

Abstract
Dilated cardiomyopathy (DCM) frequently affects relatively young, economically, and socially active adults, and is an important cause of heart failure and transplantation. DCM is a complex disease and its pathological architecture encounters many genetic determinants interacting with environmental factors. The old perspective that every pathogenic gene mutation would lead to a diseased heart, is now being replaced by the novel observation that the phenotype depends not only on the penetrance—malignancy of the mutated gene—but also on epigenetics, age, toxic factors, pregnancy, and a diversity of acquired diseases. This review discusses how gene mutations will result in mutation-specific molecular alterations in the heart including increased mitochondrial oxidation (sarcomeric gene e.g. TTN), decreased calcium sensitivity (sarcomeric genes), fibrosis (e.g. LMNA and TTN), or inflammation. Therefore, getting a complete picture of the DCM patient will include genomic data, molecular assessment by preference from cardiac samples, stratification according to co-morbidities, and phenotypic description. Those data will help to better guide the heart failure and anti-arrhythmic treatment, predict response to therapy, develop novel siRNA-based gene silencing for malignant gene mutations, or intervene with mutation-specific altered gene pathways in the heart.
This article is part of the Mini Review Series from the Varenna 2017 meeting of the Working Group of Myocardial Function of the European Society of Cardiology.
Keywords: Dilated Cardiomyopathy, Genetics, Genome-environment interaction
1. Dilated cardiomyopathy: an overlapping phenotype for multiple conditions
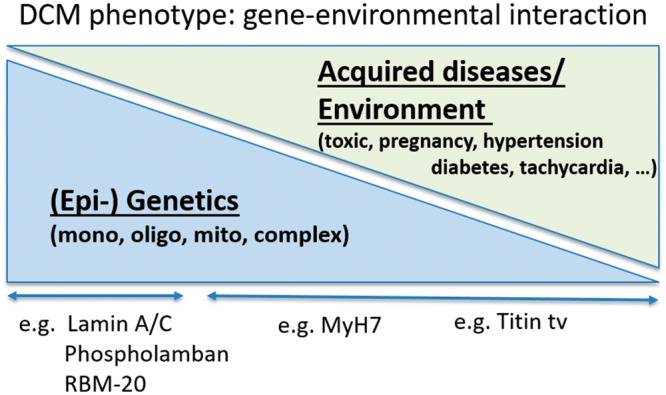
Dilated cardiomyopathy (DCM) encompasses aetiologically heterogeneous myocardial disorders that are defined by the presence of left ventricular (LV) or bi-ventricular dilation and depressed myocardial performance, in the absence of overloading (hypertension, valvular, or congenital heart disease), or chronic ischaemic conditions.1 Interactions between the genetic and non-genetic factors may influence the severity and the outcome of DCM cases, including heart failure, rhythmic disorders, stroke, and the need for cardiac transplantation.1 The early identification of the causative gene mutation or the acquired cause is an important step to implement pre-symptomatic interventions, as some gene mutations, e.g. LAMIN A/C or RBM20, are highly penetrant and can be pro-arrhythmogenic.1,2 In this review, we describe the most advanced scientific achievements on genetic and cellular defects, diagnostic assessment, and future therapeutic perspectives for this heterogeneous disease.
Modern epidemiological studies considering disease heterogeneity and variable clinical presentation are lacking regarding DCM, and it is now recognized that DCM prevalence was underestimated in the initial population studies.3–5 Based on heart failure prevalence, considering LV dysfunction as a surrogate for DCM, and extrapolating data from hypertrophic cardiomyopathy (HCM), it is now admitted that DCM could be as prevalent as HCM and reach 1/250–500 in clinical practice.2 Also, in the European registry on cardiomyopathies, the DCM proportion was unexpectedly high (almost 40% of all cardiomyopathies) supporting that DCM prevalence in Europe could be close to the one of HCM.6 Recent and reliable population data are required to strengthen this statement. Overall, the recognition of a complex architecture for DCM not only in terms of gene-environment interaction, but also regarding its clinical presentation (with early stages of the disease being now recognized and defined by the presence of hypokinetic and non-DCM, preceding overt dilatation, and heart failure) has made of DCM the most common cardiomyopathy.2,7
A positive familial history can be detected in up to 30–50% of DCM cases,7 and a genetic determinant can be identified following a screen of more than 40 genes in up to 40% of DCM cases.1,2,8 Sarcomeric, mitochondrial, and neuromuscular disorders are frequent aetiologies in the presence of a ‘familial’ history, where additional acquired conditions like exposure to toxics, diabetes, arrhythmia, myocarditis, and pregnancy also contribute to the phenotype and outcome. Often, genetic determinants increase the susceptibility or are modifying factors in the presence of an external cause for DCM. The overlap between a genetic disorder and acquired disease becomes more and more ambiguous (Figures 1 and 2), raising complexity in the assessment of these patients, but also opening new avenues in understanding and treating this aetiologically heterogeneous disease.
Figure 1.
Gene-environmental interaction in dilated cardiomyopathies. DCM is a complex multi-factorial disease related to genetic determinants interfering with environmental factors.
Figure 2.

Interplay between genes and environmental factors in cardiomyopathies. Heart cartoon is reproduced from Wilde et al.9 with permission from Nature Publishing Group.
2. The tunnel perspective: DCM seen as an inherited cardiac condition
Familial DCM is genetically heterogeneous and generally inherited as a monogenic trait transmitted in a dominant negative fashion, with incomplete penetrance (Table 1).1,2,10,11 Autosomal recessive, X-linked inheritance and mitochondrial inheritance are less frequent.1,2,10,11
Table 1.
Main genes in DCM, isolated, and syndromic
| Gene symbol | Inheritance | Cardiomyopathy overlap | Clinical characteristics |
|---|---|---|---|
| Cytoskeletal | |||
| ACTN2 | AD | HCM | |
| ANKRD1 | AD | HCM | |
| CSRP3 | AD | HCM | |
| LDB3 | AD | HCM, LVNC | Myofibrillar myopathy Type 4 |
| MYPN | AD, AR | HCM, RCM | Frequent overlapping phenotypes, Nemaline myopathy type 11 (AR) |
| TCAP | AD, AR | HCM | Limb-girdle muscular dystrophy 2G (AR) |
| TTN | AD, AR | HCM, RCM, ARVC | Most frequent, early onset myopathy, peripartum cardiomyopathy, truncated variants with rhythm disorders, and limb-girdle muscular dystrophy 2J (AR) |
| VCL | AD | HCM | |
| Sarcomeric | |||
| ACTC1 | AD | HCM, RCM, LVNC | Atrial septal defect |
| MYBPC3 | AD | HCM, RCM, LVNC | |
| MYH7 | AD, AR | HCM, LVNC | Peripheral myopathy |
| TNNC1 | AD | HCM | |
| TNNI3 | AD, AR | RCM, HCM | |
| TNNT2 | AD | RCM, HCM, LVNC | |
| TPM1 | AD | HCM, LVNC | |
| Conduction disease | |||
| DES | AD, AR | HCM, RCM | DES deposition, Limb-girdle muscular dystrophy 2R (AR), and myofibrillar myopathy (AR/AD) |
| LMNA | AD, AR | HCM | Congenital muscular dystrophy, Emery-Dreifuss muscular dystrophy, and limb-girdle muscular dystrophy 1B |
| SCN5A | AD, AR | ARVC | Brugada syndrome, sick sinus syndrome (AR), LQT3, atrial fibrillation, ventricular fibrillation, heart block |
| Arrhythmia | |||
| DSC2 | AD, AR | ARVC | Mild palmoplantar keratoderma and woolly hair |
| DSG2 | AD | ARVC | |
| DSP | AD, AR | ARVC | Epidermolysis bullosa, keratodermia, woolly hairs |
| FLNC | AD | HCM, RCM, ARVC | Rhythmic disorders unrelated to LV dysfunction, myofibrillar myopathy 5 |
| PLN | AD | HCM | |
| PKP2 | AD | ARVC | |
| RBM20 | AD | Atrial fibrillation | |
| RYR2 | AD | ARVC | Catecholaminergic polymorphic ventricular tachycardia |
| TTN truncated | AD | HCM, RCM, ARVC | |
| Mitochondrial | |||
| CTF1 | AD, AR | ||
| DNAJC19 | AR | 3-methylglutaconic aciduria, type V, syndromic (DCMA) recurring in some populations (Canadian Hutterite population) | |
| MT-TL1 | Mito | HCM→ DCM-like evolution | MELAS |
| SDHA | AD, AR, Mito | Leigh syndrome (AR/Mito) and mitochondrial respiratory chain complex II deficiency (AR) | |
| Metabolic/Neuromuscular | |||
| BAG3 | AD | RCM | Progressive myofibrillar myopathy |
| CPT2 | AR, AD | Carnitine palmitoyltranferase III deficiency | |
| CRYAB | AD, AR | RCM | Myofibrillar myopathy 2 (AR) |
| DMD | XLR | Duchenne/Becker muscular dystrophy | |
| DOLK | AR | ||
| EMD | XLR | Emery-Dreifuss muscular dystrophy | |
| HADHA | AR | HCM | AR syndromic phenotypes with cardiomyopathy |
| HFE | AR | HCM, RCM | Iron Overload |
| LAMP2 | XLD | HCM | Danon Disease |
| SGCD | AD, AR | Limb-girdle muscular dystrophy (delta-sarcoglycanopathy) | |
| SLC22A5 | AR | HCM | Carnitine deficiency |
| SYNE1 | AD, AR | Emery-Dreifuss muscular dystrophy (AD), Spinocerebellar ataxia (AR) | |
| TAZ | XLR | Endocardial fibroelastosis, BARTH Syndrome | |
| TMPO | AD | Reclassified as non-pathogenic | |
| Others | |||
| EYA4 | AD | Non-syndromic hearing loss and deafness | |
| GATAD1 | AR | ||
| NEXN | AD | HCM | |
| PSEN1 | AD | Familial Alzheimer disease | |
| PSEN2 | AD | Familial Alzheimer disease | |
Genes presenting a statistically significant excess burden over controls in ExAC are depicted in bold.12 Figure content is adapted from Burke et al.10 and Lee et al.11
AD, autosomal dominant; AR, autosomal recessive; ARVC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular non-compaction; Mito, mitochondrial; RCM, restrictive cardiomyopathy; XLD, X-linked dominant; XLR, X-linked recessive.
Autosomal dominant forms of DCM are linked to variants in more than 40 genes encoding proteins with functions in cardiac muscle contraction and relaxation (Table 1). Although those variants have been associated with DCM, the pathogenic role in DCM remains uncertain for many of them.12
Mutations in the TITIN (TTN) gene coding for titin, the giant protein functioning as a nano-spring, is perceived as the most common cause for DCM, although with incomplete penetrance and variable, often milder phenotypes.13,14 Importantly, TTN-truncating variants in the presence of acquired diseases may be related to more ventricular arrhythmias in two studies,15,16 enhanced cardiac fibrosis,15 reduced hypertrophy,15 and pronounced alterations in cardiac mitochondrial function.15 Analysis in other cohorts is required to verify and better understand those novel phenotyping findings.
