

Abstract
Aims
GTP cyclohydrolase I catalyses the first and rate-limiting reaction in the synthesis of tetrahydrobiopterin (BH4), an essential cofactor for nitric oxide synthases (NOS). Both eNOS and iNOS have been implicated in the progression of atherosclerosis, with opposing effects in eNOS and iNOS knockout mice. However, the pathophysiologic requirement for BH4 in regulating both eNOS and iNOS function, and the effects of loss of BH4 on the progression of atherosclerosis remains unknown.
Methods and results
Hyperlipidemic mice deficient in Gch1 in endothelial cells and leucocytes were generated by crossing Gch1fl/flTie2cre mice with ApoE–/– mice. Deficiency of Gch1 and BH4 in endothelial cells and myeloid cells was associated with mildly increased blood pressure. High fat feeding for 6 weeks in Gch1fl/flTie2CreApoE–/– mice resulted in significantly decreased circulating BH4 levels, increased atherosclerosis burden and increased plaque macrophage content. Gch1fl/flTie2CreApoE–/– mice showed hallmarks of endothelial cell dysfunction, with increased aortic VCAM-1 expression and decreased endothelial cell dependent vasodilation. Furthermore, loss of BH4 from pro-inflammatory macrophages resulted in increased foam cell formation and altered cellular redox signalling, with decreased expression of antioxidant genes and increased reactive oxygen species. Bone marrow chimeras revealed that loss of Gch1 in both endothelial cells and leucocytes is required to accelerate atherosclerosis.
Conclusion
Both endothelial cell and macrophage BH4 play important roles in the regulation of NOS function and cellular redox signalling in atherosclerosis.
Keywords: GCH, Tetrahydrobiopterin, Atherosclerosis, Macrophages, Endothelial cells
1. Introduction
GTP cyclohydrolase I (GTPCH) catalyses the first and rate-limiting reaction in the synthesis of tetrahydrobiopterin (BH4), an essential cofactor for the aromatic amino acid hydroxylases, alkylglycerol mono-oxygenase and the three nitric oxide synthases (NOS1–3).1 BH4 regulates NOS function, by coupling the haem-oxygen intermediate to l-arginine oxidation, thus controlling the generation of either nitric oxide (NO) or, in conditions of low BH4, superoxide, due to NOS uncoupling. In vascular disease states, BH4 bioavailability is decreased due to reduced biosynthesis,2 oxidative loss of BH4, forming dihydrobiopterin (BH2), and attenuated recycling of BH4 from BH2 by dihydrofolate reductase.3,4 In vascular disease states such as atherosclerosis, augmentation of endothelial cell BH4 levels, either by genetic over-expression of GTPCH or pharmacologic supplementation, can restore endothelial NOS function and reduce atherosclerotic plaque progression.5,6 However, the pathophysiologic requirement for endothelial cell BH4 biosynthesis in atherosclerosis, as distinct from the effects of BH4 supplementation, remain unknown.
A further important consideration in the role of NOS regulation by BH4 in atherosclerosis is the effect of BH4 on inducible NOS (iNOS) function. In atherosclerotic lesions iNOS co-localises with areas of lipid oxidation and protein nitration.7 iNOS expression exacerbates atherosclerotic plaque progression, with decreased atherosclerosis found in hyperlipidemic iNOS–/– mice.8–11 This pro-inflammatory environment is predominantly driven by monocyte/macrophage infiltration, demonstrated in bone marrow chimera studies in iNOS–/– mice in which leucocyte iNOS was shown to be pro-atherogenic.12 Inflammatory stimulation typically induces both iNOS expression, and increased BH4 biosynthesis.13,14 Patients with established atherosclerotic disease have reduced vascular BH4 levels, associated with impaired endothelial function, whereas plasma biopterins are increased, associated with biomarkers of inflammation.15,16 Thus, the requirement for BH4 in iNOS function in leucocytes is a novel and important consideration in the pathogenesis of atherosclerosis. However, previous studies in iNOS knockout mice only address the overall requirement for iNOS, and cannot address the importance of BH4 in iNOS function and the consequences of NOS uncoupling and redox dysregulation. Indeed, the opposing actions of endothelial cell eNOS vs. leucocyte iNOS in atherosclerosis, and the effects of BH4 in eNOS and iNOS function, have important implications for the potential of BH4 as a therapeutic target in vascular disease states.
We have recently generated a novel mouse model with conditional deletion of Gch1, leading to cell-specific loss of BH4 synthesis in endothelial cells and leucocytes. We sought to understand the requirement for BH4 in regulating both eNOS and iNOS, in endothelial and inflammatory cells, and the effects of loss of BH4 on the progression of atherosclerosis.
2. Methods
2.1 Animals
Endothelial and myeloid cell Gch1 knockout mice on a hyperlipidemic (ApoE knock out; ApoE–/–) background were generated by crossing Gch1fl/flTie2cre mice13,17 with ApoE–/– mice (Jackson Laboratories, Bar Harbor, USA). Matched litters of Gch1fl/flTie2CreApoE–/– and Gch1fl/flApoE–/– were generated by breeding male Gch1fl/flTie2CreApoE–/– with female Gch1fl/flApoE–/– females. Animals were housed in individually ventilated cages with 12-hour light/dark cycle and controlled temperature (20–22°C). Mice were fed standard chow (B&K Ltd, UK) up to 16 weeks of age or up to 10 weeks of age when they were switched to high fat diet (HFD; SDS 829108 Western RD diet, UK) for 6 weeks. Water and food were available ab libitum at all times.
Chimeric mice were generated in a manner similar to that described previously.18 Briefly, donor Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice were killed and a single-cell suspension of bone marrow prepared. Six-week-old Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice received a lethal dose of whole body irradiation (2 × 5 Gy) followed by an intravenous injection of 1 × 107 bone-marrow cells in 0.2 mL phosphate-buffered saline from either Tie2cre-positive or negative donor mice. Four weeks (10 weeks of age) after bone marrow transplant chimerization was confirmed by PCR of tail blood and mice were switched to an HFD for 6 weeks. DNA was extracted from bone marrow at the time of harvest and the absence or presence of Gch1 was assessed using PCR to confirm bone marrow reconstitution.
Genotyping of experimental mice was performed by standard PCR techniques. All mice were culled by exsanguination under terminal anaesthetic (isoflurane >3% in 95% O2 5% CO2). All animal procedures were approved and carried out in accordance with the University of Oxford ethical committee and the UK Home Office Animals (Scientific Procedures) Act 1986. All procedures conformed with the Directive 2010/63/EU of the European Parliament.
2.2 Tissue collection
Tissue for histological analysis was collected from mice perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde, tissue for biochemical analysis was collected from mice perfused with PBS only and was snap frozen in liquid nitrogen and stored at –80°C until analysis. Primary endothelial cells were isolated from lungs by immunoselection with CD31 antibody (BD Biosciences, Wokingham, UK) coated magnetic beads as described previously.19 Bone-marrow–derived macrophages (BMDMs) were obtained as follows. Bone marrow was obtained by flushing the femur and tibia of adult mice with PBS. A single cell suspension was prepared by passing the bone marrow through a 70 mm cell strainer. Cells were cultured in 10 cm non-tissue culture treated dishes for 7 days in DMEM: F12 (Invitrogen, Loughborough, UK) supplemented with 100 U/mL penicillin and 100 ng/mL streptomycin (Sigma, Gillingham, UK), 10% (v/v) foetal bovine serum (PAA Laboratories, Loughborough, UK), 5 mM l-glutamine (Sigma), and 10–15% (v/v) L929 conditioned medium. The differentiation of the cells was confirmed using flow cytometry using a CyAn ADP (Beckton Coulter, High Wycombe, UK) for data acquisition and Flow Jo (TreeStar Inc., Wokingham, UK) for analysis. Macrophages were defined as being CD11b (PerCP conjugated) and F4:80 (APC conjugated, both Biolegend, London, UK) positive cells, as judged against isotype controls conjugated with the same fluorochromes (Biolegend). Following differentiation, cells were harvested and plated into 6- or 96-well plates containing serum-free media [Optimem supplemented with 100 U/mL penicillin and 100 ng/mL streptomycin and 0.2% (w/v) low-endotoxin bovine serum albumin (Sigma)]. Cells were stimulated with 10 ng/mL IFNγ (Peprotech EC) and 100 ng/mL LPS (Sigma) with or without acetylated LDL (20 μg/mL; Invitrogen) for 16 h; parallel wells were left unstimulated. After 16 h cell pellets and cell culture supernatants were collected, or the cells subjected to biochemical analysis.
