolic function as indicated by % ejection fraction and E/A ratio, respectively, or LV posterior wall (LVPW) thickness, or LV mass compared to controls (Table 1). Interestingly, despite the lack of changes in cardiac function or morphology as observed by echocardiography after a 5-week recovery period, DOX treatment led to a significant reduction in heart weight at 1 week after the last DOX injection (Figure 2A), suggesting that DOX acutely impaired physiological cardiac growth or promoted cardiac atrophy.
Table 1.
Cardiac function and morphology in mice at 5 weeks after the last DOX injection
| Control | DOX | |
|---|---|---|
| HR (bpm) | 417 ± 9 | 432 ± 28 |
| Cardiac function | ||
| EF (%) | 61.0 ± 4.4 | 60.1 ± 4.1 |
| Mitral E/A ratio | 1.60 ± 0.08 | 1.55 ± 0.19 |
| SV (µL) | 49.7 ± 6.4 | 43.7 ± 9.6 |
| CO (mL/min) | 22.7 ± 3.0 | 21.7 ± 5.0 |
| Morphology | ||
| LVPW-diastole (mm) | 0.74 ± 0.03 | 0.70 ± 0.07 |
| LVPW-sytole (mm) | 1.11 ± 0.13 | 0.91 ± 0.06 |
| LVID-diastole (mm) | 4.27 ± 0.35 | 4.03 ± 0.46 |
| LVID-systole (mm) | 2.92 ± 0.43 | 2.81 ± 0.43 |
| LVEDV (µL) | 82.3 ± 15.7 | 72.2 ± 19.5 |
| LVESV (µL) | 33.6 ± 12.0 | 30.8 ± 11.3 |
| LV mass (mg) | 101.0 ± 9.1 | 88.0 ± 16.0 |
Values are mean ± SEM (n = 3–4). HR, heart rate; EF, ejection fraction; SV, stroke volume; CO, cardiac output; LVID, LV internal diameter; LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume.
Figure 2.
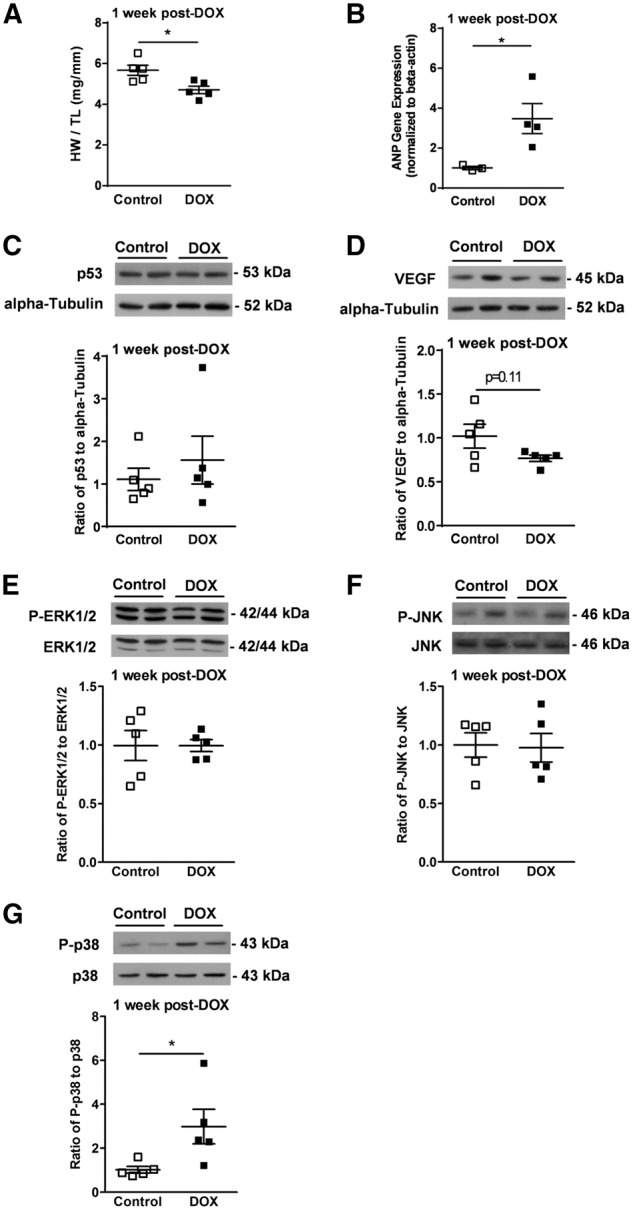
Low-dose DOX regimen causes ‘sub-clinical’ cardiac injury. (A) Heart weight normalized to tibia length (HW/TL) at 1 week after the last DOX injection (n = 5). (B) Gene expression of ANP normalized to beta-actin (n = 3–4). Representative immunoblots (n = 5): (C) p53 normalized to alpha-Tubulin; (D) VEGF normalized to alpha-Tubulin; (E) phospho (Thr202/Try204)-ERK1/2 normalized to total-ERK 1/2; (F) phospho (Thr183/Tyr185)-JNK normalized to total-JNK; and (G) phospho (Thr180/Try182)-p38 normalized to total-p38. *P < 0.05 vs. control. P-indicates phospho-. Values are presented as mean ± SEM and analysed by the unpaired Student’s two-tailed t-test.