RBM20, the regulator of TTN splicing, has also been described as a cause of familial DCM with approximate frequency of 2–3%.17,18 Outcome of RBM20 mutation carriers may be worse, with higher frequency of atrial fibrillation and progressive heart failure. Also, mutations in the LMNA gene (lamin A/C),19–22 in FLNC (filamin C),23DES (desmin),24,25PLN (phospholamban),26–28 and SCN5A1,29 have been identified as a malignant cause of DCM. These mutations are associated with a typical time-course of rhythm disturbances ranging from atrioventricular blocks (LMNA, DES, and SCN5A) to supraventricular and ventricular malignant arrhythmias (LMNA, FLNC, PLN, and SCN5A). Importantly, ventricular arrhythmias can precede systolic dysfunction conferring a risk of sudden cardiac death irrespective of LV dysfunction.1,19,30,31 This implicates the use of genetic information in sudden cardiac death risk estimation, and careful stratification of arrhythmogenic risk in DCM patients. In the presence of easy to assess clinical factors, prophylactic defibrillator implantation should be considered for these patients.32,33 In summary, DCM patients with mutations in LMNA, PLN, RBM20, FLNC, DES, or SCN5A are at risk for worse prognosis and/or a higher rate of malignant arrhythmia, the latter being the case in TTN-truncated variants,15,34,35 and creating an overlap with arrhythmogenic cardiomyopathy.
In DCM with recessive inheritance, the mutated genes encode the proteins cardiac troponin I3 (TNNI3), titin (TTN), desmoplakin (DSP), dolichol kinase (DOLK), GATA zinc finger domain-containing protein 1 (GATAD1), and flavoprotein (SDHA). X-linked diseases associated with DCM include neuromuscular disorders (such as Becker and Duchenne muscular dystrophies). DCM occurs in both patients with mitochondrial diseases and inherited storage disorders (such as haemochromatosis) as the end-phenotype of early HCM.7,36,37 Mitochondrial cardiomyopathies demonstrate clinical and genetic heterogeneity, with variable syndromic manifestations.
Primary DCM patients with a positive family history for cardiomyopathies or with distinct hints for a genetic cause of the disease (red flags, e.g. conduction disease) should be offered the possibility of genetic testing and counselling.7,32,38,39
2.1 Sarcomeric mutations and DCM: from the contractile apparatus to the dilated phenotype
The sarcomere plays a central role in cardiomyocyte function and metabolism and the molecular link between sarcomeric mutations and the dilated phenotype has increasingly been elucidated. Mutations in beta-myosin heavy chain (MYH7) as well as in alpha-myosin heavy chain (MYH6) may cause familial HCM and DCM, with sometimes features of restrictive cardiomyopathy (RCM).40–43 Thin-filament mutations have highlighted a key role for calcium-dependent tension in the disease phenotype: these mutations are only a small fraction (∼5–10%) of all HCM mutations and often cause an increase in myofilament calcium sensitivity, whereas thin-filament mutations associated with DCM frequently show reduced calcium sensitivity of myofilament force development (Figure 3).44 Hence, identification of the thin-filament sarcomeric mutation, which causes the early contractile alteration, would predict the development of hypertrophic vs. DCM. Importantly, sarcomeric gene mutations may disrupt the highly tuned balance between mechanical force generation and Ca2+-cycling dynamics. A recent study tried to differentiate thin-filament mutations that alter calcium-dependent tension (increase or decrease) into the spectrum of hypertrophic vs. DCM.44 Altering myofilament Ca2+ sensitivity in mice with specific TNNC1 gene variants resulted in the activation of both calcineurin and MEK1-ERK1/2 signalling in the presence of a variant, which increased calcium-sensitivity, resulting in concentric hypertrophy (Figure 3).44 In contrast, inhibition of MEK1-ERK occurred in the presence of a DCM-related variant, which reduced calcium-sensitivity and resulted in cell elongation, suggesting that the combination of calcineurin activation and MEK1-ERK inhibition underlies the eccentric (dilated) remodelling. High-resolution structural information about multi-protein may help to characterize the baseline function and structure of the Tropomyosin overlap region of the thin filament, as mutations with differential phenotypes exert opposite effects on the Tropomyosin − Troponin overlap.45
Figure 3.

Cardiomyopathies: from altered calcium sensitivity to disease phenotype. Altered calcium sensitivity related to sarcomeric mutations lead to either hypertrophic cardiomyopathy (increased calcium sensitivity/tension with lower free cytosolic calcium) or DCM (decreased calcium sensitivity/tension with increased free cytosolic calcium).44 Heart cartoons are reproduced from Wilde et al.9 with permission from Nature Publishing Group.
Increased myofilament Ca2+ sensitivity and improved cardiac contractility is also seen in a tropomyosin (TM-E54K) mutant mouse treated with a β-arrestin 2-biased ligand of the angiotensin II receptor.46 The increase in myofilament Ca2+ sensitivity resulted from enhanced phosphorylation of myosin light chain-2.46
The open question is how different defects in different proteins may cause a similar end-phenotype. Recent studies in human DCM samples demonstrated that a sarcomeric TNNI3 (TROPONIN I) mutation impaired length-dependent activation (i.e. Frank–Starling mechanism), and a TNNT2 (TROPONIN T) mutation increased passive stiffness, while a non-sarcomeric LMNA mutation did not cause direct sarcomere changes, but was associated with reduced force generation due to the disease-related cellular hypertrophy and reduced myofibril density.47 A basic integrated-tension index could be used to differentiate DCM from HCM and, as a consequence, to understand the phenotype and to choose the best pharmacological treatment.44
2.2 DCM, arrhythmogenic cardiomyopathy, and channelopathies
The electrical instability observed in some DCM cases creates a clinical overlap between DCM and arrhythmogenic cardiomyopathy and channelopathies. The above-mentioned LMNA mutations are associated with high-risk malignant ventricular arrhythmias, regardless of the severity of LV dysfunction and dilatation.39,48 Other genes linked to arrhythmogenic phenotypes in DCM are the gene encoding for filamin C (FLNC),23 RBM20,17,18 phospholamban (PLN),26,27 and more recently TTN-truncating variants.15,16 Also, mutations in genes for desmosomal proteins, which are usually associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) have now emerged as a cause of DCM with a propensity to ventricular arrhythmia too.49–52
Importantly, DCM may be caused also by genes encoding for/or modulating ion channels (i.e. PLN, RYR2, and SCN5A).39 Indeed, mutations involving sodium-, calcium-, and potassium-related proteins support an alternative disease mechanism of dilation primarily triggered by dysfunction in electrical excitability rather than a structural defect.1,28,53 In particular, in familial DCM, missense mutations in SCN5A, that are also linked to long QT and Brugada syndromes, carry a higher risk for arrhythmias.3 The different roles of SCN5A in the myocardium and the conduction system produce a combination of arrhythmia and cardiomyocyte dysfunction, as witnessed by transgenic-directed inducible expression of the F1759A mutation in the Scn5A gene that leads to atrial fibrillation and persistent sodium currents in atria and ventricles and reduced ejection fraction.1,54
PLN mutations lead to variable phenotype with early onset DCM.26,55 In order to assess mechanisms and impact of these mutations, induced pluripotent stem cells (iPSCs) with the R14del PLN mutation were generated.56 Aberrant Ca2+ handling after caffeine and abnormal Ca2+ transients, which could be reversed upon correction of the primary mutation by gene editing could be described in these cells,56 whereas reduced developed force that could be improved after genetic correction was observed in three-dimensional human cardiac tissue.57
Although those variants have been associated with DCM, their pathogenic role in DCM has to be reconsidered, with many of them being represented as much in large population controls than in DCM cases.12
2.3 Possible genetic basis and mechanisms of tachy-induced cardiomyopathy
The development of cardiomyopathy due to arrhythmia may take months to years, but recurrent arrhythmia can lead to LV dysfunction and heart failure.58 Tachy-induced cardiomyopathy can also be associated with genetic factors. Serum and glucocorticoid-regulated kinase 1 (SGK1) is a component of the cardiac phosphatidylinositol three-kinase signalling pathway that has proarrhythmic effects linked to biochemical and functional changes in the Na+ channel. These effects can be reversed by the late Na+ current inhibitor, ranolazine.59 Accordingly, inhibition of SGK1 in the heart protects against fibrosis, LV dysfunction, and Na+ channel alterations after haemodynamic stress.59
Whether tachyarrhythmia unmasks a latent cardiomyopathy, or whether genetic factors can induce both arrhythmia and DCM is still under debate. Recent advances in our understanding of channelopathies and the co-existence of arrhythmic and dilated phenotypes in some patients seems to foster this theory.29,60,61 Mutations in the cardiac Na+ channel and in the ryanodine receptor can indeed lead to arrhythmia, but also to LV dysfunction.58,61
2.4 Mitochondrial dysfunction in the complex road to dilated cardiomyopathies
Mitochondrial cardiomyopathy is a part of a heterogeneous group of multi-systemic diseases that develop as a consequence to mutations in nuclear or mitochondrial genes.62 Inheritance follows matrilineal rule for mtDNA mutations and Mendelian rules for nuclear gene defects.63 The cardiac phenotype is characterized by LV hypertrophy that commonly evolves through LV dilation and dysfunction.64 A typical example of DCM associated with nuclear gene defects is Barth Syndrome (BTHS), an X-linked recessive disorder characterized by LV non-compaction (LVNC) cardiomyopathy, skeletal myopathy, neutropenia, growth retardation, and 3-methylglutaconic acidurea.65 BTHS is caused by mutations in the TAFAZZIN (TAZ) gene that lead to a defective phospholipid transacylase, termed tafazzin. Defective tafazzin activity leads to alterations in the content and composition of cardiolipin and the appearance of monolysocardiolipin.66,67 mtDNA defects may range from large rearrangements (deletions) of the mtDNA to point mutations,68–70 that prevalently affect oxydative phosphorylation (OXPHOS) complexes. Moreover, a defective mtDNA repair system is evident in DCM human hearts, which is accompanied by activation of PGC-1α, abnormal mitogenesis, and increase in mtDNA deletion mutations.71 This maladaptive compensatory response further contributes to the progression of the disease.
mtDNA alterations are common in human populations72 and often lead to the replacement of amino acid residues without pathological significance. When affecting evolutionarily conserved residues, mitochondrial protein synthesis, and specific respiratory enzyme activities mtDNA mutations can have pathogenic significance.70 Defects in the mechanism of mtDNA repair can promote accumulation of mtDNA defects with wide variability in the age of onset and severity of the phenotype.73 It is also needed to consider that nuclear genes, which further increase the complexity of the phenotypic effects of the gene-to-gene interactions, control mechanisms of mtDNA repair. Genetic mutations disturbing mitochondria dynamics are responsible for neurodegenerative conditions and cardiac failure.74,75 Thus, defects involving the complex machinery of mitochondria repair/replacement can promote mtDNA defects and facilitate the occurrence of cardiac dysfunction. These considerations might be relevant in specific environmental contexts, for example in presence chronic exposure to the very low dose of ionizing radiation for occupational or health purpose (Figure 4).76,77 Epigenetic or inherited defects in mitochondria dynamic and/or function, in particular, those with low penetrance and expressivity can lead to a cardiac phenotype when interacting with a specific environment, which may explain the sizeable phenotypic variability of mitochondrial cardiomyopathy.
Figure 4.

The interaction between DNA defects and failing of mitochondrial repair systems promotes the genesis of mitochondrial DCM. The exposure of cardiomyocytes to ionizing radiation (IR), antiblastic drugs, alcohol metabolites, and many other sources of DNA damage leads to mutations in both mitochondrial and nuclear genes coding for mitochondrial proteins. When the number of gene defects overcomes cellular tolerance, mitochondria can recruit a complex pathway of self-repair and regeneration. As the above mechanisms fail, defects tend to accumulate further, leading at the end to mitochondrial dysfunction.