Nuclear fractions were extracted from a total of 6 × 106 macrophage using a nuclear fraction isolation kit (Cayman Chemicals, Ann Arbor, USA). Protein concentration in nuclear fractions was assessed using a modified Bradford assay. Nrf2 transcription activity of nuclear fractions (6 μg total nuclear protein) was quantified by assessing transcription factor binding activity (Cayman Chemicals).20
Total RNA was extracted using the Ambion Pure Link kit. Reverse transcription was carried out using QuantiTect reverse transcription kit (Qiagen, Hilden, Germany, UK) on 1 μg total cell RNA. Quantitative real-time RT–PCR was performed with an iCycler IQ real-time detection system (BioRad Laboratories, Hercules, USA) using primers and probes from the TaqMan Gene Expression Assay system (Life Technologies, Loughborough, UK). Gene expression data were normalized to GAPDH with the exception of BMDM when β-actin was used.
2.3 Western blotting
Western blotting was carried out on aorta, primary endothelial cells, BMDM homogenates (15 µg protein), liver (5 µg protein), and macrophage nuclear fraction (5 µg nuclear protein) using standard techniques and iNOS (BD Pharmigen, Wokingham, UK, 610329), anti-GTPCH (custom made, a gift from Dr S. Gross), GAPDH (Millpore, Watford, UK MAB374), TBP (Abcam, Cambridge, UK; ab818), and Nrf2 (Abcam, Cambridge, UK; ab137550) antibodies.
2.4 Isometric tension vasomotor studies
Vascular rings were isolated from thoracic aorta of female chow and HFD mice and mounted on a wire myograph (MultiMyogrph 610M, Danish Myo Technology, Aarhus, Denmark) containing Krebs-Henseleit buffer. Vessel viability was tested using 60 mM KCl. Concentration–response contraction curves were established to phenylephrine and acetylcholine in the absence and presence of 10 µM, L-NAME or 10 µM sepiapterin.
2.5 Determination of BH4 levels
BH4 levels in tissue, cells, and plasma were determined by high-performance liquid chromatography (HPLC) followed by electrochemical and fluorescent detection, as previously described.4
2.6 Quantification of superoxide production
Superoxide production from primary endothelial cells and BMDM from 16-week-old chow fed mice was measured by quantifying the accumulation of 2-hydroxyethidium by HPLC as previously described.4
2.7 Blood pressure and pulse measurements in conscious mice
Heart rate and systolic blood pressure were measured using an automated computerized tail-cuff system in 16-week-old chow and HFD fed conscious mice, as described previously (Visitech BP2000, Visitech Systems, Inc., Apex, USA).21
2.8 Lipid and lipoprotein analysis
Biochemical analyses of plasma lipids were performed on heparinized blood plasma using enzymatic assays. BMDM were differentiated into macrophages as above. Cells were cultured in Optimem containing 0.2% fatty acid free serum and labelled with [3 H] cholesterol (2 µCi/mL) overnight. Cells were washed and equilibrated overnight and reverse cholesterol transport to pooled ApoE–/– plasma (2.5%) assessed after 6 h by scintillation counting of media and cellular [H3] cholesterol. For lipid uptake assays, cells BMDM were either unstimulated or stimulated with LPS/IFNγ as above. After stimulation overnight, cells were treated with 20 µg/mL of Dil AcLDL (Invitrogen, Loughborough, UK) for 8 h. Fixed cells were imaged using the Operetta high content imaging system (Perkin Elmer, Seer Green, UK). The fluorescent intensity resulting from cellular uptake of DiI AcLDL was quantified using Harmony image analysis software.
2.9 Histology and immunohistochemistry of atherosclerotic aortic root sections
Lesion size was assessed in paraffin-embedded aortic root sections stained with Masson’s-Goldner (Merck, Darmstadt, Germany). Average lesion size was calculated from six sections taken at 45 µm intervals starting from the section showing all three aortic cusps. Aortic lesion macrophage content was analysed using anti-Gal-3 (BD Pharmingen, Wokingham, UK) immunostaining. Plaque collagen content was assessed using Sirius red staining. Mast cells within the plaque and perivascular area of the aortic root were assessed by Toludine Blue staining. Lesion area, Gal-3, and collagen positive areas were quantified from digitized microscopic images using Image-Pro Plus.
2.10 Nitrite determination and analysis of NO synthesis
Nitrite accumulation was measured in samples of cell culture medium using the Griess assay with colorimetric detection in 96-well plates, as described.22 NO synthesis activity was assessed by conversion of [14C]l-arginine to citrulline, in the presence and absence of N-methyl-Larginine, as described previously.23
2.11 Quantification of plasma leucocytes and peritoneal macrophage recruitment
Cell populations in whole blood (EDTA) from high fat fed mice were stained with monoclonal antibodies directed against CD45+, CD11b+, Ly6C+, Ly6G– (monocytes) and CD45+, CD11b+, Ly6C+, Ly6G+ (neutrophils). For in vivo peritoneal recruitment experiments mice were injected ip with 4% thioglycolate, 4 days later mice were killed and the peritoneal cavity was lavaged with 5 mL of PBS containing 2 mM EDTA. Peritoneal exudate cells were stained with antibodies against CD45 (all leucocytes), CD11b and F4:80 (macrophages). All flow cytometry was performed using a DAKO CyAn cytometer and Summit software (Beckton Coulter, High Wycombe, UK). Data was analysed using Flow Jo software (TreeStar Inc, Wokingham, UK). Where cells were enumerated an absolute count protocol was used, where cells were quantified by ratio to a known number of fluorescent beads which were spiked into the sample prior to analysis.
2.12 Statistical analysis
Data are presented as mean ± SEM. Normality was tested using D’Agostino and Pearson omnibus normality test. Groups were compared using the Mann–Whitney U test for non-parametric data or an un-paired Student’s t-test for parametric data. When comparing multiple groups data were analysed by analysis of variance (ANOVA) with Newman–Keuls post-test for parametric data or Kruskal–Wallis test with Dunns post-test for non-parametric data. When more than two independent variables were present a two-way ANOVA with Tukey’s multiple comparisons test was used. When within subject repeated measurements were present a repeated measures (RM) ANOVA was used. A value of P < 0.05 was considered statistically significant.
3. Results
3.1 Leucocyte and endothelial cell Gch1 deficiency leads to decreased circulating BH4 and increased arterial blood pressure after high fat feeding
Genomic PCR demonstrated efficient excision of the floxed Gch1 allele in endothelial and myeloid cell-rich tissues such as aorta, lungs, heart and spleen from Gch1fl/flTie2CreApoE–/– mice, compared with minimal excision in liver (Figure 1A). Endothelial and leucocyte specific Gch1 deletion resulted in a significant reduction in aortic Gch1 expression (>90%; P < 0.05; Figure 1B) and GTPCH protein (Figure 1C). Accordingly, BH4 levels were reduced by more than 80% in whole aortas (16.39 ± 1.38 vs. 3.96 ± 0.80 pmol/mg protein, P < 0.05, Figure 1D). Interestingly, a significant increase in aortic BH4 and BH2 content was observed in HFD Gch1fl/flApoE–/– mice, whereas no difference in BH4 or BH2 content was observed in aortas from HFD vs. chow fed Gch1fl/flTie2CreApoE–/– (Figure 1D and Supplementary material online, Figure S1A).
Figure 1.

Characterization of the Gch1fl/flTie2CreApoE–/– mouse. (A) PCR genomic DNA in tissues from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice. (B) Real time qRT–PCR showing a significant decrease in expression in aortas from 16-week-old non-HFD female Gch1fl/flTie2CreApoE–/– mice compared with aortas from Gch1fl/flApoE–/– littermates (un-paired T-test, n = 8 per group). (C) Representative immunoblot showing decreased expression of GTPCH in aortas from Gch1fl/flTie2CreApoE–/– mice. LPS/IFNγ stimulated BMDMs are used as a positive control. (D) BH4 in aortas of chow or HDF mice showing a significant decrease in BH4 levels in Gch1fl/flTie2CreApoE–/– and a significant increase in BH4 levels in Gch1fl/flApoE–/– after HFD (two-way ANOVA, n = 7–12 per group). (E) Systolic blood pressure was increase in chow fed Gch1fl/flTie2CreApoE–/– mice compared with Gch1fl/flApoE–/– (un-paired T-test; n = 7–9 per group). (F) Systolic blood pressure was increase in male HFD Gch1fl/flTie2CreApoE–/– male mice but not females (un-paired T-test, n = 7–14 per group). BH4, BH2, and the ratio of BH4:BH2 and B in plasma from chow and HFD male (G) and female (H) mice. No difference in plasma levels between genotypes was observed in chow few mice, but a significant decrease in plasma BH4 and BH2 levels were observed in Gch1fl/flTie2CreApoE–/– mice after HFD (two-way ANOVA; n = 5–10 per group). Data are expressed as the mean ± SEM. *P < 0.05 between genotypes of the same treatment. $P < 0.05 between treatments of the same genotype. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–.