To determine if there were indications of cardiotoxicity induced by DOX, a cohort of mice was euthanized at 1 week after the last DOX injection. Biological markers of cardiotoxicity were not extensively studied, largely due to the fact that the mice did not display overt signs of cardiotoxcity, which was an intentional design of our model. In the absence of overt biological changes, we therefore investigated ‘sub-clinical’ signs of cardiotoxicity using molecular markers. There was a significant increase in ANP gene expression in hearts of DOX-treated mice (Figure 2B), demonstrating ‘sub-clinical’ cardiac damage in hearts of DOX-treated young mice. In addition, we examined whether DOX treatment–induced additional molecular changes known to be involved in DOX-induced cardiac stress, such as increased protein expression of p53, decreased protein expression of vascular endothelial growth factor (VEGF), and increased activity of mitogen-activated protein kinase (MAPK) family.17–21 Interestingly, our DOX regimen did not alter protein expression of p53 and VEGF, or activate MAPK family such as ERK1/2 and JNK (Figure 2C–F), suggesting only modest cardiotoxic injury. Although activation of the MAPKs ERK1/2 and JNK was not altered, phosphorylation of p38 MAPK at Threonine180 and Tyrosine182 (P-p38) was significantly increased in hearts of DOX-treated mice (Figure 2G), suggesting that p38 activation may be an important component of ‘sub-clinical’ DOX-induced cardiotoxicity.
3.2 Mice with previous DOX treatment do not undergo adaptive cardiac remodelling during adult-onset hypertension
Since hypertension is a risk factor for anthracycline-induced cardiotoxicity in adult cancer patients,22 we determined whether or not juvenile mice with molecular evidence of DOX-induced cardiotoxicity have an increased susceptibility to developing an overt hypertension-induced cardiomyopathy in adulthood. To investigate this, mice were treated as described previously and also administered either a low dose of Ang II (1.4 mg/kg/day) or a high dose of Ang II (2.8 mg/kg/day) via osmotic pumps for an additional 2 weeks in order to induce hypertension and cardiac hypertrophy (Figure 3A).10,11 Following this, we first set out to demonstrate that the low dose of Ang II caused hypertension in all of the mice. Our data show that following 2 weeks of Ang II (1.4 mg/kg/day) administration, both control and DOX-treated mice displayed a significant rise in blood pressure (Figure 3B). Interestingly, even prior to Ang II infusion, mice that have been previously treated with DOX at a younger age also showed elevated blood pressure compared to control, indicating that DOX treatment at a young age can elicit a hypertensive response later in life.
Figure 3.

DOX-treated juvenile mice are susceptible to hypertension induced by Ang II infusion later in life. (A) Scheme of study design for investigating the effect of the hypertension induced by Ang II. (B) SBP immediately before and 2 weeks after the low dose of Ang II infusion (n = 4–7). (C) LVPW thickness in diastole (LVPWd, n = 9–15), (D) HW/TL (n = 6–14), (E) representative pictures of haematoxylin–eosin staining of the heart with scale bars of 25 µm, (F) cross-sectional area of the heart (n = 2–4), (G) stroke volume (n = 9–15), and (H) cardiac output (n = 9–15) after the low dose of Ang II infusion. (I) Survival rate of mice that were treated with DOX as juveniles and were infused with the high dose of Ang II later in life (n = 3). †P < 0.05 vs. before Ang II infusion, #P < 0.05 vs. saline-infused group, *P < 0.05 between groups indicated, §P < 0.05 vs. control group. 2xAngII indicates high dose (2.4 mg/kg/day) of Ang II. Values are presented as mean ± SEM and analysed by two-way ANOVA with Bonferroni post hoc test.