2.5 DCM and primary restrictive cardiomyopathy
A broad overlap exists between cardiomyopathies, and for some genes clinical presentation ranges from DCM to HCM, arrhythmogenic, or RCM, with sometimes features of non-compaction. RCM is a rare disorder characterized by increased cardiac filling pressure related to enhanced wall stiffness, leading to severe atrial dilatation, despite a limited increase in cardiac chamber dimensions and wall thickness.78,79 RCM is seen in primary myocardial disease, in infiltrative disorders, or can be associated with autoimmune diseases or metabolic disorders.78,79
Sarcomeric proteins play a central role in controlling myocardial tension and relaxation, and calcium-sensitivity is a key player in controlling this equilibrium (Section 2.1).44 In a series of 1226 HCM patients, 2, 3% had a restrictive phenotype, with one half of the RCM probands having mutations in MYH7 or TNNI3 genes.43 Underlying this phenotypical overlap, previous linkage analysis have revealed that TNNI3 mutations, a gene involved in HCM and DCM, are also associated with RCM,80 and further analysis confirmed the role of TNNI3 in RCM.81,82 Cardiomyocytes of cTnI R193H mice showed shortened cell length and impaired relaxation.83 Multiple phenotypes ranging from RCM, HCM, and DCM were also observed in the same family in the presence of a I79N TNNT2 mutation.84 Reconsidering the old view that DCM presentation could be the end-stage presentation of several cardiac diseases, those results support a genetic base for overlapping phenotypes.
Another example of phenotypical overlap is given by non-sarcomeric genes. Variants in TTN, DES, FLNC, and MYOPALLADIN (MPN) are also related to DCM, HCM, and RCM.78,79 Desmin accumulation leads to clinical phenotypes affecting peripheral and/or cardiac muscles, with sometimes predominant cardiac or peripheral involvement.25 Altering cellular trafficking, cardiac desminopathy can present as DCM, HCM, or RCM with frequent involvement of the conduction system leading to AV blocks.24 Other genes linked to rhythmic disorders in DCM including FLNC, RBM20, PLN, and TTN are also associated with RCM,78,85 supporting the existence of a genetic base for this phenotypic continuum between cardiomyopathies.
Iron overload cardiomyopathy (IOC) usually presents with a myocardial concentric or asymmetric non-extreme hypertrophy with progressive LV remodelling and dysfunction.86 After an asymptomatic period, patients with in IOC present with mild heart failure symptoms, because of restrictive physiology and may mimic heart failure with preserved ejection fraction. Later, ejection fraction regresses with the presence of end-stage heart failure.87
2.6 From genes to clinical tools: the noisy background in DCM genetics
The development of population genetics databases like the Exome Aggregation Consortium (ExAC) and the Genome Aggregation Database (gnomAD) have offered an unprecedented view of rare variation in the human genome.88 These resources have revealed that the frequency of rare variation in many genes is much higher than hitherto appreciated, which has implications for variants and genes that have been previously associated with disease. A substantial proportion of variants reported as causative for DCM and other cardiomyopathies are present in these (clinically uncontrolled) population databases.89,90 Comparing the total burden of rare variation in genes associated with DCM between ExAC and large cohorts of DCM cases has revealed that a significant excess in cases is observed in only a small number of well characterized DCM genes such as TTN, LMNA, and the sarcomeric genes.12 These findings suggest that the pleiotropic effects of variants associated with cardiac disease and the genetic overlap between DCM and other inherited cardiac conditions may have been overestimated.
The lack of an overall case excess does not necessarily preclude a role in DCM. In fact, pathogenic variants can be restricted to specific residues or small domains, and therefore, masked by the background benign variation in the rest of the gene, e.g. the DCM mutation hotspot found in the titin splicing regulator RBM20.91 However, aside from founder mutations (such as the PLN R14del in 10–15% of DCM patients in the Netherlands),27 variants in genes without a case excess are likely at best to be rarely causative in DCM. In order to reduce the uncertainty associated with standard clinical genetic testing, and the danger of false positive results, panels should be restricted to genes with strong evidence of pathogenicity. Initiatives such as ClinGen have been established to curate these gene to disease associations and identify panels of validated and interpretable genes for clinical genetic testing.92 However, as shown by a recent study assessing the evidence for genes implicated in HCM,93 it is likely that genes with an excess of rare variants in case series will account for the vast majority of pathogenic variants detected in patients. This could be explained by a higher frequency of HCM, and a smaller proportion of asymptomatic mutation carriers in HCM as compared to DCM.
The extent of rare benign variation in the population is particularly prevalent for interpreting missense variants, particularly in genes that were implicated in DCM based on limited evidence and are regularly included in clinical panels (such as MYBPC3 and MYH6).12,94 Truncating variants in several different genes have now been shown to be key determinants in DCM genetics and have been validated through burden testing between cases and controls (for TTN,13,95DSP,12,94 and FLNC23) or segregation in large families (BAG396,97). Although variant interpretation guidelines produced by the American College of Medical Genetics (ACMG)98 deem a truncating variant as actionable if it is sufficiently rare and a loss of function mechanism has been proven for the affected gene in the disease being tested, such variants in novel or poorly characterized DCM genes should not be assumed to be pathogenic given that each individual is estimated to harbour at least 100 truncating variants, of which approximately 20 will be rare.88
2.7 Deciphering missing heritability and incomplete penetrance in DCM
Although restricting genetic testing to validated genes is a prudent strategy in the clinic, whole exome, or genome sequencing of large patient cohorts and family pedigrees now offers the opportunity for the discovery of novel genetic factors underlying DCM. This may include protein-coding variation in novel genes, non-coding, and regulatory variants affecting known DCM genes, and the assessment of oligogenic inheritance which has been hypothesized as possible mode of transmission in DCM, factors that may underlie the low penetrance of this disease.1,7 Also the complex interaction between common and rare variation with other environmental and medical factors may be responsible for the clinically observed heterogeneity of DCM even in families carrying the same pathogenic variant. However, even when taking all knowledge about the impact of multiple high-penetrance variants in single patients and common variants identified in genome-wide association studies (GWAS) into account,99,100 there is still a large fraction of heritability and a considerable phenotypic variability that cannot be explained.101 The deciphering of epigenetic alterations could provide new clues to this obstacle: epigenetic changes can occur due to intrinsic and environmental factors and can, in variable degree, be transmitted to the progeny. Already during cardiac development, DNA methylation of gene bodies and post-translational modifications of histones of developmental and sarcomeric genes can be detected that also occur during heart failure.102–104 In DCM, a first study was able to map the genome-wide DCM methylome and identify putative new players in its pathophysiology, such as LY75.105 In a recent study, embarking on a multi-omics design including whole-genome sequencing, transcriptome sequencing and high-density DNA methylation profiling, Meder et al.106 could delineate a significant role of DNA methylation patterns on cardiac gene expression and DCM. They further raised a new paradigm, the cross-tissue conservation of methylation alterations, rendering epigenetics a novel class of cardiac biomarkers, and potential therapeutic target.107
3. The broader view: DCM as a phenotype at the intersection between genes and acquired conditions
3.1 Inflammation in genetic cardiomyopathy, viral infections, and myocarditis: complex interactions
In DCM, the pathogenicity of a gene mutation is modulated by interfering factors: besides age and hormonal context, inflammation (i.e. virus infection, cardiac inflammation, and systemic disease), hypertension, mitochondrial alterations, and other environmental triggers (like toxic exposure) are strong candidates to modulate the expression of genetic determinants and the prognostic of the disease.13,75,76
In particular, low-grade chronic cardiac inflammation mediated by the innate immune system is often seen in genetic cardiomyopathies.108 Importantly, autoimmune diseases, viral infections, or toxic stimuli that trigger innate immunity may increase heart inflammation in a subject with a ‘genetic’ predisposition to cardiomyopathy, worsening the prognosis.109
Not unfrequently, LV dysfunction can be caused by viral, bacterial, fungal, parasitic, rickettsial, and spirotricheal infections. In viral myocarditis, parvovirus B19, adenovirus, coxsackievirus B, and other enteroviruses, influenza A, human herpes virus 6, cytomegalovirus, Epstein–Barr virus, herpes simplex virus Type 1, and hepatitis C virus are the most common viruses identified by means of endomyocardial biopsies,110,111 with coxsackievirus being predominant in the 1980s, adenovirus in the 1990s, and parvovirus B19 since 2000.112
TLRs are involved in early activation of the innate immune response against viruses and other infections. So far, specific roles in inflammatory cardiomyopathy and myocarditis have been described for TLR2, TLR3, TLR4, TLR7, and TLR9 along with their downstream adaptors MyD88 and TRIF.113–117 In particular, enteroviruses, activate TLR3 signalling: animals lacking TLR3 exhibit enhanced mortality after being infected with enteroviruses.118 Importantly, subjects affected by enteroviral myocarditis/cardiomyopathy present with a common TLR3 polymorphism.119 Upon viral infection, patients who develop myocarditis have elevated TLR4 transcripts, whereas TLR4 deficient mice had heart inflammation in CVB3 myocarditis.108
On the other way, animal studies have shown that gene mutations may trigger inflammation, independent of other acquired immune activators, but more studies are needed in order to confirm this hypothesis.108 In the presence of myocardial derangements, such as the ones that occur in DCM, inflammation enhances fibrosis in the left ventricle and promotes fibrofatty degeneration in the right ventricle.108 Parallel to this, stimulation of the immune system will lead to the appearance of circulating cardiac auto-antibodies, which are potential therapeutic targets.108,120 Also, it is very likely that myocyte hypertrophy in DCM may act on macrophages via enhanced oxidative stress, that in turn favours further myocyte hypertrophy and fibrosis, hence letting cardiac disease to progress.108
3.2 The genetic influence on development of toxic cardiomyopathies
Alcohol and anthracyclines are well-known cardio-toxic triggers sharing the ability to induce a broad spectrum of cardiac impairments, going from asymptomatic cardiac remodelling and subclinical dysfunction to severe heart failure.121–123 Recent studies highlight the potential role of genetic factors interacting with toxic triggers and potentially contributing to the inter-individual variability of cardiac phenotype.124–128
In alcoholic cardiomyopathy, alcohol metabolism and accumulation of its main metabolite acetaldehyde could contribute to the myocardial damage through alterations of mitochondria, of sarcoplasmic reticulum calcium transient and Ca2+ sensitivity of myofibrils, through increased apoptosis, alterations in lipid energetic metabolism and protein synthesis, increased oxidative stress, and activation of renin-angiotensin and sympathetic systems.121 SNPs in genes involved in ethanol metabolisms such as ADH1B (A/A), ALDH2 (A/G or A/A), and CYP2E1(T/C or T/T) increase genetic susceptibility to ethanol-induced cardiac dysfunction. In particular, fast ADH1B (A/A) induces a more rapid conversion of ethanol into acetaldehyde, whose degradation can be delayed due to the reduced enzyme activity of ALDH2 (A/G or A/A), and resulting in a toxic accumulation. CYP2E1(T/C or T/T) further worsens the impairment of ethanol metabolism inducing greater accumulation of both acetaldehyde and reactive oxygen species (ROS).124
Anthracyclines cardiotoxicity (ACT) represents the most critical dose-limiting side effect of these wide-used antineoplastic drugs. Novel mechanisms of cardio-toxicity have been recently identified and explored in patients with malignancy treated with anthracyclines. First, anthracyclines can induce dsDNA break and cell death through reticence of Topoisomerase 2-beta (Top2β).122 The SNP Ser427Leu in RARG is highly associated with ACT because this variant alters the ability to downregulate Top2β transcription, leading to its accumulation in cardiomyocytes where it is targeted by anthracyclines.125 A potential role in the ACT might be played by the reduction of anthracyclines to cardiotoxic alcohol metabolites through carbonyl reductases (CBR). Indeed, individuals with CBR3 V244M homozygous G genotypes (CBR3 G/G) show a higher cardiomyopathy risk, since this genetic variant increases synthesis of these metabolites.126 The ACT pathophysiology involves unbalancing between ROS generation and intrinsic antioxidant species. This leads to impairment of calcium homeostasis, mitochondrial bio-energetic disruption, activation of the ubiquitin-proteasome system leading to sarcomeric structure dysregulation, activation of NFκB signalling and immune system, and senescence of cardiac- and endothelial progenitor cells.122 Anthracyclines accumulation within the cardiomyocytes is the essential step to determine their oxidative potential. Accordingly, many of the other SNPs associated with higher risk of the ACT are located in genes able to influence anthracycline pharmacokinetics (SLC28A3, SLC28A1, SLC10A2, ABCB1, ABCB4, ABCC1, UGT1A).127,129 Genetic susceptibility to ACT can also affect cardiac protective mechanisms. Indeed, the rs2232228 AA genotype in HAS3 gene reduces hyaluronan synthesis leading to inadequate tissue remodelling and insufficient protection of the heart from ROS-mediated injury.130 ACT also shows coexistence of two or more TNNT2 (cTNT) splicing variants, which lead to a temporally split myofilament response to increasing Ca2+ concentrations and could decrease myocardial contractility.131 Individuals homozygous for the CELF4 rs1786814 G allele are more likely to co-express the embryonic and adult TNNT2 variants and, thus, to possibly enhance cardiotoxicity risk in response to anthracyclines.128
Recently, a further mechanism of doxorubicin-mediated cardiotoxic effects has been identified based on the down-regulation of the crucial circular RNA regulator Quaking.132 Doxorubicin leads to a significant down-regulation of Quaking in the heart, thus making it more susceptible for cardiac apoptosis. In a therapeutic approach, AAV-mediated cardiac overexpression of Quaking rescued the heart from Doxorubicin-mediated toxicity thus presenting a potential future entry point of novel cardiac anti-toxic treatments.132
Antineoplastic drugs can also cause cardiomyopathy by an interference with the innate immune system.133 In particular, toll-like-receptors (TLRs) can ‘sense’ anthracycline-mediated damage, with TLR2 KO mice showing partially preserved cardiac function and improved survival compared to WT animals in a model of acute doxorubicin administration.108,134 Other TLRs exhibit a controversial behaviour: for instance TLR4 KO mice showed protection from anthracycline-induced cardiac toxicity135; whereas anti-TLR4 antibodies had opposite effects.108,136
Of notice, the innate and adaptive immune system has a pivotal role in the recognition and elimination of tumour cells. One of the supposed mechanisms by which anthracyclines are effective in cancer is the stimulation of an anti-tumour immunity by inducing immunogenic cell death. This effect is achieved via innate immune receptors, including TLR3 and 4.137 Intuitively, the immune system is involved also in cardiotoxicity developed after oncological treatments with immune checkpoint inhibitors, that can cause, among other immune related adverse events, cardiac immune-related adverse effects such myocarditis and pericarditis.108,138,139