We next assessed how endothelial cell and leucocyte-specific Gch1 deficiency altered blood pressure in hyperlipidemic ApoE–/– mice. At 16 weeks of age systolic blood pressure was slightly higher in both male (103 ± 1.5 vs. 109 ± 1.3 mmHg) and female (97 ± 2 vs. 105 ± 2.3 mmHg) chow fed Gch1fl/flTie2CreApoE–/– mice compared with their control littermates (P < 0.05, Figure 1E). This increase in blood pressure was preserved in HFD male mice, however, in HFD female mice the increase in blood pressure observed in chow fed Gch1fl/flTie2CreApoE–/– was no longer apparent (Figure 1F). There was no difference in heart rate in either chow or HFD males or female mice (Supplementary material online, Figure S2). In mice fed a standard chow diet, circulating levels of BH4 and BH2 in male and female Gch1fl/flTie2CreApoE–/– were not significantly different compared with their matched littermates. However, after 6 weeks on an HFD plasma levels of both BH4 and BH2 levels were significantly reduced in both male (Figure 1G) and female (Figure 1H) Gch1fl/flTie2CreApoE–/– mice (P < 0.05). The similar reduction in BH4 and BH2 in Gch1fl/flTie2CreApoE–/– mice, resulted in no change in the ratio of BH4 to BH2 and B. As the liver is hypothesized to be the major site of circulating BH4, we assessed the impact of loss of Gch1 in endothelial cells and leucocytes on liver levels. The absence of Gch1 did not impact on either GTPCH protein or BH4 levels in the livers of chow fed mice (Supplementary material online, Figure S1B and C). However, after 6 weeks of HFD a significant reduction in BH4 levels was observed in livers from Gch1fl/flTie2CreApoE–/– mice (Supplementary material online, Figure S1C). In contrast, a significant increase in liver BH2 was observed after HFD in both groups, with a greater increase observed in livers from Gch1fl/flApoE–/– mice (Supplementary material online, Figure S1D).
3.2 Deficiency in Gch1 in leucocytes and endothelial cells results in increased atherosclerosis burden
We next aimed to assess how Gch1 deletion and BH4 deficiency in endothelial cells and leucocytes altered the development of atherosclerosis. Male and female Gch1fl/flTie2CreApoE–/– and Gch1fl/flApoE–/– littermates were fed an HFD for 6 weeks from 10 to 16 weeks of age. There was no difference in body weight between the two groups (Table 1). Plasma lipid analysis revealed that loss of Gch1 in endothelial cells and leucocytes did not alter circulating levels of either total cholesterol, LDL cholesterol, triglycerides, or HDL. Furthermore, there was no change in circulating leucocytes between groups, with no difference observed in numbers of monocytes (CD45+, CD11b+, Ly6C+, and Ly6C–) or neutrophils (CD45+, CD11b+, Ly6C+, and Ly6G+; Table 1).
Table 1.
Plasma lipid levels and circulating leucocyte levels in the high fat fed Gch1fl/flApoE–/– and Gch1fl/flTies2CreApoE–/– mice
| Males |
Females |
|||
|---|---|---|---|---|
| Gch1fl/flApoE–/– | Gch1fl/flTie2Cre ApoE–/– | Gch1fl/flApoE–/– | Gch1fl/flTie2Cre ApoE–/– | |
| Body weight (g) | 36.7 ± 1.0 | 37.9 ± 1.3 | 25.6 ± 0.8 | 25.3 ± 1.0 |
| Total cholesterol (mmol/L) | 46.64 ± 2.09 | 43.76 ± 1.40 | 38.44 ± 1.79 | 33.13 ± 1.52 |
| LDL cholesterol (mmol/L) | 31.66 ± 1.20 | 30.83 ± 0.84 | 26.08 ± 1.66 | 24.37 ± 1.00 |
| Triglycerides (mmol/L) | 4.17 ± 0.28 | 3.65 ± 0.26 | 2.08 ± 0.29 | 2.67 ± 0.35 |
| HDL cholesterol (mmol/L) | 2.95 ± 0.11 | 2.88 ± 0.09 | 2.13 ± 0.07 | 2.24 ± 0.07 |
| Monocytes (cells/mL blood) | 2.558 × 105 ± 1.073 × 105 | 5.365 × 105 ± 1.388 × 105 | 1.958 × 105 ± 2.067 × 104 | 2.491 × 105 ± 2.193 × 104 |
| Neutrophils (cells/mL blood) | 2.787 × 105 ± 5.161 × 104 | 4.109 × 105 ± 7.988 × 104 | 2.010 × 105 ± 7.995 × 103 | 2.364 × 105 ± 1.144 × 104 |
Data are presented as the mean ± SEM, n = 5 for leucocytes analysis and 6–10 per group for lipid analysis.
Analysis of Masson-Goldner stained aortic roots (average of six sections, taken 45 µm apart) demonstrated a significant increase in plaque burden in aortic roots from Gch1fl/flTie2CreApoE–/– mice in both males (0.040 ± 0.003 vs. 0.055 ± 0.006 mm2, P < 0.05) and females (0.100 ± 0.009 vs. 0.141 ± 0.017 mm2, P < 0.05; Figure 2A and B). Although absolute plaque size was different in males vs. females, a similar percentage increase in plaques size was observed in both sexes (28% and 27%, respectively). Collagen content, assessed with Sirius red staining, was not significantly different in plaques from Gch1fl/flTie2CreApoE–/– mice and their littermate controls (Figure 2A and C). Total mast cell content of the aortic root, assessed in Toluline Blue stained sections, was not significantly different between groups. In addition, there was no difference in the number of granulated or de-granulated mast cells in aortic roots between Gch1fl/flTie2CreApoE–/– mice and their littermate controls (Supplementary material online, Figure S3).
Figure 2.

Loss of Gch1 in endothelial cells and leucocytes causes increased atherosclerosis. Quantification of aortic root atherosclerosis in Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice at 16 weeks after 6 weeks of HFD (10–16 weeks of age). (A) Representative picture of aortic roots stained with Masson’s Goldner for plaque quantification, Sirius red for collagen content and Galectin-3 for macrophage content (macrophages stained red). (B) A significant increase in plaque was observed in both male and female Gch1fl/flTie2CreApoE–/– compared with Gch1fl/flApoE–/– (P < 0.05) as assessed by Masson’s Goldner staining. (C) Sirius red staining was not different between genotypes (P > 0.05). (D) A significant increase in plaque macrophage content was observed in aortic roots from both male and female Gch1fl/flTie2CreApoE–/– mice (P < 0.05). (E) A significant increase was observed in percentage macrophage content in plaques from male Gch1fl/flTie2CreApoE–/– but not female mice (P < 0.05). Data are expressed as the mean ± SEM, un-paired T-test between different genotypes of the same sex; n = 12–18 per group *P < 0.05 between genotypes of the same treatments. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–, black scale bar = 0.5 mm.
Immunostaining for macrophages (Galectin-3; Gal-3) indicated that the increase in plaque size was accompanied by an increase in plaque macrophage content in Gch1fl/flTie2CreApoE–/– mice in both males (0.014 ± 0.002 vs. 0.030 ± 0.004 mm2, P < 0.05) and females (0.023 ± 0.04 vs. 0.047 ± 0.008 mm2, P < 0.05; Figure 2A and D). In female mice, there was no difference in the percentage of macrophage content in the plaques, indicating that the increase in macrophage content was accompanied by a proportionate increase in plaque size. However, in male mice there was a significant increase in the percentage macrophage content of the plaques (31.51 ± 3.15 vs. 40.47 ± 2.61%, P < 0.05) indicating that plaques from male Gch1fl/flTie2CreApoE–/– mice were more inflamed than plaques from Gch1fl/flApoE–/– (Figure 2E).
3.3 Endothelial cell deficiency in BH4 leads to enhanced vasoconstriction, impaired vasodilation, and endothelial cell dysfunction
We next evaluated the requirement for BH4 in primary endothelial cells from Gch1fl/flTie2CreApoE–/– mice. Both Gch1 RNA expression and GTPCH protein levels were significantly reduced (P < 0.05; Figure 3A and B), leading to greatly reduced BH4 and BH2 levels in endothelial cells from Gch1fl/flTie2CreApoE–/– compared with Gch1fl/flApoE–/– (25.83 ± 6.33 vs. 6.13 ± 0.40, Figure 3C and Supplementary material online, Figure S1E).
Figure 3.