As expected, following a rise in blood pressure at 2 weeks after a low dose of Ang II infusion, control mice developed compensatory cardiac hypertrophy indicated by increases in LV posterior wall thickness (Figure 3C), heart weight to tibia length ratio (Figure 3D), and cardiomyocyte cross-sectional area (Figure 3E and F). Conversely, mice that had previous DOX treatment displayed none of these compensatory cardiac adaptions to elevated blood pressure (Figure 3C–F). In accordance with the inability of hearts from DOX-treated mice to adapt to elevated afterloads, there was a decrease in stroke volume (Figure 3G) and cardiac output (Figure 3H) in DOX-treated mice following 2 weeks of Ang II infusion compared to controls. Interestingly, although a decrease in stroke volume and cardiac output may be related to systolic and/or diastolic dysfunction, we found no impairment in % ejection fraction, fractional shortening or E/A ratio in DOX-treated mice following 2 weeks of Ang II infusion (see Supplementary material online, Table S1). However, after 2 weeks of Ang II infusion, DOX-treated mice displayed a reduced LV internal diameter in diastole that resulted in a reduced LV volume at end-diastole (see Supplementary material online, Table S1). These results suggest that the reduction in LV volume and LV internal diameter in diastole, stroke volume, and cardiac output in DOX-treated mice following 2 weeks of Ang II infusion was mainly due to a failure to develop compensatory hypertrophy, resulting in smaller hearts in DOX-treated mice. Of importance, this inability of the DOX-treated mice to adapt to elevated blood pressure was further demonstrated with the high dose of Ang II administration, which increases blood pressure more dramatically,23 and resulted in significant morbidity and mortality of the DOX-treated mice compared to the control mice (Figure 3I). Upon necropsy, there was no obvious sign of aortic dissection or aneurysm and due to ethical issues, these experiments were not repeated in order to thoroughly examine the cause of death.
3.3 Resveratrol prevents DOX-induced hypertension and cardiac p38 activation
Since resveratrol has been shown to protect against DOX-induced cardiotoxicity in adult animals,12,24–26 we examined whether or not resveratrol co-administration with DOX could protect against the development of hypertension or the development of molecular signs of cardiotoxicity in this juvenile mouse model. To do this, 4-week-old mice were placed on a regular chow diet or a chow diet containing 0.4% resveratrol for 4 weeks, which overlapped with the DOX treatment regimen (Figure 4A). To investigate whether or not resveratrol had any protective effects on DOX-induced hypertension, blood pressure was measured at 1 week after the last DOX injection. Although DOX administration–induced hypertension, resveratrol co-administration with DOX attenuated this increase in blood pressure (Figure 4B), demonstrating the protective effects of resveratrol on the loss of blood pressure control in DOX-treated mice. Interestingly, while DOX treatment alone elevated blood pressure in mice without resveratrol co-treatment (Figure 4B), the heart weight of DOX-treated mice was modestly, but significantly, reduced compared to controls, showing a lack of hypertrophic compensation even at young age (Figure 2A, Figure 4C). However, resveratrol co-treatment with DOX restored normal heart weight gain (Figure 4C). Since we identified earlier that p38 phosphorylation was significantly increased by DOX treatment (Figure 2G) and previous studies have shown that resveratrol can modulate p38 in the heart,27 we investigated whether or not resveratrol prevented the phosphorylation of p38 induced by juvenile DOX treatment. Interestingly, while the activation of p38 was shown as a molecular marker of cardiotoxicity, the levels of P-p38 were completely normalized by resveratrol co-administration (Figure 4D), suggesting reduced cardiotoxicity.
Figure 4.

Resveratrol co-administration prevents DOX-induced hypertension and cardiac p38 activation. A cohort of mice did not undergo the full treatment protocol and were terminated early at 8 weeks of age. (A) Scheme of study design for investigating the acute effect of resveratrol co-administration with DOX. (B) SBP (n = 5–10), (C) HW/TL (n = 7), and (D) representative immunoblots of phospho (Thr183/Tyr185)-p38 normalized to total-p38 from hearts removed from mice at 8 weeks of age (n = 7). *P < 0.05. DOX + R indicates resveratrol co-administration with DOX; P-indicates phospho-. Values are presented as mean ± SEM and analysed by two-way ANOVA with Bonferroni post hoc test.