3.3 Genetic predisposition to peripartum heart failure: what about hormones?
Most genetic DCMs only become penetrant after puberty, indicating an hormonal change as a requirement to develop the phenotype. Peripartum cardiomyopathy (PPCM) is an example of interaction between hormonal changes and genetic predisposition. PPCM has been defined as ‘an idiopathic cardiomyopathy presenting with heart failure secondary to LV systolic dysfunction towards the end of pregnancy or in the months following delivery, where no other cause of heart failure is found’.140 PPCM is a severe complication of pregnancy with a high morbidity and mortality.141,142 Meanwhile, it is considered more a syndrome with different underlying disorders. Among those, genetic forms of cardiomyopathy seem to account for some cases since for example the disease is more frequent in certain geographical regions, Africa (1 in 100 to 1 in 1000 pregnancies) or Haiti (1 in 299 pregnancies), and since there are reports from PPCM patients with a clear family history of heart failure.143,144 Indeed, more recently, several studies discovered mutations associated with familial forms of DCM in PPCM patients, i.e. mutations in TTN, MYBPC3, MYH6, MYH7, PSEN2, SCN5A, TNNC1, and TNNT2.145–147 Beside the presence of mutations associated with familial forms of DCM in PPCM patients, it is also likely that additional mutations or polymorphism may be present in these patients and contribute to their susceptibility to peripartum heart failure. Extrinsic factors like viral infections could also mediate peripartum heart failure and it has been shown that virus including Epstein–Barr virus, human cytomegalovirus, human herpes virus 6, and parvovirus B19 could be identified in up to 30% of cardiac samples in PPCM,148 but their exact pathogenic role still needs to be defined.149
Interestingly, it seems that genetic and non-genetic forms of PPCM may merge on a common pathway including unbalanced oxidative stress and subsequent cleavage of the nursing hormone prolactin into an angiostatic fragment that initiates and drives progression of PPCM.141,142 This discovery led to a disease specific treatment concept combining heart failure treatment with the prolactin blocker bromocriptine (BR), which is named ‘the BOARD therapy regime’ (BR, Oral heart failure drugs, Anticoagulation, Diuretics) for PPCM.150
Although current data suggest that patients with PPCM of different aetiologies seem to profit from the BOARD therapy, epidemiological studies suggest that PPCM in women with a familial history of cardiomyopathies have a poorer prognosis—a feature that might affect risk stratification and clinical management of these patients.142 Therefore, further analysis of underlying pathophysiology is important for more personalized therapy regimes. Moreover, genetic counselling in patients with a family history of (peripartum) heart failure should be considered, which may contribute to a better understanding of the disease pathophysiology and for better and earlier treatment and management of relatives at risk.
3.4 Metabolic triggers in DCM: diabetes and DCM
In diabetic patients, cardiovascular diseases represent a major cause of morbidity and mortality. In addition to increased coronary artery disease and hypertension, there is compelling evidence that diabetes has a direct negative effect on the heart,151 being an independent risk factor for heart failure even after adjusting for age, sex, race, and hypertension.152–154 Mechanisms of such deleterious cardiac effects include mitochondrial dysfunction, oxidative stress, shift in energetic substrate utilization (increase in fatty acid oxidation and decrease in glucose metabolism), disturbed calcium homeostasis, and neuro-humoral activation.151,155
Among the described gene mutations associated with DCM, mutations in LMNA gene, encoding nuclear intermediate filament proteins lamins A/C, more specifically the variant p.G602S was recently associated with Type 2 diabetes.156 Further molecular and physiological studies regarding this variant are needed to understand its predictive value in DCM.156 Taken into consideration the increasing prevalence of diabetes, and its associated complications, an analysis of the correlation between genotypic and phenotypic patient characteristics deserves further investigations, intending to establish a disease-gene mutation association and apply an early intervention in disease progression. Therefore, the presence of diabetes and hypertension or other metabolic conditions should be included in the patient phenotyping, and in order to assess gene-environmental interactions in DCM.
4. How can imaging help in DCM phenotyping?
Echocardiography remains the cornerstone for routine and quick assessment LV function in DCM patients. In addition to confirming the clinical diagnosis, routine echocardiography provides additional prognostic information such as the severity of ventricular dysfunction, the presence of any concurrent valvular disease, and the pulmonary arterial pressure.157 More sophisticated echocardiography reliant on tissue deformation parameters may provide additional risks stratification and/or prognostic information, such as global longitudinal strain, circumferential, and radial strain.158 Those parameters will help clinicians in deciphering the disease heterogeneity associated with DCM.
Cardiac magnetic resonance (CMR) imaging is currently embedded in routine clinical practice with respect to follow-up assessment of LV volumes and accurate ejection fraction, due to its high reproducibility and accuracy.116,117 CMR imaging provides information on tissue characterization, such as the presence or absence of fibrosis, storage diseases and inflammation, and informs on the prognosis, including the risk of developing subsequent malignant arrhythmias.33,159–161
5. New nosological frontiers for myocardial diseases: the MOGE(S) classification system
Myocardial diseases include all those conditions in which the myocardium is not the victim of flow restrictions or does not have to adapt to abnormal flow or pressure loads, such as in valvular or coronary disease and hypertension. Integrating morphology as a cornerstone,162 but also deciphering heterogeneity regarding organ involvement, genetic predisposition and functional consequences, a new classification system for cardiomyopathies called the ‘MOGES’ classification was raised in 2013.163–165 MOGE(S) is composed of four main (M, O, G, E) and one optional (S) attributes163–165 and is supported by a dedicated and renewed app that is now linked with the ICD-10 coding system and workable for transfer individual data in any sort of database. The five attributes describe the morpho-functional cardiac phenotype (M),166 the possible involvement of other organs and tissues (O), the genetic or non-genetic origin of the disease (G), the specific aetiology (E) describing gene and mutation in case of hereditary diseases, or inflammation and infectious agents in case of myocarditis, or autoimmune causes or toxic causes in case of non-genetic diseases. The S attribute is dynamic and optional: it describes the functional status of the affected heart including both NYHA class and AHA stage. The application of MOGE(S) imposes a clinical work-up that starts with the phenotypic characterization of the disease and progresses with the investigation of involvement of extra-cardiac organs/tissues, the evaluation of the genetic/familial or non-familial disease through the clinical screening of the relatives of the patients and the identification, where possible, of the causes (Figure 5).
Figure 5.

Three examples of MOGE(S) application in ACM (A), familial DCM (B), and sporadic DCM phenotype caused by chronic myocarditis (C). The left column shows the pedigrees of three families. The right column shows how family members are described by the MOGE(S) system including the imaging view of the proband (B) and the pathology features in endomyocardial biopsy of the index patient of family C (C). For arrhythmogenic cardiomyopathy, descriptors of the phenotype can detail the combination of major and minor criteria or define family members as diagnosed with definite, possible, and borderline cardiomyopathy according to the 2010 Task Force. For dilated cardiomyopathy, the age dependency of the phenotype explains the absence of clinical signs in the two children (IV:1 and IV:5) of the fourth generation. The p.(Arg190Trp) in LMNA is a recurrent, highly penetrant, malignant, and validated mutation (https://www.ncbi.nlm.nih.gov/clinvar/variation/66908/).
Since its first description, MOGE(S) has attracted the attention of experts in cardiomyopathies for possible applications in clinical practice: numerous implementations have been proposed to accommodate a synthetic but precise description of phenotypes and causes.167–169 In 2015, Hazebroek et al.109 integrated multiple aetiological attributes to assess the possible predictive role of their combination on the clinical outcome; they observed that familial DCM associated with additional aetiological-environmental factors such as significant viral load, immune-mediated factors, rhythm disturbances, or toxic triggers, had worse outcome. The novelty from their proposal was the idea of applying the MOGE(S) classification to assess clinical prognosis and risk stratification in patients with DCM.170 Westphal et al.171 further considered those cardiomyopathies whose phenotype may not be unequivocally recognizable by echocardiography and highlighted the dynamic level of impairment of the left and/or right ventricular systolic (and diastolic) function during the course of myocardial diseases, including baseline and post-treatment evolution. For phenocopies of cardiomyopathies, MOGE(S) describes the possible multi-organ/system involvement and summarizes big raw clinical and genetic data, in a short string of essential data that can facilitate the collection of large database.172 A similar implementation can be easily obtained for genetic cardiomyoapthies.173 Finally, the hypokinetic non-dilated cardiomyopathy recently proposed by the Working Group on Myocardial and Pericardial Diseases of the ESC perfectly fits with the need of describing the increasingly recognized probands’ relatives presenting with early phenotypes in familial DCM.7 Based on the same need of describing early phases of the disease, the recent guidelines for HCM of the ESC first introduced the concept of early diagnosis that applies to relatives of probands with HCM, when the maximal LV thickness is <15 mm.174 MOGE(S) may appear complex, but the ‘complexity’ reflects the modern diagnostics of myocardial diseases, providing a uniform tool to describe disease heterogeneity beyond a given phenotype, integrating genetic, and acquired determinants.165
6. Therapeutic prospects
The treatment of a genetic DCM will depend on the (i) malignancy (or penetrance) of the gene mutation, (ii) the phenotypic presentation, (iii) the underlying molecular mechanisms leading cardiac involvement, and (iv) the gene-environmental interactions determining the DCM phenotype.