Characterization of endothelial cells from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/–mice. (A) Real time qRT–PCR showing a significant decrease in Gch1 expression in primary endothelial cells from Gch1fl/flTie2CreApoE–/– mice (Mann–Whitney U test; n = 3–4 per group). (B) Representative immunoblot of GTPCH levels in primary endothelial cells showing decreased expression of GTPCH in endothelial cells from Gch1fl/flTie2CreApoE–/– mice. (C) BH4 levels in primary endothelial cells were significant reduced in endothelial cells from Gch1fl/flTie2CreApoE–/– mice (Mann–Whitney U test, n = 4–6 per group). (D) Superoxide generation in primary lung endothelial cells from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice, endothelial cells from Gch1fl/flTie2CreApoE–/– mice had a significant increase in 2-hydroxyethidium (two-way ANOVA; n = 5–7 per group). (E) SOD (100 U/mL) caused a significant decrease in 2-hydroxyethidium production in both groups with a greater inhibitable fraction observed in endothelial cells from Gch1fl/flTie2CreApoE–/– (Mann–Whitney U test; n = 6–7 per group). (F) Quantification of arginine to citrulline conversion in primary endothelial cells from HFD mice. A significant decrease in conversion was observed in cells from Gch1fl/flTie2CreApoE–/– (un-paired T-test, n = 6–8 in KHB, n = 2 representative aortas in the LNMMA group). (G) Endothelial dependent vasodilation to acetylcholine (Ach) was significantly reduced in aortas from chow fed female Gch1fl/flTie2CreApoE–/– mice, incubation with L-NAME completely inhibited relaxation in both groups (RM ANOVA; n = 7 per group). (H) HFD caused a significant decrease in endothelial dependent vasodilation to Ach in aortas from female Gch1fl/flTie2CreApoE–/– mice, ex vivo incubation with sepiapterin restored the dilator response to wild type levels (RM ANOVA, n = 8 per group). *P < 0.05 between genotypes of the same treatments. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–, KHB = Krebs Henseleit buffer.
To determine the effects of endothelial cell-specific BH4 deficiency on endothelial cell function, we first measured basal O2– production in isolated primary endothelial cells. Generation of 2-hydroxyethidium from hydroxyethidine was significantly higher in primary endothelial cells from Gch1fl/flTie2CreApoE–/– mice (209 ± 20 vs. 340 ± 28; Figure 3D). Using superoxide dismutase (SOD) to confirm the specificity of the 2-hydroxyethidium product for O2–; we also observed a greater SOD inhabitable fraction of 2-hydroxyethidium in endothelial cells from Gch1fl/flTie2CreApoE–/– (P < 0.05; Figure 3E).
We next assessed NOS activity using radiolabelled l-arginine to l-citrulline conversion. Primary endothelial cells from HFD Gch1-deficient mice showed a significant decrease in l-arginine to l-citrulline conversion compared with endothelial cells from their control littermates. The specificity for NOS activity was confirmed using the NOS inhibitor LNNMA, in both groups (P < 0.05; Figure 3F).
We next investigated how endothelial cell BH4 deficiency in ApoE–/– mice affected vascular function. Isometric tension studies in isolated aortas from chow fed Gch1fl/flTie2CreApoE–/– mice demonstrated a significant increase in phenylephrine-induced vasoconstriction and a significant reduction in endothelial-dependent vasorelaxation to acetylcholine. The relaxation response to acetylcholine in both groups was completely abolished in the presence of L-NAME, confirming NOS as the major source of vasodilators (Figure 3G and Supplementary material online, Figure S4A). Endothelial cell dependent relaxation response to acetylcholine was significantly blunted in aortas from HFD Gch1fl/flTie2CreApoE–/– mice, but this effect was reversed in the presence of sepiapterin (Figure 3H). The enhanced constrictor response observed in chow fed Gch1fl/flTie2CreApoE–/– mice was not observed in HFD mice. There was no difference in constrictor response between the two genotypes after treatment with sepiapterin (Supplementary material online, Figure S4B).
We next assessed how deficiency in endothelial cell Gch1 altered aortic expression of pro-atherogenic genes. Aortic Vcam1 expression was significantly higher in chow fed Gch1fl/flTie2CreApoE–/– mice (0.1 ± 0.01 vs. 0.17 ± 0.03; P < 0.05 Figure 4A). Deficiency in Gch1 in chow fed mice did not lead to a compensatory change in eNOS levels, nor did it not alter expression of either CCL2 or TNFα (Figure 4A). Vcam1 levels were also significantly elevated in aortas from HFD Gch1fl/flTie2CreApoE–/– mice and, in contrast to aortas from chow fed mice, a significant increase in both TNF-α and eNOS was observed along with a non-significant trend towards an increase in CCL2 (P = 0.053, Figure 4B).
Figure 4.
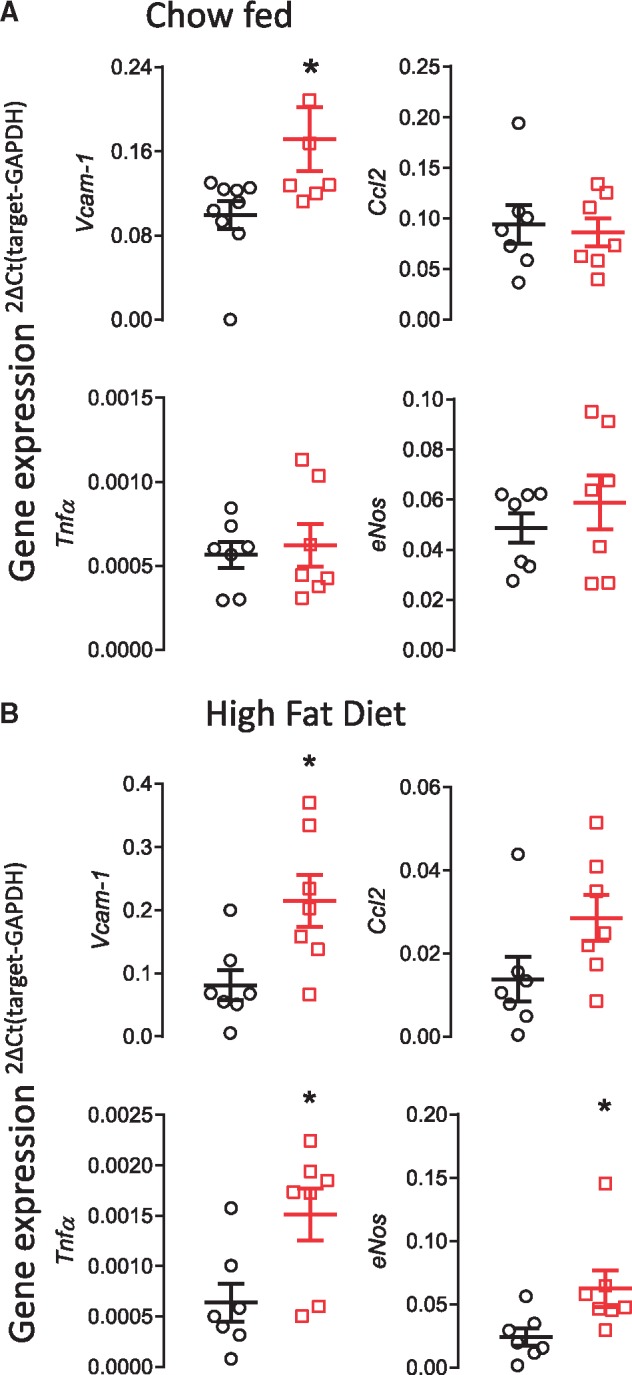
Loss of Gch1 leads to increased aortic expression of atherosclerosis related genes. (A) Increase in VCAM-1expression in aortas from 16-week-old chow fed female Gch1fl/flTie2CreApoE–/– mice compared with their Gch1fl/flApoE–/– littermates. There was no difference between genotype in expression of either CCL2, eNOS or TNF-α. (B) A significant increase in VCAM-1, eNOS and TNF-α was observed in aortas from HFD Gch1fl/flTie2CreApoE–/–. No significant difference in CCL2 was observed (P = 0.053). Data are expressed as the mean ± SEM, Mann–Whitney U test, n = 6–9 per group, *P < 0.05 between genotypes of the same treatment. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–.
3.4 Macrophages from Gch1fl/flTie2CreApoE–/– mice have altered iNOS function and increased ROS generation
We next sought to determine the requirement for Gch1, and BH4, in macrophages from ApoE–/– mice. Deletion of Gch1 resulted in a greater than 90% loss of Gch1 mRNA and no detectable GTPCH protein in BMDMs (Figure 5A and B). Stimulation of BMDM for 24 h with LPS/IFNγ resulted in upregulation of Gch1 in macrophages from control Gch1fl/flApoE–/– mice, which was accompanied by a robust increase in iNOS protein (Figure 5B). However, despite iNOS being similarly upregulated in macrophages from both groups, Gch1 mRNA remained significantly decreased in macrophages from Gch1fl/flTie2CreApoE–/– mice (0.034 ± 0.0022 vs. 0.002 ± 0.0004, P < 0.05) with GTPCH protein levels undetectable by western blot (Figure 5B and C). Consequently, BH4 levels in both unstimulated and stimulated and BH2 levels in stimulated macrophages from Gch1fl/flTie2CreApoE–/– mice were strikingly reduced (P < 0.05; Figure 5D and Supplementary material online, Figure S1F). We next assessed how this deficiency in Gch1 and BH4 altered the ability of the macrophages to generate nitric oxide. Nitrite levels were undetectable in unstimulated macrophages from either genotype. However, upon stimulation with LPS/IFNγ, macrophages from control mice accumulated very high levels of nitrite in the medium, whereas BH4-deficient macrophages from Gch1fl/flTie2CreApoE–/– mice generated minimal nitrite (31.8 ± 5.4 vs. 1.9 ± 00.9, P > 0.05; Figure 5E). A similar result was observed using radiolabelled l-arginine to l-citrulline conversion, in order to directly quantify NOS enzymatic activity. LPS/IFNγ-stimulated macrophages from Gch1 deficient mice showed minimal l-arginine to l-citrulline conversion compared with macrophages from their control littermates (Figure 5F). The specificity for NOS activity was confirmed using the NOS inhibitor L-NMMA, in both groups (P < 0.05; Figure 5F).