3.4 Co-administration of resveratrol with DOX in young mice attenuates detrimental late-occurring cardiovascular changes
While we showed the acute beneficial effects of resveratrol on the pathogenesis of juvenile DOX-induced cardiotoxicity, little is known about long-term effects of resveratrol after the co-administration ended. Therefore, we investigated whether or not resveratrol co-administration with DOX to young mice protected against detrimental DOX-induced cardiovascular injury that prevented hypertension-induced cardiac remodelling later in life (Figure 5A). To determine whether resveratrol co-administration with DOX at an early age could also protect from mild hypertension later in life, mice were infused with the low dose (1.4 mg/kg/day) of Ang II after a 5-week recovery period. Interestingly, the enhanced Ang II–mediated elevation in blood pressure in DOX-treated mice was completely prevented by the much earlier resveratrol treatment (Figure 5B). Importantly, much earlier co-administration of resveratrol with DOX also improved the ability of hearts from DOX-treated mice to adapt to elevated afterloads with improvements in adaptive cardiac hypertrophy (Figure 5C and D), and remodelling (Figure 5E and F) along with almost a complete restoration of stroke volume (Figure 5G) and cardiac output (Figure 5H). Together, these data show that resveratrol treatment of young mice at the time of DOX administration has lasting protective effects that allow the DOX-treated mice to more appropriately respond to adult-onset hypertension. Since apoptosis and senescence are known to play critical roles in DOX-induced cardiotoxicity,28,29 we performed an RNAseq study to measure the downstream effectors of p53, a key regulator of both pathways.28 Using the KEGG pathway analysis, we could not identify significant differences in the expression of p53 effector genes in myocardial tissues of DOX-treated or Ang-II–treated mice (Figure 5I). These results suggest that neither DOX nor Ang II alone is sufficient to modulate the signalling pathway downstream of p53. Conversely, the combined treatment of DOX followed by Ang II was sufficient to upregulate several genes including: cyclin-dependent kinase inhibitor 1A (p21), insulin-like growth factor binding protein 3 (Igfbp3), growth arrest and DNA damage inducible gamma (Gadd45g), TP53 apoptosis effector (Perp), and tumour necrosis factor receptor superfamily member 6 (Fas). Importantly, in keeping with its beneficial physiological effects,30 resveratrol corrected the upregulation of all the genes with the exception of Fas (Figure 5I and Supplementary material online, Figure S1).
Figure 5.

Early resveratrol co-administration with DOX results in beneficial effects later in life. (A) Scheme of study design for investigating the long-term effects of resveratrol co-administration after Ang II–induced hypertension later in life. (B) SBP (n = 5–6), (C) LVPWd (n = 5–8), (D) HW/TL (n = 6–7), (E) LV internal dimension in diastole (LVDd, n = 5–8), (G) LV end-diastolic volume (LVEDV, n = 5–8), (F) stroke volume (n = 5–8), and (H) cardiac output (n = 5–8) after the low dose of Ang II. *P < 0.05 between groups indicated. Values are presented as mean ± SEM and analysed by two-way ANOVA with Bonferroni post hoc test. (I) Heat map illustrating the changes in expression of p53 effector genes by DOX alone or resveratrol co-administration with DOX at 1 week after the last DOX injection (1 week post-DOX) and after 2 weeks of continuous Ang II infusion (after Ang II). CON indicates control; D + R indicates resveratrol co-administration with DOX.
4. Discussion
Since there are very limited data about the pathophysiology of sub-clinical cardiac injury seen in long-term cancer survivors who previously received low-dose anthracycline therapy during their childhood, we studied this phenomenon using a novel juvenile DOX-induced cardiotoxicity mouse model. An important distinction of the model used herein is that we used clinically relevant low doses of DOX that resulted in no cardiac dysfunction during the time of administration and the age of the mice can be considered to be close to age of paediatric patients. This is in contrast to previous models that either administered doses of DOX that rapidly damaged the heart29 or used mice that were injected with DOX at post-natal day 5.21 Although important studies, both of these previous scenarios are not as clinically relevant as the model that we employed and thus limit the translation of the findings to paediatric cancer patients.