The penetrance of gene mutations varies upon the different genes. E.g. a LAMIN A/C mutation is highly penetrant, and therefore, would rather require gene correction through silencing of the mutated allele. SiRNA-mediated silencing of the toxic allele may allow treating more malignant gene mutations. In contrast, more benign gene mutations—such as TTN truncating mutations—with a high probability of improvement or recovery of cardiac function upon heart failure treatment would not benefit from gene silencing strategies. Also the phenotypic presentation of a gene mutation will determine the therapy. Anti-arrhythmic therapies are more often to be considered in LMNA- or RBM20-gene mutated DCM patients, whereas classical heart failure therapy may be sufficient in other more benign genetic DCM cases.
The underlying molecular mechanisms identified in experimental models and in the cardiac samples of genetic DCM results in novel therapeutic approaches. One example is the recently started Phase 3 trial using p38 MAPK inhibitors in LMNA-related DCM, since loss of functional lamin proteins results in activation of the p38 MAPK pathway, and secondary cardiomyocyte apoptosis and interstitial fibrosis.175 In TTNtv patients anti-oxidative therapies such as SS-31 or elamipretide could be considered, in view of the extreme hyperactivation of mitochondrial oxidative pathways.176 In sarcomeric gene mutated DCM patients, interventions on calcium sensitivity may help to improve myofilament force development, thereby prevent cardiomyocyte dysfunction.44
Finally, the knowledge how gene mutated hearts would react to environmental—additionally acquired diseases or hormonal factors during life-factors, may help to guide our therapeutic approach. E.g. TTNtv mutated patients do have a benign functional phenotype, but are prone to develop arrhythmias upon additional cardiac stress, such as alcohol, chemotherapy, or pregnancy. Therefore, an isolated more benign gene mutation without additional environmental factors would require more conservative therapeutic approach, whereas a more penetrant LMNA mutations in multi-morbid person would require a more aggressive medical attitude.
7. Conclusion
In the last 20 years, growing evidence supported that DCM is the final morpho-functional phenotype for various diseases, for which a spectrum of genetic determinants plays either as a causative role (as in case of overt familial disease), or acts as disease modulators regarding another condition (like in ‘sporadic’ cases and ‘acquired’ DCM). By combining clinical characteristics, identifying acquired/environmental triggers, using multi-modal imaging and genetics, disease heterogeneity becomes progressively a clinical reality, allowing improved management of those patients and personalized risk stratification. Genetic data provides more and more cues to understand the molecular mechanisms underlying this complex disease and its heritability. Nevertheless, caution has to be raised as the use of broad genetic screen exposes clinicians to noisy background in DCM, requiring permanent evaluation of variant pathogenicity. Actual data support that inheritance is often complex, with many disease modifiers—going from causal diseases (e.g. anthracyclines) to modifying co-morbidities (e.g. diabetes) having to be taken into account including other genes, epigenetics, and lifestyle. Deep molecular comprehension of DCM improves its assessment, and opens new avenues for personalized medicine and the development of new therapeutic strategies.
Conflict of interest: B.M. filed patents for biomarkers using genetic, epigenetic and transcriptomic information. C.G.T. received speaking fees from Alere. T.T. filed and licensed patents in the field of non-coding RNAs and is founder of Cardior Pharmaceuticals GmbH. And all other authors have none to declare.
Funding
The authors acknowledge the support from the Netherlands Cardiovascular Research Initiative, with support of the Dutch Heart Foundation, CVON2011-ARENA, CVON2018-ARENA PRIME, CVON2016-Early HFPEF, and CVON 2017-ShePREDICTS to S.H.
S.H. received funding from the European Union Commission’s Seventh Framework programme under grant agreement N° 305507 (HOMAGE), N° 602904 (FIBROTARGETS), and FP7-Health-2013-Innovations-1 N° 602156 (HECATOS). E.A. received support from the EU funded project INHERITANCE (n°241924), from the Italian Ministry of Health, from the Cariplo Foundation, and from the Magica Onlus Charity. A.B. received support from the Erasme Foundation (ULB) and from the Belgian Cardiac Surgery Foundation. M.C. from the Funds for Basic Research-University of Salerno (FARB2016-2017). A.L. was supported by the Project DOCnet (NORTE-01-0145-FEDER-000003), by the Norte Portugal Regional Operational Program (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF), and by the project NETDIAMOND (POCI-01-0145-FEDER-016385), supported by European Structural and Investment Funds, Lisbon's Regional Operational Program 2020 and Portuguese funds from the Portuguese Foundation for Science and Technology. B.M. received funding from European Union (FP7 INHERITANCE, BestAgeing, ERA-CVD Detectin-HF), from the Klaus Tschira Foundation, and from CaRNAtion. C.G.T. was supported by a Federico II University grant ‘Ricerca di Ateneo’. J.V.D.V. received a grant from the Dutch Heart Foundation (CVON-Dosis 2014-40). N.H. was supported by a Deutsche Forshungsgemeinschaft (DFG) grant (HA 7512/2-1). R.W. was supported by the National Institute for Health Research (NIHR) Cardiovascular Biomedical Research Unit based at Royal Brompton & Harefield NHS Foundation Trust and Imperial College London, the Wellcome Trust (107469/Z/15/Z) and the British Heart Foundation (SP/10/10/28431). T.T. received funding from European Union, ERANET (Expert) and the ERC Consolidator grant Longheart. E.U. funded project INHERITANCE (n°241924) and the Italian Ministry of Health Grants to E.A.
References
- 1. McNally EM, Mestroni L.. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hershberger RE, Hedges DJ, Morales A.. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 3. Williams DG, Olsen EG.. Prevalence of overt dilated cardiomyopathy in two regions of England. Br Heart J 1985;54:153–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Codd MB, Sugrue DD, Gersh BJ, Melton LJ.. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation 1989;80:564–572. [DOI] [PubMed] [Google Scholar]
- 5. Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D.. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol 2011;108:1171–1176. [DOI] [PubMed] [Google Scholar]
- 6. Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L, Tendera M, Maupain C, Laroche C, Rubis P, Jurcut R, Calò L, Heliö TM, Sinagra G, Zdravkovic M, Kavoliūnienė A, Felix SB, Grzybowski J, Losi M-A, Asselbergs FW, García-Pinilla JM, Salazar-Mendiguchia J, Mizia-Stec K, Maggioni AP; EORP Cardiomyopathy Registry Investigators. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J 2018;29:270–210. [DOI] [PubMed] [Google Scholar]
- 7. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, De Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio ALP, Charron P.. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016;37:1850–1858. [DOI] [PubMed] [Google Scholar]
- 8. Wolf MJ, Noeth D, Rammohan C, Shah SH.. Complexities of genetic testing in familial dilated cardiomyopathy. Circ Cardiovasc Genet 2016;9:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilde AAM, Behr ER.. Genetic testing for inherited cardiac disease. Nat Rev Cardiol 2013;10:571–583. [DOI] [PubMed] [Google Scholar]
- 10. Burke MA, Cook SA, Seidman JG, Seidman CE.. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, Chung WK, Jefferies JL, Rossano JW, Castleberry CD, Addonizio LJ, Lal AK, Lamour JM, Miller EM, Thrush PT, Czachor JD, Razoky H, Hill A, Lipshultz SE.. Pediatric cardiomyopathies. Circ Res 2017;121:855–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV; Exome Aggregation Consortium. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2016;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJR, Cook SA, Mestroni L, Seidman JG, Seidman CE.. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerull B, Gramlich M, Atherton J, McNabb M, Trombitás K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S, Frenneaux M, Thierfelder L.. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 2002;30:201–204. [DOI] [PubMed] [Google Scholar]
- 15. Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiarán Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca H-P, Brunner HG, Krapels IPC, Heymans SRB.. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 2018;10:531–510. [DOI] [PubMed] [Google Scholar]
- 16. Tayal U, Newsome S, Buchan R, Whiffin N, Halliday B, Lota A, Roberts A, Baksi AJ, Voges I, Midwinter W, Wilk A, Govind R, Walsh R, Daubeney P, Jarman JWE, Baruah R, Frenneaux M, Barton PJ, Pennell D, Ware JS, Prasad SK, Cook SA.. Phenotype and clinical outcomes of titin cardiomyopathy. J Am Coll Cardiol 2017;70:2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE.. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci 2010;3:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Refaat MM, Lubitz SA, Makino S, Islam Z, Frangiskakis JM, Mehdi H, Gutmann R, Zhang ML, Bloom HL, MacRae CA, Dudley SC, Shalaby AA, Weiss R, McNamara DM, London B, Ellinor PT.. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012;9:390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor MRG, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li W-LP, Stetler GL, Muntoni F, Bristow MR, Mestroni L, Familial DCRRG.. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771–780. [DOI] [PubMed] [Google Scholar]
- 20. Sébillon P, Bouchier C, Bidot LD, Bonne G, Ahamed K, Charron P, Drouin GV, Millaire A, Desrumeaux G, Benaïche A, Charniot J-C, Schwartz K, Villard E, Komajda M.. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet 2003;40:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, Edvardsen T, Haugaa KH.. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J 2017;15:376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, Crijns HJGM, Pinto YM.. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med 2005;83:79–83. [DOI] [PubMed] [Google Scholar]
- 23. Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padrón-Barthe L, Duro-Aguado I, Jiménez-Jáimez J, Hidalgo-Olivares VM, García-Campo E, Lanzillo C, Suárez-Mier MP, Yonath H, Marcos-Alonso S, Ochoa JP, Santomé JL, García-Giustiniani D, Rodríguez-Garrido JL, Domínguez F, Merlo M, Palomino J, Peña ML, Trujillo JP, Martín-Vila A, Stolfo D, Molina P, Lara-Pezzi E, Calvo-Iglesias FE, Nof E, Calò L, Barriales-Villa R, Gimeno-Blanes JR, Arad M, García-Pavía P, Monserrat L.. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 2016;68:2440–2451. [DOI] [PubMed] [Google Scholar]
- 24. Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, Repetto A, Bellini O, Azan G, Scaffino M, Campana C, Piccolo G, Viganò M, Tavazzi L.. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail 2006;8:477–483. [DOI] [PubMed] [Google Scholar]
- 25. Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatsellas G, Tsikitis M.. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol 2015;32:113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan G-C, Tsiapras D, Parekh RR, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG.. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 2006;103:1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Zwaag PA, van Rijsingen IAW, Asimaki A, Jongbloed JDH, van Veldhuisen DJ, Wiesfeld ACP, Cox MGPJ, van Lochem LT, de Boer RA, Hofstra RMW, Christiaans I, van Spaendonck-Zwarts KY, Lekanne dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJH, Hauer RNW, Saffitz JE, Wilde AAM, van den Berg MP, van Tintelen JP.. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail 2012;14:1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE.. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003;299:1410–1413. [DOI] [PubMed] [Google Scholar]