Figure 5.

Characterization of BMDM from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice. (A) Real time qRT–PCR expression of Gch1 in unstimulated macrophages showing a significant decrease in Gch1 expression in Gch1fl/flTie2CreApoE–/– macrophages (Mann–Whitney U test, n = 5 per group). (B) Representative immunoblot showing iNOS and GTPCH levels in stimulated (LPS/IFNγ) and unstimulated macrophages. (C) Real time qRT–PCR expression of Gch1 in LPS/IFNγ stimulated macrophages showing a significant decrease in Gch1 expression in Gch1fl/flTie2CreApoE–/– macrophages (Mann–Whitney U test, n = 5 per group). (D) BH4 levels in unstimulated and LPS/IFNγ stimulated macrophages, a significant reduction in BH4 levels was observed in macrophages from Gch1fl/flTie2CreApoE–/– mice (two-way ANOVA, n = 8 per group). (E) Nitrite accumulation in the cell culture media over 18 h of cell stimulation using the Greiss assay. Stimulation with LPS/IFNγ cause a significant increase in nitrite in macrophages from Gch1fl/flApoE–/– mice however levels were only just detectable in macrophages from Gch1fl/flTie2CreApoE–/– mice (two-way ANOVA, n = 8 per group). (F) Quantification of arginine to citrulline conversion in LPS/IFNγ stimulated macrophages. A significant decrease in conversion was observed in macrophages from Gch1fl/flTie2CreApoE–/–, LNNMA cause a significant decrease in arginine to citrulline conversion in both groups (two-way ANOVA, n = 3–6 per group). (G) Superoxide generation in LPS/IFNγ stimulated macrophages from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice, macrophages from Gch1fl/flTie2CreApoE–/– mice had a significant increase in 2-hydroxyethidium production. SOD (100 U/mL) caused a significant decrease in 2-hydroxyethidium production in both groups (two-way ANOVA, n = 6–7 per group) with a significantly greater inhabitable fraction observed in macrophages from Gch1fl/flTie2CreApoE–/– (H, Mann–Whitney U test, n = 6–7 per group). Data are expressed as the mean ± SEM, *P < 0.05 between genotypes of the same treatment, $P > 0.05 between treatments of the same genotype. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–.
To investigate how deletion of Gch1, loss of BH4 and altered iNOS activity affects macrophage ROS production, we next measured O2– in LPS/IFNγ stimulated macrophages. 2-Hydroxyethidium production was significantly increased in Gch1 deficient macrophages, indicating elevated O2– production (214 ± 30.5 vs. 317 ± 19.8, P < 0.05; Figure 5G). In addition the SOD inhabitable fraction was significantly greater in macrophages from Gch1fl/flTie2CreApoE–/– mice (70.0 ± 7.8 vs. 123.1 ± 13.8, P < 0.05; Figure 5H).
3.5 Loss of BH4 increases macrophage foam cell formation and modifies cellular redox signalling
We next sought to determine the effects of BH4-dependent iNOS function in macrophages, in order to explore the mechanisms underlying increased atherosclerotic plaque formation in Gch1fl/flTie2CreApoE–/– mice. We first assessed in vivo chemotaxis in to the peritoneal cavity in response to an acute inflammatory response (4% thioglycollate). There was no difference in the number of CD45+ cells between Gch1fl/flTie2CreApoE–/– and control littermates (P > 0.05; Figure 6A). We next characterized the recruited cells, using CD11b and F4:80 to identify macrophages. We found no significant difference in the number of peritoneal macrophages between the two groups (P > 0.05; Figure 6A), suggesting that loss of BH4, and NO production, in LPS/IFNγ stimulated macrophages does not significantly alter chemotactic responses in vivo.
Figure 6.

Loss of Gch1 in BMDM leads to increased foam cell formation and anti-oxidant signalling. (A) Representative flow cytometry dot plots showing side scatter and cell surface expression of CD45 and CD11b and F4:80. Quantification of CD45+ cells and CD11b+ and F4:80+ macrophages in peritoneal lavage from thioglycolate treated mice showed no difference between the two genotypes (Mann–Whitney U test, n = 4–5). (B) Reverse cholesterol transport to pooled ApoE–/– plasma in unstimulated BMDM from Gch1fl/flApoE–/– and Gch1fl/flTie2CreApoE–/– mice, no significant differences were observed between groups (Mann–Whitney U test, n = 3–4 per group). (C) Foam cell formation in unstimulated and LPS/IFNγ stimulated BMDM, no difference between groups was observed in unstimulated macrophages; however, a significant increase in foam cell formation was observed in LPS/IFNγ stimulated macrophages from Gch1fl/flTie2CreApoE–/– mice (two-way ANOVA, n = 5 per group). (D) Real time qRT–PCR in unstimulated and LPS/IFNγ stimulated macrophages fed AcLDL (20 µg/mL). No difference in heme oxygenase expression was observed in unstimulated macrophages, stimulation with LPS/IFNγ resulted in a significant increase expression in macrophages from Gch1fl/flApoE–/–; however, this augmentation was significantly reduced in macrophages from Gch1fl/flTie2CreApoE–/– mice (two-way ANOVA, n = 4–5 per group). Real time qRT–PCR to Prdx1 (E), Nqo1 (F), and Cxcl5 (G) in LPS/IFNγ stimulated macrophages with or without AcLDL (20 µg/mL). For all genes, a significant reduction in expression was observed in AcLDL treated macrophages from Gch1fl/flTie2CreApoE–/– mice (two-way ANOVA, n = 4–5 per group). (H) Nrf2 nuclear transcription assay in LPS/IFNγ stimulated macrophages with or without AcLDL (20 µg/mL). No significant difference was observed in nuclear Nrf2 activity between groups (one-way ANOVA, n = 5 per group). (I) Representative western blot showing Nrf2 levels in unstimulated (C) and LPS/IFNγ ± AcLDL stimulated BMDMs. Data are expressed as the mean ± SEM, *P < 0.05 between genotypes of the same treatment, $P > 0.05 between control and treatment of the same genotype. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–.
To investigate how Gch1 deficiency affects lipid handling, we assessed the capacity of macrophages from Gch1fl/flTie2CreApoE–/– mice to form foam cells and efflux cholesterol. There was no difference in reverse cholesterol transport from macrophages to plasma between Gch1fl/flTie2CreApoE–/– and Gch1fl/flApoE–/– mice (P > 0.05, Figure 6B). We next used fluorescently labelled acetylated low-density lipoprotein (AcLDL) to assess foam cell formation. Foam cell formation in unstimulated macrophages was unchanged in macrophages from Gch1fl/flTie2CreApoE–/– mice. However, in inflammatory LPS/IFNγ stimulated macrophages Gch1 deficiency resulted in a significant increase in foam cell formation (P < 0.05; Figure 6C).
Dysregulation of the oxidative pathway have previously been shown to alter foam cell formation. We used qRT–PCR to quantify expression of anti-oxidant genes in unstimulated and LPS/IFNγ stimulated macrophages. We found that treatment with LPS/IFNγ caused a significant up regulation of heme oxygenase in both genotypes. However, upregulation of heme oxygenase was strikingly reduced in macrophages from Gch1 deficient mice compared with control Gch1fl/flApoE–/– mice (0.19 ± 0.048 vs. 0.039 ± 0.010, P < 0.05). Treatment of macrophages with AcLDL did not alter the expression of heme oxygenase (Figure 6D). Reduced expression of Prdx1 and Nqo1 was also observed in Gch1 deficient macrophages (Figure 6E and F). In addition, we also observed a striking absence in the expression of the chemokine receptor Cxcl5 in Gch1-deficient LPS/IFNγ stimulated macrophages treated with LPS/IFN-γ and AcLDL (Figure 6G). In order to assess whether the differences observed in anti-oxidant gene expression was due to alteration in the translocation of Nrf2 to the nucleus, we assessed Nrf2 transcription activity in nuclear fractions from LPS/INF-γ and LPS/IFN-γ and AcLDL treated macrophages. We observed no differences in Nrf2 reporter activity in nuclear fractions from either LPS/IFN-γ or LPS/IFN-γ and AcLDL treated macrophages between Gch1 deficient and control macrophages (Figure 6H). Western blot analysis showed a similar finding to the activity assay with no difference between genotypes observed (Figure 6I).