Using the model that we described, we show that DOX resulted in slower weight gain in DOX-exposed young mice during the period of DOX administration, which is consistent to previous reports.21,29 In addition, DOX caused a significant decrease in heart weight in young mice in a similar manner that has been observed in anthracycline-treated childhood cancer survivors.31 Intriguingly, the reduction in heart weight was observed only one week after the last DOX injection, and was absent at later time points. While the reasons for this are unknown, our findings are in agreement with an earlier study of juvenile DOX-induced cardiotoxicity.29 Although our DOX protocol did not affect cardiac function, there were signs of cardiac injury at the molecular level including a significant induction of the cardiac stress marker, ANP and a significant increase in the phosphorylation of p38 MAPK, a conserved MAPK mostly implicated in stress signalling.32 This activation of p38 MAPK is important given that activation of the p38 MAPK pathway can have several detrimental effects on the heart,33,34 which is consistent with DOX-induced cardiotoxicity. Of importance, we demonstrated that resveratrol co-administration with DOX prevented DOX-induced activation of p38 MAPK. This is in agreement with previous work showing that resveratrol attenuates p38 activation in other models of cardiac disease, such as diabetes-induced cardiac dysfunction27 and ischemia/reperfusion-induced cardiomyocytes toxicity.35 Thus, we speculate that the cardioprotective effect of resveratrol in this model may be mediated, in part via p38 MAPK suppression.
Another important observation from this study is that exposure of juvenile mice to DOX caused a significant increase in their systolic blood pressure (SBP). While previous work has suggested that DOX-induced decrease in blood pressure is secondary to DOX-induced decrease in cardiac output and the general poor health status of the experimental animals,12 this was obviously not the case in our study. In fact, given that exposure of the vascular bed of rats to DOX led to a marked blunting of the aortic response to the endothelium-dependent vasodilator, acetylcholine,36 we propose that there is a direct detrimental effect on the vasculature of juvenile mice receiving DOX, which can persist into adulthood. This is entirely consistent with the report that there was a strong association between higher anthracycline doses and endothelial dysfunction in anthracycline-treated paediatric cancer patients.37 Although we did not confirm these proposed mechanisms in the current work, we offer a valuable animal model that can be used to elucidate the mechanisms of DOX-induced increase in blood pressure in future studies. Despite not identifying the molecular mechanisms involved in disturbed blood pressure control induced by DOX, our findings are extremely important as pre-existing hypertension has been suggested to be a risk factor for anthracycline-induced cardiotoxicity in adult cancer patients22 and that hypertension significantly increases the risk of heart failure in childhood cancer survivors.8 Equally important is that co-administration of resveratrol at the time of DOX treatment could prevent this elevation of blood pressure and thus may have significant clinical utility in paediatric cancer patients receiving anthracyclines.
Given the fact that: (i) we observed no overt cardiac dysfunction in mice administered DOX in our study but did observe molecular signs of ‘sub-clinical’ injury and (ii) hypertension is a risk factor for adult onset cardiomyopathy or heart failure, we postulated that juvenile mice with latent DOX-induced cardiotoxicity would have increased susceptibility to developing an overt, hypertension-induced cardiomyopathy in adulthood. Our data show that Ang II infusion for 2 weeks failed to induce cardiac hypertrophy in adult mice pre-exposed to DOX as juveniles despite the higher magnitude of SBP elevation observed in these mice. Surprisingly, adaptive cardiac hypertrophy in response to elevated blood pressure was not observed in mice previously treated with DOX, demonstrating a disconnect between elevated blood pressure and enhanced cardiac growth in these mice. Although cardiac hypertrophy is traditionally perceived as a pathological process,38 it is still an integral part of the physiological response to elevated blood pressure. In the current study, prevention of Ang