- 29. McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L; Familial Cardiomyopathy Registry Research Group. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol 2011;57:2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ehlermann P, Lehrke S, Papavassiliu T, Meder B, Borggrefe M, Katus HA, Schimpf R.. Sudden cardiac death in a patient with lamin A/C mutation in the absence of dilated cardiomyopathy or conduction disease. Clin Res Cardiol 2011;100:547–551. [DOI] [PubMed] [Google Scholar]
- 31. Hasselberg NE, Edvardsen T, Petri H, Berge KE, Leren TP, Bundgaard H, Haugaa KH.. Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace 2014;16:563–571. [DOI] [PubMed] [Google Scholar]
- 32. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP.. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011;13:1077–1339. [DOI] [PubMed] [Google Scholar]
- 33. Priori SG, Blomström Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck K-H, Hernández-Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ; Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Europace 2015;17;1601–1687. [DOI] [PubMed] [Google Scholar]
- 34. Hershberger RE, Siegfried JD.. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2011;57:1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zaklyazminskaya E, Dzemeshkevich S.. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta 2016;1863:1799–1805. [DOI] [PubMed] [Google Scholar]
- 36. Hershberger RE, Morales A, Siegfried JD.. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med 2010;12:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tayal U, Prasad S, Cook SA.. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med 2017;9:20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mogensen J, van Tintelen JP, Fokstuen S, Elliott P, van Langen IM, Meder B, Richard P, Syrris P, Caforio ALP, Adler Y, Anastasakis A, Gimeno JR, Klingel K, Linhart A, Imazio M, Pinto Y, Newbery R, Schmidtke J, Charron P.. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: a viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur Heart J 2015;36:1367–1370. [DOI] [PubMed] [Google Scholar]
- 39. Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G.. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail 2018;20:228–239. [DOI] [PubMed] [Google Scholar]
- 40. Klos M, Mundada L, Banerjee I, Morgenstern S, Myers S, Leone M, Kleid M, Herron T, Devaney E.. Altered myocyte contractility and calcium homeostasis in alpha-myosin heavy chain point mutations linked to familial dilated cardiomyopathy. Arch Biochem Biophys 2017;615:53–60. [DOI] [PubMed] [Google Scholar]
- 41. Carniel E, Taylor MRG, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, Graw SL, Feiger J, Zhu XZ, Dao D, Ferguson DA, Bristow MR, Mestroni L.. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005;112:54–59. [DOI] [PubMed] [Google Scholar]
- 42. Møller DV, Andersen PS, Hedley P, Ersbøll MK, Bundgaard H, Moolman-Smook J, Christiansen M, Køber L.. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet 2009;17:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y, McKenna WJ.. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol 2007;49:2419–2426. [DOI] [PubMed] [Google Scholar]
- 44. Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, Seidman CE, Seidman JG, Regnier M, Metzger JM, Wu JC, Molkentin JD.. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 2016;165:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McConnell M, Tal Grinspan L, Williams MR, Lynn ML, Schwartz BA, Fass OZ, Schwartz SD, Tardiff JC.. Clinically divergent mutation effects on the structure and function of the human cardiac tropomyosin overlap. Biochemistry 2017;56:3403–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ryba DM, Li J, Cowan CL, Russell B, Wolska BM, Solaro RJ.. Long-term biased β-arrestin signaling improves cardiac structure and function in dilated cardiomyopathy. Circulation 2017;135:1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bollen IAE, Schuldt M, Harakalova M, Vink A, Asselbergs FW, Pinto JR, Krüger M, Kuster DWD, van der Velden J.. Genotype-specific pathogenic effects in human dilated cardiomyopathy. J Physiol 2017;595:4677–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Rijsingen IAW, Arbustini E, Elliott PM, Mogensen J, Ast JFH-V, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AAM, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM.. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers. J Am Coll Cardiol 2012;59:493–500. [DOI] [PubMed] [Google Scholar]
- 49. Elliott P, O'Mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A, McKenna WJ.. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2010;3:314–322. [DOI] [PubMed] [Google Scholar]
- 50. Spezzacatene A, Sinagra G, Merlo M, Barbati G, Graw SL, Brun F, Slavov D, Di Lenarda A, Salcedo EE, Towbin JA, Saffitz JE, Marcus FI, Zareba W, Taylor MRG, Mestroni L; The Familial Cardiomyopathy Registry; Arrhythmogenic phenotype in dilated cardiomyopathy: natural history and predictors of life-threatening arrhythmias. J Am Heart Assoc 2015;4:e002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ.. Left-dominant arrhythmogenic cardiomyopathy. J Am Coll Cardiol 2008;52:2175–2187. [DOI] [PubMed] [Google Scholar]
- 52. Corrado D, Link MS, Calkins H.. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2017;376:61–72. [DOI] [PubMed] [Google Scholar]
- 53. Asahi M, Nakayama H, Tada M, Otsu K.. Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med 2003;13:152–157. [DOI] [PubMed] [Google Scholar]
- 54. Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO.. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest 2016;126:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu G-S, Morales A, Vafiadaki E, Lam CK, Cai W-F, Haghighi K, Adly G, Hershberger RE, Kranias EG.. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res 2015;107:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karakikes I, Stillitano F, Nonnenmacher M, Tzimas C, Sanoudou D, Termglinchan V, Kong C-W, Rushing S, Hansen J, Ceholski D, Kolokathis F, Kremastinos D, Katoulis A, Ren L, Cohen N, Gho JMIH, Tsiapras D, Vink A, Wu JC, Asselbergs FW, Li RA, Hulot J-S, Kranias EG, Hajjar RJ.. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun 2015;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stillitano F, Turnbull IC, Karakikes I, Nonnenmacher M, Backeris P, Hulot J-S, Kranias EG, Hajjar RJ, Costa KD.. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur Heart J 2016;37:3282–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gopinathannair R, Etheridge SP, Marchlinski FE, Spinale FG, Lakkireddy D, Olshansky B.. Arrhythmia-induced cardiomyopathies: mechanisms, recognition, and management. J Am Coll Cardiol 2015;66:1714–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Das S, Aiba T, Rosenberg M, Hessler K, Xiao C, Quintero PA, Ottaviano FG, Knight AC, Graham EL, Boström P, Morissette MR, del Monte F, Begley MJ, Cantley LC, Ellinor PT, Tomaselli GF, Rosenzweig A.. Pathological role of serum- and glucocorticoid-regulated kinase 1 in adverse ventricular remodeling. Circulation 2012;126:2208–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beckermann TM, McLeod K, Murday V, Potet F, George AL.. Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm 2014;11:1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM.. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 62. Meyers DE, Basha HI, Koenig MK.. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J 2013;40:385–394. [PMC free article] [PubMed] [Google Scholar]
- 63. Holmgren D, Wahlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M.. Cardiomyopathy in children with mitochondrial disease;clinical course and cardiological findings. Eur Heart J 2003;24:280–288. [DOI] [PubMed] [Google Scholar]
- 64. Finsterer J, Kothari S.. Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol 2014;177:754–763. [DOI] [PubMed] [Google Scholar]
- 65. Dudek J, Maack C.. Barth syndrome cardiomyopathy. Cardiovasc Res 2017;113:399–410. [DOI] [PubMed] [Google Scholar]
- 66. Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D.. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 1996;12:385–389.8630491 [Google Scholar]
- 67. Johnston J, Kelley RI, Feigenbaum A, Cox GF, Iyer GS, Funanage VL, Proujansky R.. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am J Hum Genet 1997;61:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brunel-Guitton C, Levtova A, Sasarman F.. Mitochondrial diseases and cardiomyopathies. Can J Cardiol 2015;31:1360–1376. [DOI] [PubMed] [Google Scholar]
- 69. Bannwarth S, Procaccio V, Lebre AS, Jardel C, Chaussenot A, Hoarau C, Maoulida H, Charrier N, Gai X, Xie HM, Ferre M, Fragaki K, Hardy G, Mousson de Camaret B, Marlin S, Dhaenens CM, Slama A, Rocher C, Paul Bonnefont J, Rötig A, Aoutil N, Gilleron M, Desquiret-Dumas V, Reynier P, Ceresuela J, Jonard L, Devos A, Espil-Taris C, Martinez D, Gaignard P, Le Quan Sang K-H, Amati-Bonneau P, Falk MJ, Florentz C, Chabrol B, Durand-Zaleski I, Paquis-Flucklinger V.. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet 2013;50:704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marin-Garcia J, Goldenthal MJ, Moe GW.. Mitochondrial pathology in cardiac failure. Cardiovasc Res 2001;49:17–26. [DOI] [PubMed] [Google Scholar]
- 71. Ahuja P, Wanagat J, Wang Z, Wang Y, Liem DA, Ping P, Antoshechkin IA, Margulies KB, Maclellan WR.. Divergent mitochondrial biogenesis responses in human cardiomyopathy. Circulation 2013;127:1957–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li YY, Maisch B, Rose ML, Hengstenberg C.. Point mutations in mitochondrial DNA of patients with dilated cardiomyopathy. J Mol Cell Cardiol 1997;29:2699–2709. [DOI] [PubMed] [Google Scholar]
- 73. Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM, Bohr VA.. Protecting the mitochondrial powerhouse. Trends Cell Biol 2015;25:158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T.. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015;350:aad0116. [DOI] [PubMed] [Google Scholar]
- 75. Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW.. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016;540:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Franco A, Ciccarelli M, Sorriento D, Napolitano L, Fiordelisi A, Trimarco B, Durante M, Iaccarino G.. Rays sting: the acute cellular effects of ionizing radiation exposure. Transl Med UniSa 2016;14:42–53. [PMC free article] [PubMed] [Google Scholar]
- 77. Franco A, Sorriento D, Gambardella J, Pacelli R, Prevete N, Procaccini C, Matarese G, Trimarco B, Iaccarino G, Ciccarelli M.. GRK2 moderates the acute mitochondrial damage to ionizing radiation exposure by promoting mitochondrial fission/fusion. Cell Death Discov 2018;4:10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pereira NL, Grogan M, Dec GW.. Spectrum of restrictive and infiltrative cardiomyopathies part 2 of a 2-part series. J Am Coll Cardiol 2018;71:1149–1166. [DOI] [PubMed] [Google Scholar]
- 79. Pereira NL, Grogan M, Dec GW.. Spectrum of restrictive and infiltrative cardiomyopathies part 1 of a 2-part series. J Am Coll Cardiol 2018;71:1130–1148. [DOI] [PubMed] [Google Scholar]
- 80. Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, Gimeno JR, Elliott P, McKenna WJ.. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003;111:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kostareva A, Gudkova A, Sjöberg G, Mörner S, Semernin E, Krutikov A, Shlyakhto E, Sejersen T.. Deletion in TNNI3 gene is associated with restrictive cardiomyopathy. Int J Cardiol 2009;131:410–412. [DOI] [PubMed] [Google Scholar]