3.6 Deficiency in BH4 in both endothelial cells and leucocytes is required to increase atherosclerosis burden
Given the changes observed in both endothelial cell and macrophage biology in the absence of Gch1 we next sought to determine the contributions of these cell types to the observed increase in atherosclerosis in Gch1fl/flTie2CreApoE–/– mice. Three groups of bone marrow chimeric animals were generated: (1) Gch1fl/flTie2CreApoE–/– were given bone marrow from Gch1fl/flApoE–/– to assess the impact of loss of Gch1 from endothelial cells on atherosclerosis progression; (2) Gch1fl/flApoE–/– received Gch1fl/flTie2CreApoE–/– bone marrow to assess the role of leucocyte Gch1; or (3) Gch1fl/flApoE–/– received Gch1fl/flApoE–/– bone marrow, as a control group.
BMDMs from chimeric mice were assessed for BH4 levels and nitrite production to confirm appropriate reconstitution with donor marrow. As expected, both BH4 and nitrite levels in BMDM followed the genotype of the bone marrow donor. Gch1fl/flApoE–/– mice which received bone marrow from Gch1fl/flTie2CreApoE–/– mice had a significant reduction in both BH4 and nitrite, whereas BH4 and nitrite levels in BMDM from Gch1fl/flTie2CreApoE–/– which received Gch1fl/flApoE–/– bone marrow where similar to the levels observed in the control mice (Gch1fl/flApoE–/– which received Gch1fl/flApoE–/– bone marrow, Figure 7A and B). We next quantified plasma biopterin levels in chimeric mice. In male mice, loss of Gch1 from leucocytes caused a significant reduction in plasma BH2 levels but with preserved BH4 levels (Figure 7C). The preservation of BH4 levels and the reduction in both BH2 and B (data not shown) resulted in a significant increase in BH4:BH2 + B ratio in both endothelial cells and leucocyte knockout mice. In contrast, no difference in either BH4, BH2 or the ratio was observed between any of the groups of female mice (Figure 7C).
Figure 7.

Loss of Gch1 in both endothelial cells and leucocytes is required to increase atherosclerosis. (A) BH4 levels and (B) nitrite accumulation in LPS/IFNγ stimulated BMDM, a significant reduction in BH4 and nitrite levels was observed in macrophages from Gch1fl/flApoE–/– recipient mice who received bone marrow from a Gch1fl/flTie2CreApoE–/– donor (Kruskal–Wallis test, n = 5–10 per group). (C) BH4, BH2, and the ratio of BH4:BH2 and B in plasma from chimeric male (Kruskal–Wallis test) and female (one-way ANOVA) mice. No difference in plasma BH4 levels were observed between genotypes. In male mice a significant reduction in BH2 was observed in mice with a leucocyte deficiency in Gch1, with no difference observed between groups in female mice. In males a significant increase in the ratio of BH4:BH2 and B was observed in both endothelial cells and leucocytes Gch1 deficient mice, no difference were observed between groups in female mice (n = 5–11 per group). Quantification of aortic root atherosclerosis at 16 weeks after 6 weeks of HFD in male (D, Kruskal–Wallis test) and female (E, one-way ANOVA) mice, no difference in plaque area was observed between groups in either males or females (n = 7–12 per group). *P < 0.05 compared with control. Black symbols = Gch1fl/flApoE–/–, red symbols = Gch1fl/flTie2CreApoE–/–.
We next assessed the impact of loss of Gch1 in either endothelial cells or leucocytes on atherosclerotic plaque in aortic root sections. Loss of Gch1 from either endothelial cells or leucocytes alone did not alter atherosclerotic plaque burden in either male or female mice, indicating that deficiency of Gch1 from both endothelial cells and leucocytes is required to increase atherosclerosis burden (Figures 7D and E and 8).
Figure 8.
Loss of Gch1 in both endothelial cells and monocyte/macrophages is required to increase atherosclerosis burden.
4. Discussion
We have generated a novel mouse model of endothelial cell and leucocyte-targeted Gch1 deletion, crossed with the hyperlipidemic ApoE–/– mouse, to test the cellular requirement for BH4 in the pathology of atherosclerosis. This novel mouse model reveals several new and important findings. First, deficiency in endothelial cell and leucocyte BH4 results in an increase in atherosclerotic plaque formation. Second, Gch1fl/flTie2Cre ApoE–/– mice display hallmarks of endothelial cell dysfunction characterized by loss of NO-mediated effects. Third, loss of BH4 in inflammatory macrophages alters lipid handling, leading to increased foam cell formation and alteration in anti-oxidant dependent gene expression. Finally, loss of Gch1 in both endothelial cells and leucocytes was required to increase atherosclerotic burden. Our study highlights the consequences of loss of both endothelial cell and macrophage BH4 in the regulation of NOS function and cellular redox signalling in atherosclerosis.
The generation of a new mouse model for targeted Gch1 deletion in atherosclerosis enables important new questions to be addressed on the requirement for BH4. Genetic or pharmacological increases in BH4 have shown that supra-physiological levels of BH4 can inhibit plaque progression. However, it is unknown whether BH4 deficiency alone is sufficient to alter plaque biology. Previous models of BH4 deficiency are either limited by very extreme global phenotypes24,25 or embryonic lethality26 which prevents the study of atherosclerosis. The Hph-1 mouse, which has variable systemic BH4 deficiency in different cell types,27,28 has not been assessed for atherosclerosis and hence to date the impact of loss of BH4 on plaque progression is unknown. We have now shown that cell-specific deficiency in endothelial cell and leucocyte BH4 is sufficient to accelerate atherosclerosis in ApoE–/– mice. In order to evaluate the contribution of specific cell types in the progression of atherosclerosis, we generated bone marrow chimeras in which Gch1 was selectively deleted in either endothelial or leucocytes. We found that loss of Gch1 from either cell type alone was insufficient to accelerate atherosclerosis, indicating that Gch1 from both endothelial cells and leucocytes was required for accelerated plaque formation. BH4 levels and nitrate production in BMDM from the chimeric mice confirmed correct bone marrow reconstitution. Although this demonstrates that all cells derived from the recipient bone marrow after chimerization were derived from the donor mice, we cannot exclude the possibility that the selection pressures of bone marrow reconstitution may impact plaque progression.
Loss of eNOS function and the resultant loss of NO in the eNOS–/– mouse leads to endothelial cell dysfunction, increased macrophage recruitment via increased vascular VCAM-1 expression and ultimately accelerated atherosclerosis.29 In the current study we now show a similar pattern of endothelial cell dysfunction with an enhanced contractile function in chow fed mice driven by a decrease in NOS dependent relaxation and blunted endothelial cell mediated dilation in the aorta. HFD feeding resulted in an even more pronounced blunting in endothelial cell mediated dilation in Gch1fl/flTie2CreApoE–/– mice which was fully reversible with ex vivo incubation with the BH4 precursor, sepiapterin, indicating that this defect was mediated by BH4 deficiency. Increased aortic VCAM-1 expression was also observed in both chow and HFD fed Gch1fl/flTie2CreApoE–/– mice. This is in keeping with previous studies where we demonstrated that pharmacological supplementation of BH4 decreased aortic VCAM-1 expression.27 Furthermore, in HFD Gch1fl/flTie2CreApoE–/– expression of the pro-atherogenic cytokine TNFα was significantly increased, indicating a more atherogenic environment. Interestingly, eNOS mRNA expression was also increased in HFD mice, possibly due to a compensatory upregulation mediated by H2O2.30,31 Increased blood pressure was observed in both male and female chow fed Gch1fl/flTie2CreApoE–/–. The endothelial cell dysfunction observed in these mice could in part contribute to increase in blood pressure. However, in HFD mice the increase in blood pressure was observed only in male not female Gch1fl/flTie2Cre ApoE–/–, whereas endothelial cell dysfunction was clearly apparent in female HFD Gch1fl/flTie2CreApoE–/– mice making it unlikely that changes in endothelial function are solely responsible for the change in blood pressure. Blood pressure control is dependent not only on vascular function but also baroreceptor function, sensitivity of the renin–angiotensin–aldosterone system, central control and kidney function. It is possible that alteration in one of these alternative control systems has resulted in a compensatory normalization in blood pressure.