II–induced cardiac hypertrophy by previous DOX treatment was associated with lower stroke volume and cardiac output, suggesting a detrimental rather than a beneficial effect. While we studied the relative acute effects of elevated blood pressure, since hypertension is a life-long condition, the effect of longer periods of hypertension would be important to be studied in future experiments. Nevertheless, our data suggest that ‘sub-clinical’ cardiac injury may render the heart refractory to a normal physiological adaptation to elevated workloads and may be an important clinical parameter to monitor in cancer survivors. Equally important as identifying a potential new clinical endpoint for physicians to be aware of is the fact that we were also able to partially restore the ability of the heart to beneficially remodel in response to elevated blood pressure using resveratrol. Indeed, co-administration of resveratrol at the time of DOX treatment also confers long-lasting protection against DOX-induced cardiovascular toxicity even after discontinuation. Mechanistically, we demonstrated that Ang II–induced hypertension upregulated a number of p53 effector genes (p21, Perp, Igfbp3, Gadd45b, and Fas) in hearts of DOX-treated mice, suggesting senescence and/or apoptosis as plausible mechanisms of Ang II–induced unmasking of ‘sub-clinical’ DOX-induced cardiotoxicity. Importantly, co-administration of resveratrol with DOX to young mice prevented Ang II–induced upregulation in p21, Perp, Igfbp3, and Gadd45b, corroborating the known senolytic and anti-apoptotic effects of resveratrol.39
In conclusion, we have developed a new preclinical model of occult DOX-induced cardiovascular toxicity in young mice. Utilizing this model, we have shed more light into the important pathophysiologal mechanisms of DOX-induced sub-clinical cardiotoxicity commonly detected in childhood cancer survivors who received low-dose anthracycline therapy. In addition, we demonstrated that juvenile exposure to DOX makes mice more susceptible to the detrimental effects of Ang II–induced hypertension later in life. Importantly, we showed that co-administration of resveratrol with DOX protected the young mice from occult cardiotoxicity, and conferred a long-lasting protection against the detrimental effects of Ang II–induced hypertension in adult mice pre-exposed to DOX as juveniles. Based on this, we suggest that resveratrol could be attempted in clinical trials involving paediatric cancer patients receiving low-dose anthracyclines. Given this suggestion, it is important to point out that previous preclinical work has shown that resveratrol can also augment the chemotherapeutic effect of DOX.40–42 Thus, resveratrol may hold great promise as an adjunct therapy to anthracycline chemotherapy where it protects the heart while also improving cancer treatment.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors would like to thank Suresh C. Bairwa for his technical assistance.
Conflict of interest: none declared.
Funding
Canadian Institute of Health Research (CIHR) [MOP119336 to J.R.B.D.]; the Women & Children’s Health Research Institute (WCHRI) through the generous support of Stollery Children's Hospital Foundation (SCHF) and a Hair Massacure/WCHRI Grant to J.R.B.D. National Center for Advancing Translational Sciences of the National Institutes of Health [UL1TR000114 to B.N.Z]; and Rally Foundation for Childhood Cancer Research [508200 to B.N.Z.]. I.M.R was supported by postdoctoral fellowships from CIHR and Alberta Innovates Health Solutions (AIHS). A.B. was supported by the Fesler-Lampert Chair in Aging Studies, Center on Aging, University of Minnesota.
Footnotes
Time for primary review: 44 days
References
- 1. Lipshultz SE, Diamond MB, Franco VI, Aggarwal S, Leger K, Santos MV, Sallan SE, Chow EJ.. Managing chemotherapy-related cardiotoxicity in survivors of childhood cancers. Pediatr Drugs 2014; 16:373–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lipshultz SE, Sambatakos P, Maguire M, Karnik R, Ross SW, Franco VI, Miller TL.. Cardiotoxicity and cardioprotection in childhood cancer. Acta Haematol 2014; 132:391–399. [DOI] [PubMed] [Google Scholar]
- 3. Blaauwbroek R, Stant AD, Groenier KH, Kamps WA, Meyboom B, Postma A.. Health-related quality of life and adverse late effects in adult (very) long-term childhood cancer survivors. Eur J Cancer 2007; 43:122–130. [DOI] [PubMed] [Google Scholar]