- 82. Kostareva A, Kiselev A, Gudkova A, Frishman G, Ruepp A, Frishman D, Smolina N, Tarnovskaya S, Nilsson D, Zlotina A, Khodyuchenko T, Vershinina T, Pervunina T, Klyushina A, Kozlenok A, Sjöberg G, Golovljova I, Sejersen T, Shlyakhto E.. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One 2016;11:e0163362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Du J, Liu J, Feng H-Z, Hossain MM, Gobara N, Zhang C, Li Y, Jean-Charles P-Y, Jin J-P, Huang X-P.. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol 2008;294:H2604–H2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, Herron KJ, Nelson SM, Rodeheffer RJ, Olson TM.. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet 2008;74:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Burke M, Cook S, Seidman JG, Seidman CE.. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Díez-López C, Comín-Colet J, González-Costello J.. Iron overload cardiomyopathy: from diagnosis to management. Curr Opin Cardiol 2018;33:1–340. [DOI] [PubMed] [Google Scholar]
- 87. Kremastinos DT, Farmakis D.. Iron overload cardiomyopathy in clinical practice. Circulation 2011;124:2253–2263. [DOI] [PubMed] [Google Scholar]
- 88. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG.. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Norton N, Robertson PD, Rieder MJ, Züchner S, Rampersaud E, Martin E, Li D, Nickerson DA, Hershberger RE; National Heart, Lung and Blood Institute GO Exome Sequencing Project. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet 2012;5:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunsø S, Svendsen JH, Olesen MS.. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet 2013;21:918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM.. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009;54:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, Wright MW, Milko LV, Cherry JM, Giovanni MA, Murray MF, O’Daniel JM, Ramos EM, Santani AB, Scott AF, Plon SE, Rehm HL, Martin CL, Berg JS.. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet 2017;100:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, Prasad S, Barton PJ, Funke B, Watkins H, Ware JS, Cook SA.. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J 2017;38:3461–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH.. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014;16:601–608. [DOI] [PubMed] [Google Scholar]
- 95. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JAL, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O'Regan DP, San TR, de Marvao A, Dawes TJW, Gulati A, Birks EJ.. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chami N, Tadros R, Lemarbre F, Lo KS, Beaudoin M, Robb L, Labuda D, Tardif J-C, Racine N, Talajic M, Lettre G.. Nonsense mutations in BAG3 are associated with early-onset dilated cardiomyopathy in French Canadians. Can J Cardiol 2014;30:1655–1661. [DOI] [PubMed] [Google Scholar]
- 97. Toro R, Pérez-Serra A, Campuzano O, Moncayo-Arlandi J, Allegue C, Iglesias A, Mangas A, Brugada R.. Familial dilated cardiomyopathy caused by a novel frameshift in the BAG3 gene. PLoS One 2016;11:e0158730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meder B, Rühle F, Weis T, Homuth G, Keller A, Franke J, Peil B, Lorenzo Bermejo J, Frese K, Huge A, Witten A, Vogel B, Haas J, Völker U, Ernst F, Teumer A, Ehlermann P, Zugck C, Friedrichs F, Kroemer H, Dörr M, Hoffmann W, Maisch B, Pankuweit S, Ruppert V, Scheffold T, Kühl U, Schultheiss H-P, Kreutz R, Ertl G, Angermann C, Charron P, Villard E, Gary F, Isnard R, Komajda M, Lutz M, Meitinger T, Sinner MF, Wichmann H-E, Krawczak M, Ivandic B, Weichenhan D, Gelbrich G, El-Mokhtari N-E, Schreiber S, Felix SB, Hasenfuß G, Pfeufer A, Hübner N, Kääb S, Arbustini E, Rottbauer W, Frey N, Stoll M, Katus HA.. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J 2014;35:1069–1077. [DOI] [PubMed] [Google Scholar]
- 100. Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, Ruppert V, Arbustini E, Wichter T, Germain M, Dubourg O, Tavazzi L, Aumont M-C, DeGroote P, Fauchier L, Trochu J-N, Gibelin P, Aupetit J-F, Stark K, Erdmann J, Hetzer R, Roberts AM, Barton PJR, Regitz-Zagrosek V, Aslam U, Duboscq-Bidot L, Meyborg M, Maisch B, Madeira H, Waldenström A, Galve E, Cleland JG, Dorent R, Roizes G, Zeller T, Blankenberg S, Goodall AH, Cook S, Tregouet DA, Tiret L, Isnard R, Komajda M, Charron P, Cambien F.. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J 2011;32:1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim W-K, Zhao X, Fradkin D, Köhler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjær H, Jørgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RHLD, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Mörner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B.. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2015;36:1123–135a. [DOI] [PubMed] [Google Scholar]
- 102. Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, Zhou B, Chang C-P.. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010;466:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sergeeva IA, Hooijkaas IB, Ruijter JM, van der Made I, de Groot NE, van de Werken HJG, Creemers EE, Christoffels VM.. Identification of a regulatory domain controlling the Nppa-Nppbgene cluster during heart development and stress. Development 2016;143:2135–2146. [DOI] [PubMed] [Google Scholar]
- 104. Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A, Kurz K, Carell T, Angius A, Latronico MVG, Papait R, Greco CM, Condorelli G.. DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun 2016;7:12418–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, Marquart S, Sedaghat-Hamedani F, Kayvanpour E, Köhler D, Wolf NM, Hassel S, Nietsch R, Wieland T, Ehlermann P, Schultz J-H, Dösch A, Mereles D, Hardt S, Backs J, Hoheisel JD, Plass C, Katus HA, Meder B.. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med 2013;5:413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Meder B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Frese K, Lai A, Nietsch R, Scheiner C, Mester S, Bordalo DM, Amr A, Dietrich C, Pils D, Siede D, Hund H, Bauer A, Holzer DB, Ruhparwar A, Mueller-Hennessen M, Weichenhan D, Plass C, Weis T, Backs J, Wuerstle M, Keller A, Katus HA, Posch AE.. Epigenome-wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation 2017;136:1528–1544. [DOI] [PubMed] [Google Scholar]
- 107. Mayer SC, Gilsbach R, Preissl S, Monroy Ordonez EB, Schnick T, Beetz N, Lother A, Rommel C, Ihle H, Bugger H, Rühle F, Schrepper A, Schwarzer M, Heilmann C, Bönisch U, Gupta SK, Wilpert J, Kretz O, Elverfeldt von D, Orth J, Aktories K, Beyersdorf F, Bode C, Stiller B, Krüger M, Thum T, Doenst T, Stoll M, Hein L.. Adrenergic repression of the epigenetic reader MeCP2 facilitates cardiac adaptation in chronic heart failure. Circ Res 2015;117:622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Frantz S, Falcao-Pires I, Balligand J-L, Bauersachs J, Brutsaert D, Ciccarelli M, Dawson D, de Windt LJ, Giacca M, Hamdani N, Hilfiker-Kleiner D, Hirsch E, Leite-Moreira A, Mayr M, Thum T, Tocchetti CG, van der Velden J, Varricchi G, Heymans S.. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail 2018;20:445–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hazebroek MR, Moors S, Dennert R, van den Wijngaard A, Krapels I, Hoos M, Verdonschot J, Merken JJ, de Vries B, Wolffs PF, Crijns HJGM, Brunner-La Rocca H-P, Heymans S.. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol 2015;66:1313–1323. [DOI] [PubMed] [Google Scholar]
- 110. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss H-P, McCarthy R, Hare J, Bricker JT, Bowles KR, Towbin JA.. Detection of viruses in myocardial tissues by polymerase chain reaction: evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol 2003;42:466–472. [DOI] [PubMed] [Google Scholar]
- 111. Bowles NE, Bowles KR, Towbin JA.. Viral genomic detection and outcome in myocarditis. Heart Fail Clin 2005;1:407–417. [DOI] [PubMed] [Google Scholar]
- 112. Jefferies JL, Towbin JA.. Dilated cardiomyopathy. Lancet 2010;375:752–762. [DOI] [PubMed] [Google Scholar]
- 113. Abston ED, Coronado MJ, Bucek A, Bedja D, Shin J, Kim JB, Kim E, Gabrielson KL, Georgakopoulos D, Mitzner W, Fairweather D.. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin Dev Immunol 2012;2012:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, Rose NR, Fairweather D.. Cutting edge: cross-regulation by TLR4 and T cell Ig Mucin-3 determines sex differences in inflammatory heart disease. J Immunol 2007;178:6710–6714. [DOI] [PubMed] [Google Scholar]
- 115. Riad A, Westermann D, Escher F, Becher PM, Savvatis K, Lettau O, Heimesaat MM, Bereswill S, Volk HD, Schultheiss HP, Tschöpe C.. Myeloid differentiation factor-88 contributes to TLR9-mediated modulation of acute coxsackievirus B3-induced myocarditis in vivo. Am J Physiol Heart Circ Physiol 2010;298:H2024–H2031. [DOI] [PubMed] [Google Scholar]
- 116. Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, Heimesaat MM, Lettau O, Lassner D, Dorner A, Poller W, Busch M, Felix SB, Schultheiss HP, Tschope C.. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol 2011;186:2561–2570. [DOI] [PubMed] [Google Scholar]
- 117. Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, Pedrazzini T, Berger CT, Dirnhofer S, Matter CM, Penninger JM, Luscher TF, Eriksson U.. Myeloid differentiation factor-88/Interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res 2009;105:912–920. [DOI] [PubMed] [Google Scholar]
- 118. Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, Kim E, Bedja D, Sung Y-K, Radtke AJ, Gabrielson KL, Mitzner W, Fairweather D.. TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. Am J Physiol Regul Integr Comp Physiol 2013;304:R267–R277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JLF, Varela J, David RM, Banks L, Huang C-H, Li H, Schultheiss H-P, Towbin JA, Vallejo JG, Bowles NE.. A role for toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem 2010;285:23208–23223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Felix SB, Beug D, Dörr M.. Immunoadsorption therapy in dilated cardiomyopathy. Expert Rev Cardiovasc Ther 2015;13:145–152. [DOI] [PubMed] [Google Scholar]
- 121. Guzzo-Merello G, Cobo-Marcos M, Gallego-Delgado M, Garcia-Pavia P.. Alcoholic cardiomyopathy. World J Cardiol 2014;6:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Renu K, V G A, P B TP, Arunachalam S.. Molecular mechanism of doxorubicin-induced cardiomyopathy—an update. Eur J Pharmacol 2018;818:241–253. [DOI] [PubMed] [Google Scholar]
- 123. Cappetta D, Rossi F, Piegari E, Quaini F, Berrino L, Urbanek K, De Angelis A.. Doxorubicin targets multiple players: a new view of an old problem. Pharmacol Res 2018;127:4–14. [DOI] [PubMed] [Google Scholar]
- 124. Hung C-L, Chang S-C, Chang S-H, Chi P-C, Lai Y-J, Wang S-W, Wu Y-J, Yeh H-I, Lin S-J, Chen C-H, Mochly-Rosen D, Wang L-Y; MAGNET Study Investigator. Genetic polymorphisms of alcohol metabolizing enzymes and alcohol consumption are associated with asymptomatic cardiac remodeling and subclinical systolic dysfunction in large community-dwelling Asians. Alcohol Alcohol 2017;52:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Aminkeng F, Bhavsar AP, Visscher H, Rassekh SR, Li Y, Lee JW, Brunham LR, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Amstutz U, Rieder MJ, Bernstein D, Carleton BC, Hayden MR, Ross CJD, Hayden MR, Carleton BC, Ross CJD, MacLeod S, Smith A, Hildebrand C, Ghannadan R, Rassekh SR, Visscher H, Aminkeng F, Miao F, Higginson M, Massah N.. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 2015;47:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Blanco JG, Sun C-L, Landier W, Chen L, Esparza-Duran D, Leisenring W, Mays A, Friedman DL, Ginsberg JP, Hudson MM, Neglia JP, Oeffinger KC, Ritchey AK, Villaluna D, Relling MV, Bhatia S.. Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes—a report from the Children's Oncology Group. J Clin Oncol 2012;30:1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Visscher H, Ross CJD, Rassekh SR, Barhdadi A, Dubé M-P, Al-Saloos H, Sandor GS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Brown AMK, Rogers PC, Phillips MS, Rieder MJ, Carleton BC, Hayden MR; Canadian Pharmacogenomics Network for Drug Safety Consortium. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol 2012;30:1422–1428. [DOI] [PubMed] [Google Scholar]