Further to observations in the eNOS–/– ApoE–/– mouse, where endothelial cell dysfunction is attributed to loss of eNOS and hence NO production,32 we now show that preservation of eNOS, but selective loss of NO generation due to BH4 deficiency is associated with increased endothelial cell superoxide and ROS generation. It is likely that both decreased NO and increased ROS generation, due to eNOS uncoupling, contributed to the increase in atherosclerosis, as shown by a previous study where endothelial overexpression of eNOS increased NO but also increased superoxide, by eNOS uncoupling, resulting in increased atherosclerosis.5 This study confirmed the pathological role of uncoupled eNOS in atherosclerosis, since treatment with BH4 or crossing with mice with endothelial cell targeted Gch1 overexpression re-coupled eNOS, resulting in a reduction in atherosclerosis.5,6
An important new finding in male Gch1fl/flTie2CreApoE–/– mice is an increased contribution of macrophages to atherosclerotic plaques. This increase could be due to an increase in either recruitment or retention of macrophages. Importantly, there was no difference in the number of circulating monocytes in male Gch1fl/flTie2CreApoE–/– mice. The difference in macrophage content of the plaques could be due to an increase in the migratory capacity of Gch1-deficient macrophages. However, this is unlikely to be the mechanism, since we found no difference in in vivo macrophage chemotaxis between the two groups. The increase could also be due to increased retention of macrophages within the plaque. We observed decreased Cxcl5 expression in LPS/IFNγ stimulated macrophages from Gch1fl/flTie2CreApoE–/– mice. Cxcl5 is important for macrophage turnover within the plaque, with a previous study showing that antibody-mediated inhibition of Cxcl5 increased macrophage retention in plaques.33 Interestingly, increased macrophage content was not observed in plaques from female Gch1fl/flTie2CreApoE–/– mice. This difference could be due to the well-known difference in the time course of plaque progression observed between male and female ApoE–/– mice. In addition to macrophages, mast cells have been demonstrated to have a role in plaque progression.34,35Gch1 expression is regulated in mast cells and has been shown to have an important role in the release of NO and serotonin.36–38 However, the increase in atherosclerosis observed in Gch1fl/flTie2CreApoE–/– mice is unlikely to be mediated by changes in mast cells, since no differences were observed in either total, granulated or de-granulated mast cell numbers.
The deletion of Gch1 in macrophages in Gch1fl/flTie2CreApoE–/– mice allows, for the first time, the selective investigation of the effects of iNOS regulation by BH4 in vivo. In contrast to eNOS, which has an atheroprotective role, iNOS has been shown to be pro-atherogenic; iNOS–/– mice are protected from atherosclerosis, with a greater impact observed in more advanced lesions.8–11 In plaques iNOS is predominantly co-localized to macrophages, with the pro-inflammatory role of iNOS ascribed to the high-level output of NO, which reacts with ROS to form reactive nitrogen species. Strikingly, we observed that inflammatory BMDMs from Gch1fl/flTie2CreApoE–/– mice had almost complete loss of NO production, as measured by nitrite production. However, this loss of NO production was accompanied by increased ROS production, revealing a key role of Gch1 in the control of macrophage cellular redox state. This finding is in contrast to BMDM from iNOS–/– mice which have reduced NO production and reduced ROS production12 and reduced 3-nitrotyrosine staining in plaques.12 These differences raise the possibility that BH4 deficiency in inflammatory macrophages results in a more pathological phenotype than that observed in macrophages from iNOS–/– mice. Alterations in macrophage ROS production has been previously shown to alter foam cell formation,39 with inhibition of iNOS altering the ability of macrophages to form foam cells.40 We observed that Gch1 deficient inflammatory LPS/IFNγ stimulated macrophages formed significantly larger foam cells than macrophages from their littermate controls. This increase was not due to alterations in cholesterol efflux from the cell as reverse cholesterol transport was similar between genotypes. In contrast to macrophages from iNOS knockout mice, macrophages from Gch1fl/flTie2CreApoE–/– mice had decreased NO and increased ROS, implicating iNOS dependent ROS in foam cell development. We observed a significant reduction in anti-oxidant genes in the absence of Gch1, HO-1, Nqo1 and Prdx1 are known to be Nrf2 related genes. However, interestingly the reduction in anti-oxidant genes was not associated with a reduction in Nrf2 nuclear translocation. This could be due to difference in the time course of Nrf2 nuclear translocation vs. gene expression or due to modulation of alternative pathways. Bach1 binds to heme-responsive elements inhibiting the binding of Nrf2,41 with global deficiency in Bach 1 associated with reduced atherosclerosis.42 Alternatively, the changes in gene expression could be mediated by an alternative pathway such as repression of Ndy1 which has been shown to modulate the expression of Nqo1.43 This significant decrease in anti-oxidant defence signalling may compound the cellular redox state caused by iNOS uncoupling, by decreasing activation of cellular redox defence mechanisms.
In this study, deficiency in endothelial cell and leucocyte BH4 was not sufficient to impact on circulating BH4 levels in chow fed mice, demonstrating that under these conditions circulating BH4 levels are determined by BH4 production by cells other than endothelial cells and leucocytes. However, after high fat feeding a significant reduction in circulating BH4 and BH2 levels was observed in both male and female Gch1 knockout mice, indicating that under these conditions endothelial cell and leucocyte BH4 make a significant contribution to circulating levels. The change in plasma BH4 was accompanied by a decrease in liver BH4 levels in Gch1fl/flTie2CreApoE–/– mice, which in turn could be a contributing factor to the reduction in circuiting biopterin levels observed in this mouse. In both genotypes, HFD increased liver BH2 levels with a greater increase observed in liver from Gch1fl/flTie2CreApoE–/– mice, suggesting increased oxidation of BH4 and/or decreased recycling of BH2 back to BH4, by DHFR. In addition to these changes in liver biopterins, aortic BH4 levels in HFD Gch1fl/flApoE–/– were increased compared with chow fed animals, which may in part explain the preservation of circulating BH4 levels in WT mice. There was no difference in circulating BH4 levels in either male of female chimeric mice with loss of Gch1 in either endothelial cells or leucocytes. This indicates that loss of Gch1 in both endothelial cells and leucocytes is required for the reduction in circulating BH4 levels in HFD animals. It might be expected that plasma biopterin levels would increase after HFD as previous studies in humans have shown a positive correlation between plasma BH4 levels and systemic inflammation.15 It could be that systemic inflammation observed in ApoE–/– mice44 is a sufficient stimulus to elevate plasma BH4 levels and that HFD is not additive to this effect.
In conclusion, we have shown for the first time a key role for endothelial cell and leucocyte Gch1 and BH4 in the progression of atherosclerosis. Our study highlights the importance of both endothelial cell and macrophage BH4 in the regulation of NOS function and cellular redox signalling in atherosclerosis. These novel findings reveal contrasting endothelial cell and leucocyte requirements for Gch1 and BH4 in the progression of atherosclerosis and identify new BH4-dependent determinants of inflammatory cell redox signalling and foam cell formation.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
British Heart Foundation Programme Grant (RG/12/5/29576), Chair award (CH/16/1/32013), and Oxford British Heart Foundation Centre of Research Excellence (RE/13/1/30181). Wellcome Trust (090532/Z/09/Z) and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre.
Supplementary Material
Footnotes
Time for primary review: 42 days
References
- 1. Werner ER, Blau N, Thony B.. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J 2011; 438:397–414. [DOI] [PubMed] [Google Scholar]
- 2. Widder JD, Chen W, Li L, Dikalov S, Thony B, Hatakeyama K, Harrison DG.. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ Res 2007; [DOI] [PubMed] [Google Scholar]
- 3. Li L, Chen W, Rezvan A, Jo H, Harrison DG.. Tetrahydrobiopterin deficiency and nitric oxide synthase uncoupling contribute to atherosclerosis induced by disturbed flow. Arterioscler Thromb Vasc Biol 2011; 31:1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crabtree MJ, Channon KM.. Dihydrofolate reductase and biopterin recycling in cardiovascular disease. J Mol Cell Cardiol 2009; 47:749–751. [DOI] [PubMed] [Google Scholar]
- 5. Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, Hirata K-I, Yasui H, Sakurai H, Yoshida Y, Masada M, Yokoyama M.. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest 2002; 110:331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takaya T, Hirata K, Yamashita T, Shinohara M, Sasaki N, Inoue N, Yada T, Goto M, Fukatsu A, Hayashi T, Alp NJ, Channon KM, Yokoyama M, Kawashima S.. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arterioscler Thromb Vasc Biol 2007; 27:1632–1637. [DOI] [PubMed] [Google Scholar]