- 4. Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, Friedman DL, Marina N, Hobbie W, Kadan-Lottick NS, Schwartz CL, Leisenring W, Robison LL, Childhood Cancer Survivor S.. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 2006; 355:1572–1582. [DOI] [PubMed] [Google Scholar]
- 5. Nysom K, Holm K, Lipsitz SR, Mone SM, Colan SD, Orav EJ, Sallan SE, Olsen JH, Hertz H, Jacobsen JR, Lipshultz SE.. Relationship between cumulative anthracycline dose and late cardiotoxicity in childhood acute lymphoblastic leukemia. J Clin Oncol 1998; 16:545–550. [DOI] [PubMed] [Google Scholar]
- 6. Leger K, Slone T, Lemler M, Leonard D, Cochran C, Bowman WP, Bashore L, Winick N.. Subclinical cardiotoxicity in childhood cancer survivors exposed to very low dose anthracycline therapy. Pediatr Blood Cancer 2015; 62:123–127. [DOI] [PubMed] [Google Scholar]
- 7. Vandecruys E, Mondelaers V, De Wolf D, Benoit Y, Suys B.. Late cardiotoxicity after low dose of anthracycline therapy for acute lymphoblastic leukemia in childhood. J Cancer Surviv 2012; 6:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Armstrong GT, Oeffinger KC, Chen Y, Kawashima T, Yasui Y, Leisenring W, Stovall M, Chow EJ, Sklar CA, Mulrooney DA, Mertens AC, Border W, Durand JB, Robison LL, Meacham LR.. Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J Clin Oncol 2013; 31:3673–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ewer MS, Ewer SM.. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol 2010; 7:564–575. [DOI] [PubMed] [Google Scholar]
- 10. Dolinsky VW, Chakrabarti S, Pereira TJ, Oka T, Levasseur J, Beker D, Zordoky BN, Morton JS, Nagendran J, Lopaschuk GD, Davidge ST, Dyck JR.. Resveratrol prevents hypertension and cardiac hypertrophy in hypertensive rats and mice. Biochim Biophys Acta 2013; 1832:1723–1733. [DOI] [PubMed] [Google Scholar]
- 11. Matsumura N, Robertson IM, Hamza SM, Soltys CM, Sung MM, Masson G, Beker DL, Dyck JR.. A novel complex I inhibitor protects against hypertension-induced left ventricular hypertrophy. Am J Physiol Heart Circ Physiol 2017; 312:H561–H570. [DOI] [PubMed] [Google Scholar]
- 12. Dolinsky VW, Rogan KJ, Sung MM, Zordoky BN, Haykowsky MJ, Young ME, Jones LW, Dyck JR.. Both aerobic exercise and resveratrol supplementation attenuate doxorubicin-induced cardiac injury in mice. Am J Physiol Endocrinol Metab 2013; 305:E243–E253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Razzoli M, Karsten C, Yoder JM, Bartolomucci A, Engeland WC.. Chronic subordination stress phase advances adrenal and anterior pituitary clock gene rhythms. Am J Physiol Regul Integr Comp Physiol 2014; 307:R198–R205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kienesberger PC, Pulinilkunnil T, Sung MM, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR.. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 2012; 32:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grant MK, Seelig DM, Sharkey LC, Zordoky BN.. Sex-dependent alteration of cardiac cytochrome P450 gene expression by doxorubicin in C57Bl/6 mice. Biol Sex Differ 2017; 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M.. KEGG: kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 1999; 27:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ.. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 2009; 119:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J, Mao W, Ding B, Liang CS.. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am J Physiol Heart Circ Physiol 2008; 295:H1956–H1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang WT, Li J, Haung HH, Liu H, Han M, Ramachandran S, Li CQ, Sharp WW, Hamann KJ, Yuan CS, Hoek TL, Shao ZH.. Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J Cell Biochem 2011; 112:2873–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thandavarayan RA, Watanabe K, Sari FR, Ma M, Lakshmanan AP, Giridharan VV, Gurusamy N, Nishida H, Konishi T, Zhang S, Muslin AJ, Kodama M, Aizawa Y.. Modulation of doxorubicin-induced cardiac dysfunction in dominant-negative p38alpha mitogen-activated protein kinase mice. Free Radic Biol Med 2010; 49:1422–1431. [DOI] [PubMed] [Google Scholar]
- 21. Huang C, Zhang X, Ramil JM, Rikka S, Kim L, Lee Y, Gude NA, Thistlethwaite PA, Sussman MA, Gottlieb RA, Gustafsson AB.. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 2010; 121:675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szmit S, Jurczak W, Zaucha JM, Drozd-Sokołowska J, Spychałowicz W, Joks M, Długosz-Danecka M, Torbicki A.. Pre-existing arterial hypertension as a risk factor for early left ventricular systolic dysfunction following (R)-CHOP chemotherapy in patients with lymphoma. J Am Soc Hypertens 2014; 8:791–799. [DOI] [PubMed] [Google Scholar]