- 128. Wang X, Sun C-L, Quiñones-Lombraña A, Singh P, Landier W, Hageman L, Mather M, Rotter JI, Taylor KD, Chen Y-DI, Armenian SH, Winick N, Ginsberg JP, Neglia JP, Oeffinger KC, Castellino SM, Dreyer ZE, Hudson MM, Robison LL, Blanco JG, Bhatia S.. CELF4Variant and anthracycline-related cardiomyopathy: a Children’s Oncology Group genome-wide association study. J Clin Oncol 2016;34:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Visscher H, Ross CJD, Rassekh SR, Sandor GSS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Rogers PC, Rieder MJ, Carleton BC, Hayden MR; CPNDS Consortium. Validation of variants in SLC28A3and UGT1A6as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr Blood Cancer 2013;60:1375–1381. [DOI] [PubMed] [Google Scholar]
- 130. Wang X, Liu W, Sun C-L, Armenian SH, Hakonarson H, Hageman L, Ding Y, Landier W, Blanco JG, Chen L, Quiñones A, Ferguson D, Winick N, Ginsberg JP, Keller F, Neglia JP, Desai S, Sklar CA, Castellino SM, Cherrick I, Dreyer ZE, Hudson MM, Robison LL, Yasui Y, Relling MV, Bhatia S.. Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the Children's Oncology Group. J Clin Oncol 2014;32:647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Anderson PAW, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK.. Molecular basis of human cardiac troponin t isoforms expressed in the developing, adult, and failing heart. Circ Res 1995;76:681–686. [DOI] [PubMed] [Google Scholar]
- 132. Gupta SK, Garg A, Bär C, Chatterjee S, Foinquinos A, Milting H, Streckfuß-Bömeke K, Fiedler J, Thum T.. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expressionnovelty and significance. Circ Res 2018;122:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Tocchetti CG, Cadeddu C,D, Lisi D, Femminò S, Madonna R, Mele D, Monte I, Novo G, Penna C, Pepe A, Spallarossa P, Varricchi G, Zito C, Pagliaro P, Mercuro G.. From molecular mechanisms to clinical management of antineoplastic drug-induced cardiovascular toxicity: a translational overview. Antioxid Redox Signal 2017. Epub ahead of print; doi:10.1089/ars.2016.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nozaki N, Shishido T, Takeishi Y, Kubota I.. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 2004;110:2869–2874. [DOI] [PubMed] [Google Scholar]
- 135. Riad A, Bien S, Gratz M, Escher F, Heimesaat MM, Bereswill S, Krieg T, Felix SB, Schultheiss HP, Kroemer HK, Tschöpe C.. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur J Heart Fail 2008;10:233–243. [DOI] [PubMed] [Google Scholar]
- 136. Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H, Wang Q, Wang Z, Yan J, Fan G, Lindsey ML, Hu Z.. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One 2012;7:e40763–e40710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira J-P, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L.. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13:1050–1059. [DOI] [PubMed] [Google Scholar]
- 138. Wang DY, Okoye GD, Neilan TG, Johnson DB, Moslehi JJ.. Cardiovascular toxicities associated with cancer immunotherapies. Curr Cardiol Rep 2017;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Varricchi G, Galdiero MR, Tocchetti CG.. Cardiac toxicity of immune checkpoint inhibitors. Circulation 2017;136:1989–1992. [DOI] [PubMed] [Google Scholar]
- 140. Sliwa K, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann E, Regitz-Zagrosek V, Schaufelberger M, Tavazzi L, Van Veldhuisen DJ, Watkins H, Shah AJ, Seferovic PM, Elkayam U, Pankuweit S, Papp Z, Mouquet F, McMurray JJV.. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail 2010;12:767–778. [DOI] [PubMed] [Google Scholar]
- 141. Hilfiker-Kleiner D, Haghikia A, Nonhoff J, Bauersachs J.. Peripartum cardiomyopathy: current management and future perspectives. Eur Heart J 2015;36:1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hilfiker-Kleiner D, Sliwa K.. Pathophysiology and epidemiology of peripartum cardiomyopathy. Nat Rev Cardiol 2014;11:364–370. [DOI] [PubMed] [Google Scholar]
- 143. Fett JD. Peripartum cardiomyopathy. Insights from Haiti regarding a disease of unknown etiology. Minn Med 2002;85:46–48. [PubMed] [Google Scholar]
- 144. Meyer GP, Labidi S, Podewski E, Sliwa K, Drexler H, Hilfiker-Kleiner D.. Bromocriptine treatment associated with recovery from peripartum cardiomyopathy in siblings: two case reports. J Med Case Rep 2010;4:80.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. van Spaendonck-Zwarts KY, van Tintelen JP, Van Veldhuisen DJ, van der Werf R, Jongbloed JDH, Paulus WJ, Dooijes D, van den Berg MP.. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010;121:2169–2175. [DOI] [PubMed] [Google Scholar]
- 146. van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IAE, Sliwa K, Alders M, Almomani R, van Langen IM, van der Meer P, Sinke RJ, van der Velden J, Van Veldhuisen DJ, van Tintelen JP, Jongbloed JDH.. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 2014;35:2165–2173. [DOI] [PubMed] [Google Scholar]
- 147. Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, Cook SA, Halder I, Prasad SK, Pisarcik J, Hanley-Yanez K, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Kealey A, Alexis J, Ramani G, Safirstein J, Boehmer J, Pauly DF, Wittstein IS, Thohan V, Zucker MJ, Liu P, Gorcsan J, McNamara DM, Seidman CE, Seidman JG, Arany Z.. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016;374:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bültmann BD, Klingel K, Näbauer M, Wallwiener D, Kandolf R.. High prevalence of viral genomes and inflammation in peripartum cardiomyopathy. Am J Obstet Gynecol 2005;193:363–365. [DOI] [PubMed] [Google Scholar]
- 149. Fett JD. Viral infection as a possible trigger for the development of peripartum cardiomyopathy. Int J Gynaecol Obstet 2007;97:149–150. [DOI] [PubMed] [Google Scholar]
- 150. Arrigo M, Blet A, Mebazaa A.. Bromocriptine for the treatment of peripartum cardiomyopathy: welcome on BOARD. Eur Heart J 2017;38:2680–2682. [DOI] [PubMed] [Google Scholar]
- 151. Ernande L, Derumeaux G.. Diabetic cardiomyopathy: myth or reality? Arch Cardiovasc Dis 2012;105:218–225. [DOI] [PubMed] [Google Scholar]
- 152. Gottdiener JS, Arnold AM, Aurigemma GP, Polak JF, Tracy RP, Kitzman DW, Gardin JM, Rutledge JE, Boineau RC.. Predictors of congestive heart failure in the elderly: the cardiovascular health study. J Am Coll Cardiol 2000;35:1628–1637. [DOI] [PubMed] [Google Scholar]
- 153. Kannel WB, Hjortland M, Castelli WP.. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 1974;34:29–34. [DOI] [PubMed] [Google Scholar]
- 154. Bertoni AG, Tsai A, Kasper EK, Brancati FL.. Diabetes and idiopathic cardiomyopathy: a nationwide case-control study. Diabetes Care 2003;26:2791–2795. [DOI] [PubMed] [Google Scholar]
- 155. Boudina S, Abel ED.. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 2010;11:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Florwick A, Dharmaraj T, Jurgens J, Valle D, Wilson KL.. LMNA sequences of 60,706 unrelated individuals reveal 132 novel missense variants in A-type lamins and suggest a link between variant p.G602S and Type 2 diabetes. Front Genet Frontiers 2017;8:1134.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Thomas DE, Wheeler R, Yousef ZR, Masani ND.. The role of echocardiography in guiding management in dilated cardiomyopathy. Eur J Echocardiogr 2009;10:iii15–iii21. [DOI] [PubMed] [Google Scholar]
- 158. Mignot A, Donal E, Zaroui A, Reant P, Salem A, Hamon C, Monzy S, Roudaut R, Habib G, Lafitte S.. Global longitudinal strain as a major predictor of cardiac events in patients with depressed left ventricular function: a multicenter study. J Am Soc Echocardiogr 2010;23:1019–1024. [DOI] [PubMed] [Google Scholar]
- 159. Dawson DK, Hawlisch K, Prescott G, Roussin I, Di Pietro E, Deac M, Wong J, Frenneaux MP, Pennell DJ, Prasad SK.. Prognostic role of CMR in patients presenting with ventricular arrhythmias. JACC Cardiovasc Imaging 2013;6:335–344. [DOI] [PubMed] [Google Scholar]
- 160. Mahida S, Sacher F, Dubois R, Sermesant M, Bogun F, Haïssaguerre M, Jaïs P, Cochet H.. Cardiac imaging in patients with ventricular tachycardia. Circulation 2017;136:2491–2507. [DOI] [PubMed] [Google Scholar]
- 161. Rodrigues P, Joshi A, Williams H, Westwood M, Petersen SE, Zemrak F, Schilling RJ, Kirkby C, Wragg A, Manisty C, Mohiddin S.. Diagnosis and prognosis in sudden cardiac arrest survivors without coronary artery disease: utility of a clinical approach using cardiac magnetic resonance imaging. Circ Cardiovasc Imaging 2017;10:e006709.. [DOI] [PubMed] [Google Scholar]
- 162. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A.. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2007;29:270–276. [DOI] [PubMed] [Google Scholar]
- 163. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J.. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 2013;62:2046–2072. [DOI] [PubMed] [Google Scholar]
- 164. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J.. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. Glob Heart 2013;8:355–382. [DOI] [PubMed] [Google Scholar]
- 165. Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M, Favalli V, Bellazzi R, Tajik JA, Bonow RO, Bonow RD, Fuster V, Narula J.. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 2014;64:304–318. [DOI] [PubMed] [Google Scholar]
- 166. Elliott PM. Classification of cardiomyopathies: evolution or revolution? J Am Coll Cardiol 2013;62:2073–2074. [DOI] [PubMed] [Google Scholar]
- 167. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J.. Reply: The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: more questions than answers? J Am Coll Cardiol 2013;8:2584–2586. [DOI] [PubMed] [Google Scholar]
- 168. Agarwal A, Yousefzai R, Jan MF, Cho C, Shetabi K, Bush M, Khandheria BK, Paterick TE, Treiber S, Sra J, Werner P, Allaqaband S, Bajwa T, Tajik AJ.. Clinical application of WHF-MOGE(S) classification for hypertrophic cardiomyopathy. Glob Heart 2015;10:209–219. [DOI] [PubMed] [Google Scholar]
- 169. Mayosi BM. MOGE(S): a standardized classification of cardiomyopathies? Nat Rev Cardiol 2014;11:134–135. [DOI] [PubMed] [Google Scholar]
- 170. Dec GW, Arbustini E.. Utilizing the MOGE(S) classification for predicting prognosis in dilated cardiomyopathy. J Am Coll Cardiol 2015;66:1324–1326. [DOI] [PubMed] [Google Scholar]
- 171. Westphal JG, Rigopoulos AG, Bakogiannis C, Ludwig SE, Mavrogeni S, Bigalke B, Doenst T, Pauschinger M, Tschöpe C, Schulze PC, Noutsias M.. The MOGE(S) classification for cardiomyopathies: current status and future outlook. Heart Fail Rev 2017;22:743–710. [DOI] [PubMed] [Google Scholar]
- 172. Favalli V, Disabella E, Molinaro M, Tagliani M, Scarabotto A, Serio A, Grasso M, Narula N, Giorgianni C, Caspani C, Concardi M, Agozzino M, Giordano C, Smirnova A, Kodama T, Giuliani L, Antoniazzi E, Borroni RG, Vassallo C, Mangione F, Scelsi L, Ghio S, Pellegrini C, Zedde M, Fancellu L, Sechi GPietro, Ganau A, Piga S, Colucci A, Concolino D, Di Mascio MT, Toni D, Diomedi M, Rapezzi C, Biagini E, Marini M, Rasura M, Melis M, Nucera A, Guidetti D, Mancuso M, Scoditti U, Cassini P, Narula J, Tavazzi L, Arbustini E.. Genetic screening of anderson-fabry disease in probands referred from multispecialty clinics. J Am Coll Cardiol 2016;68:1037–1050. [DOI] [PubMed] [Google Scholar]
- 173. Captur G, Arbustini E, Bonne G, Syrris P, Mills K, Wahbi K, Mohiddin SA, McKenna WJ, Pettit S, Ho CY, Muchir A, Gissen P, Elliott PM, Moon JC.. Lamin and the heart. Heart 2018;104:468–479. [DOI] [PubMed] [Google Scholar]
- 174. Authors/Task Force Members, Elliott P, Anastasakis A, A. Borger M, Borggrefe M, Cecchi F, Charron P, Alain Hagege A, Lafont A, Limongelli G, Mahrholdt H, J. McKenna W, Mogensen J, Nihoyannopoulos P, Nistri S, G. Pieper P, Pieske B, Rapezzi C, H. Rutten F, Tillmanns C, Watkins H.. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Russ J Cardiol 2015;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 175. Muchir A, Worman HJ.. Targeting mitogen-activated protein kinase signaling in mouse models of cardiomyopathy caused by lamin A/C gene mutations. Methods Enzymol 2016;568:557–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 2014;171:2029–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]