- 7. Cromheeke KM, Kockx MM, De Meyer GR, Bosmans JM, Bult H, Beelaerts WJ, Vrints CJ, Herman AG.. Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res 1999; 43:744–754. [DOI] [PubMed] [Google Scholar]
- 8. Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL.. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation 2001; 103:3099–3104. [DOI] [PubMed] [Google Scholar]
- 9. Chen J, Kuhlencordt P, Urano F, Ichinose H, Astern J, Huang PL.. Effects of chronic treatment with L-arginine on atherosclerosis in apoE knockout and apoE/inducible NO synthase double-knockout mice. Arterioscler Thromb Vasc Biol 2003; 23:97–103. [DOI] [PubMed] [Google Scholar]
- 10. Miyoshi T, Li Y, Shih DM, Wang X, Laubach VE, Matsumoto AH, Helm GA, Lusis AJ, Shi W.. Deficiency of inducible NO synthase reduces advanced but not early atherosclerosis in apolipoprotein E-deficient mice. Life Sci 2006; 79:525–531. [DOI] [PubMed] [Google Scholar]
- 11. Detmers PA, Hernandez M, Mudgett J, Hassing H, Burton C, Mundt S, Chun S, Fletcher D, Card DJ, Lisnock J, Weikel R, Bergstrom JD, Shevell DE, Hermanowski-Vosatka A, Sparrow CP, Chao YS, Rader DJ, Wright SD, Puré E.. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein E-deficient mice. J Immunol 2000; 165:3430–3435. [DOI] [PubMed] [Google Scholar]
- 12. Ponnuswamy P, Ostermeier E, Schrottle A, Chen J, Huang PL, Ertl G, Nieswandt B, Kuhlencordt PJ.. Oxidative stress and compartment of gene expression determine proatherosclerotic effects of inducible nitric oxide synthase. Am J Pathol 2009; 174:2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McNeill E, Crabtree MJ, Sahgal N, Patel J, Chuaiphichai S, Iqbal AJ, Hale AB, Greaves DR, Channon KM.. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Radic Biol Med 2015; 79:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimizu S, Shiota K, Yamamoto S, Miyasaka Y, Ishii M, Watabe T, Nishida M, Mori Y, Yamamoto T, Kiuchi Y.. Hydrogen peroxide stimulates tetrahydrobiopterin synthesis through the induction of GTP-cyclohydrolase I and increases nitric oxide synthase activity in vascular endothelial cells. Free Radic Biol Med 2003; 34:1343–1352. [DOI] [PubMed] [Google Scholar]
- 15. Antoniades C, Shirodaria C, Crabtree M, Rinze R, Alp N, Cunnington C, Diesch J, Tousoulis D, Stefanadis C, Leeson P, Ratnatunga C, Pillai R, Channon KM.. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 2007; 116:2851–2859. [DOI] [PubMed] [Google Scholar]
- 16. Cunnington C, Antoniades C, Shirodaria C, Van Assche T, Lee R, Kylintireas I, Lyndsay A, Lee JM, Neubauer S, Choudhury RP, Channon KM.. Systemic and vascular oxidation limits efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 2012; 125:1356–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chuaiphichai S, McNeill E, Douglas G, Crabtree MJ, Bendall JK, Hale AB, Alp NJ, Channon KM.. Cell-autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension 2014; 64:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Douglas G, Van Kampen E, Hale AB, McNeill E, Patel J, Crabtree MJ, Ali Z, Hoerr RA, Alp NJ, Channon KM.. Endothelial cell repopulation after stenting determines in-stent neointima formation: effects of bare-metal vs. drug-eluting stents and genetic endothelial cell modification. Eur Heart J 2013; 34:3378–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM.. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE−/− mice. Cardiovasc Res 2012;94:20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu M, Li XX, Wang L, Wang M, Zhang Y, Li PL.. Contribution of Nrf2 to atherogenic phenotype switching of coronary arterial smooth muscle cells lacking CD38 gene. Cell Physiol Biochem 2015; 37:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM.. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(-)/(-) mice. Cardiovasc Res 2012; 94:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Granger DL, Taintor RR, Boockvar KS, Hibbs JB. Jr.,. Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol 1996;268:142–151. [DOI] [PubMed] [Google Scholar]
- 23. de Bono JP, Warrick N, Bendall JK, Channon KM, Alp NJ.. Radiochemical HPLC detection of arginine metabolism: measurement of nitric oxide synthesis and arginase activity in vascular tissue. Nitric Oxide 2007; 16:1–9. [DOI] [PubMed] [Google Scholar]
- 24. Elzaouk L, Leimbacher W, Turri M, Ledermann B, Burki K, Blau N, Thony B.. Dwarfism and low insulin-like growth factor-1 due to dopamine depletion in Pts–/– mice rescued by feeding neurotransmitter precursors and H4-biopterin. J Biol Chem 2003; 278:28303–28311. [DOI] [PubMed] [Google Scholar]
- 25. Sumi-Ichinose C, Urano F, Kuroda R, Ohye T, Kojima M, Tazawa M, Shiraishi H, Hagino Y, Nagatsu T, Nomura T, Ichinose H.. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvoyltetrahydropterin synthase knockout mice. J Biol Chem 2001; 276:41150–41160. [DOI] [PubMed] [Google Scholar]
- 26. Douglas G, Hale AB, Crabtree MJ, Ryan BJ, Hansler A, Watschinger K, Gross SS, Lygate CA, Alp NJ, Channon KM.. A requirement for Gch1 and tetrahydrobiopterin in embryonic development. Dev Biol 2015; 399:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidt TS, Alp NJ.. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin Sci 2007; 113:47–63. [DOI] [PubMed] [Google Scholar]
- 28. Wang CH, Li SH, Weisel RD, Fedak PW, Hung A, Li RK, Rao V, Hyland K, Cherng WJ, Errett L, Leclerc Y, Bonneau D, Latter DA, Verma S.. Tetrahydrobiopterin deficiency exaggerates intimal hyperplasia after vascular injury. Am J Physiol Regul Integ Comp Physiol 2005; 289:R299–R304. [DOI] [PubMed] [Google Scholar]
- 29. Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL.. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001; 104:448–454. [DOI] [PubMed] [Google Scholar]
- 30. Cai H, Davis ME, Drummond GR, Harrison DG.. Induction of endothelial NO synthase by hydrogen peroxide via a Ca2+/calmodulin-dependent protein kinase II/janus kinase 2–dependent pathway. Arteriosc Thromb Vasc Biol 2001;21:1571–1576. [DOI] [PubMed] [Google Scholar]
- 31. Youn JY, Wang T, Blair J, Laude KM, Oak JH, McCann LA, Harrison DG, Cai H.. Endothelium-specific sepiapterin reductase deficiency in DOCA-salt hypertension. Am J Physiol Heart Circ Physiol 2012; 302:H2243–H2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ponnuswamy P, Schrottle A, Ostermeier E, Gruner S, Huang PL, Ertl G, Hoffmann U, Nieswandt B, Kuhlencordt PJ.. eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PLoS One 2012; 7:e30193.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rousselle A, Qadri F, Leukel L, Yilmaz R, xFc stem, Fontaine J-F, Sihn G, Bader M, Ahluwalia A, Duchene J.. CXCL5 limits macrophage foam cell formation in atherosclerosis. J Clin Invest 123:1343–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Clair JM-S, Shi G-P.. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med 2007; 13:719.. [DOI] [PubMed] [Google Scholar]
- 35. Bot I, de Jager SCA, Zernecke A, Lindstedt KA, van Berkel TJC, Weber C, Biessen EAL.. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E–deficient mice. Circulation 2007; 115:2516–2525. [DOI] [PubMed] [Google Scholar]
- 36. Hesslinger C, Kremmer E, Hültner L, Ueffing M, Ziegler I.. Phosphorylation of GTP cyclohydrolase I and modulation of its activity in rodent mast cells: GTP cyclohydrolase I hyperphosphorylation is coupled to high affinity IgE receptor signaling and involves protein kinase. J Biol Chem 1998; 273:21616–21622. [DOI] [PubMed] [Google Scholar]
- 37. Ziegler I, Hültner L, Egger D, Kempkes B, Mailhammer R, Gillis S, Rödl W.. In a concerted action kit ligand and interleukin 3 control the synthesis of serotonin in murine bone marrow-derived mast cells. Up-regulation of GTP cyclohydrolase I and tryptophan 5-monooxygenase activity by the kit ligand. J Biol Chem 1993; 268:12544–12551. [PubMed] [Google Scholar]
- 38. Gilchrist M, Hesslinger C, Befus AD.. Tetrahydrobiopterin, a critical factor in the production and role of nitric oxide in mast cells. J Biol Chem 2003; 278:50607–50614. [DOI] [PubMed] [Google Scholar]
- 39. Lee SH, Park DW, Park SC, Park YK, Hong SY, Kim JR, Lee CH, Baek SH.. Calcium-independent phospholipase A2beta-Akt signaling is involved in lipopolysaccharide-induced NADPH oxidase 1 expression and foam cell formation. J Immunol 2009;183:7497–7504. [DOI] [PubMed] [Google Scholar]
- 40. Smirnova IV, Kajstura M, Sawamura T, Goligorsky MS.. Asymmetric dimethylarginine upregulates LOX-1 in activated macrophages: role in foam cell formation. Am J Physiol Heart Circ Physiol 2004; 287:H782–H790. [DOI] [PubMed] [Google Scholar]
- 41. Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K.. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA 2004; 101:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watari Y, Yamamoto Y, Brydun A, Ishida T, Mito S, Yoshizumi M, Igarashi K, Chayama K, Ohshima T, Ozono R.. Ablation of the Bach1 gene leads to the suppression of atherosclerosis in Bach1 and apolipoprotein E double knockout mice. Hypertens Res 2008; 31:783.. [DOI] [PubMed] [Google Scholar]
- 43. Polytarchou C, Pfau R, Hatziapostolou M, Tsichlis PN.. The JmjC domain histone demethylase Ndy1 regulates redox homeostasis and protects cells from oxidative stress. Mol Cell Biol 2008; 28:7451–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Naura AS, Hans CP, Zerfaoui M, Errami Y, Ju J, Kim H, Matrougui K, Kim JG, Boulares AH.. High-fat diet induces lung remodeling in ApoE-deficient mice: an association with an increase in circulatory and lung inflammatory factors. Lab Invest 2009; 89:1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.