- 23. Rahman M, Kimura S, Nishiyama A, Hitomi H, Zhang G, Abe Y.. Angiotensin II stimulates superoxide production via both angiotensin AT1A and AT1B receptors in mouse aorta and heart. Eur J Pharmacol 2004; 485:243–249. [DOI] [PubMed] [Google Scholar]
- 24. Al-Harthi SE, Alarabi OM, Ramadan WS, Alaama MN, Al-Kreathy HM, Damanhouri ZA, Khan LM, Osman AM.. Amelioration of doxorubicininduced cardiotoxicity by resveratrol. Mol Med Rep 2014; 10:1455–1460. [DOI] [PubMed] [Google Scholar]
- 25. Gu J, Song ZP, Gui DM, Hu W, Chen YG, Zhang DD.. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in lymphoma nude mice by heme oxygenase-1 induction. Cardiovasc Toxicol 2012; 12:341–349. [DOI] [PubMed] [Google Scholar]
- 26. Tatlidede E, Şehirli Ö, Velioğlu-Öğünç A, Çetinel Ş, Yeğen BÇ, Yarat A, Süleymanoğlu S, Şener G.. Resveratrol treatment protects against doxorubicin-induced cardiotoxicity by alleviating oxidative damage. Free Radic Res 2009; 43:195–205. [DOI] [PubMed] [Google Scholar]
- 27. Gao Y, Kang L, Li C, Wang X, Sun C, Li Q, Liu R, Wang J.. Resveratrol ameliorates diabetes-induced cardiac dysfunction through AT1R-ERK/p38 MAPK signaling pathway. Cardiovasc Toxicol 2016; 16:130–137. [DOI] [PubMed] [Google Scholar]
- 28. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IWF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, de Keizer PLJ.. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017;169:132–147 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu W, Shou W, Payne RM, Caldwell R, Field LJ.. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr Res 2008; 64:488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zordoky BNM, Robertson IM, Dyck JRB.. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Bba-Mol Basis Dis 2015; 1852:1155–1177. [DOI] [PubMed] [Google Scholar]
- 31. Lipshultz SE, Lipsitz SR, Sallan SE, Simbre VC 2nd, Shaikh SL, Mone SM, Gelber RD, Colan SD.. Long-term enalapril therapy for left ventricular dysfunction in doxorubicin-treated survivors of childhood cancer. J Clin Oncol 2002;20:4517–4522. [DOI] [PubMed] [Google Scholar]
- 32. Ono K, Han J.. The p38 signal transduction pathway: activation and function. Cell Signal 2000; 12:1–13. [DOI] [PubMed] [Google Scholar]
- 33. Streicher JM, Ren S, Herschman H, Wang Y.. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res 2010; 106:1434–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J, Wang Y.. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation 2005; 111:2494–2502. [DOI] [PubMed] [Google Scholar]
- 35. Hayward R, Lien CY, Jensen BT, Hydock DS, Schneider CM.. Exercise training mitigates anthracycline-induced chronic cardiotoxicity in a juvenile rat model. Pediatr Blood Cancer 2012; 59:149–154. [DOI] [PubMed] [Google Scholar]
- 36. Ulu N, Buikema H, Gilst W. H V, Navis G.. Vascular dysfunction in adriamycin nephrosis: different effects of adriamycin exposure and nephrosis. Nephrol Dial Transplant 2008; 23:1854–1860. [DOI] [PubMed] [Google Scholar]
- 37. Okur A, Karadeniz C, Özhan Oktar S, Pınarlı FG, Aral A, Oğuz A.. Assessment of brachial artery reactivity, carotid intima-media thickness, and adhesion molecules in pediatric solid tumor patients treated with anthracyclines. Pediatr Hematol Oncol 2016; 33:178–185. [DOI] [PubMed] [Google Scholar]
- 38. Frey N, Olson EN.. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 2003; 65:45–79. [DOI] [PubMed] [Google Scholar]
- 39. Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Rudd JA, Siu PM.. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J Physiol 2014; 592:2535–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB.. Chemosensitization of tumors by resveratrol. Ann N Y Acad Sci 2011; 1215:150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khaleel SA, Al-Abd AM, Ali AA, Abdel-Naim AB.. Didox and resveratrol sensitize colorectal cancer cells to doxorubicin via activating apoptosis and ameliorating P-glycoprotein activity. Sci Rep 2016; 6:36855.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rai G, Mishra S, Suman S, Shukla Y.. Resveratrol improves the anticancer effects of doxorubicin in vitro and in vivo models: a mechanistic insight. Phytomedicine 2016; 23:233–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.