

Abstract
As a cofactor for numerous reactions, NAD+ is found widely dispersed across many maps of cellular metabolism. This core redox role alone makes the biosynthesis of NAD+ of great interest. Recent studies have revealed new biological roles for NAD+ as a substrate for diverse enzymes that regulate a broad spectrum of key cellular tasks. These NAD+-consuming enzymes further highlight the importance of understanding NAD+ biosynthetic pathways. In this study, we developed a chemo-enzymatic synthesis of isotopically labeled NAD+ precursor, nicotinamide riboside (NR). The synthesis of NR isotopomers allowed us to unambiguously determine that NR is efficiently converted to NAD+ in the cellular environment independent of degradation to nicotinamide, and it is incorporated into NAD+ in its intact form. The versatile synthetic method along with the isotopically labeled NRs will provide powerful tools to further decipher the important yet complicated NAD+ metabolism.
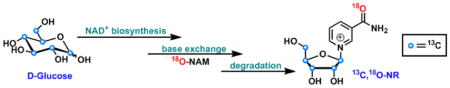
Graphical Abstract
INTRODUCTION
Nicotinamide adenine dinucleotide (NAD+) plays critical roles in various cellular events, both as a cofactor for redox reactions and as a substrate to donate ADP-ribose unit. NAD+-consuming enzymes such as Poly(ADP-ribose) polymerases (PARPs), sirtuins, and CD38 are key players in the regulation of DNA repair,1,2 stress resistance,3,4 metabolism,4,5 and signaling.6 Some of these enzymes rapidly deplete NAD+ from the cells,7 and others are regulated by NAD+ availability.3 The identification of NAD+-utilizing enzymes has stimulated interest in NAD+ biosynthesis and its regulation.
In mammals, the intracellular NAD+ level is tightly regulated by the complementary de novo,8 salvage8,9 pathways as well as the newly discovered nicotinamide riboside (NR) pathway10,11,12 (Scheme 1). The de novo pathway requires the formation of quinolinate, which is the precursor of nicotinic acid mononucleotide (NaMN). NaMN is then adenylated to nicotinic acid adenine dinucleotide (NaAD) and further acted upon by ATP-dependent NAD+ synthetase to form NAD+. Salvage pathway is known to reconstitute NAD+ from metabolites that already incorporate a complete nicotinamide or nicotinic acid ring. Nicotinamide (NAM) is directly recycled by nicotinamide phosphoribosyltransferase (NAMPT) that couples it to 5-phosphoribose-1-pyrophosphate (PRPP) to form nicotinamide mononucleotide (NMN), a mechanism first recognized by Dietrich et al in 1966.13 NMN thereby formed can then be converted to NAD+ by NMN adenylyltransferases (NMNAT). Nicotinic acid (NA) is recycled to form NaMN in what is known as the Preiss-Handler pathway,14 reaching a common intermediate of the de novo pathway.12,15 In addition, NAD+ can also be derived from a dietary compound, NR. NR is a naturally occurring substance found in milk.16 A dedicated pathway has been proposed for NR conversion to NAD+ in mammalian cells, encoded by two nicotinamide riboside kinases (NRK1 and NRK2) that convert NR to NMN (Scheme 1).10,12,17 NMN can then be fed into the Preiss-Handler pathway for the production of NAD+.
Scheme 1.

NAD+ biosynthetic pathways in mammals.
NR has been shown to be a potent NAD+ elevating agent.18,19,17,20 Increasing cellular NAD+ concentration was thought to provide beneficial effects on healthspan and lifespan extension.20,21,22,23 Repletion of the intracellular NAD+ pool using NR, therefore, has been suggested as a novel therapeutic for the treatment of metabolic and age-related diseases.24,25,26
However, the metabolism of NR is still not fully understood. It was reported that the dominant biochemical activity on NR in the cellular context was degradative.27 In mammalian hepatocyte extract, NR was not converted efficiently to NMN or NAD+, but rather to ribose-1-phosphate via the action of a phosphorylase, possibly purine nucleoside phosphorylase (PNP).27,28 Recent work from the Brenner lab confirms that in yeast NR is handled metabolically via direct phosphorylation by NR kinase as well as degradation to NAM by N-ribosylphosphorylase.11 Similar results have been observed in mouse models.19 Intraperitoneal injection of NR caused significant increases of plasma NAM level in both wildtype and NRK1 KO animals, suggesting the presence of NR degradation pathway. The potential coexistence of competing phosphorylation and degradation pathways creates significant analytical challenges in dissecting NR metabolism. The use of isotopically labeled NRs can enable the quantitative measurement of NR metabolites. However, this method is limited by the lack of NR isotopomers. Total chemical synthesis suffered from low yields, the use of environmentally harmful reagents, lengthy synthetic routes and lack of stereoselectivity.29,30 The need for a better and efficient method to create isotopically labeled NRs becomes apparent. Herein, we report an efficient chemo-enzymatic synthesis of NR with the specific incorporation of isotope labels into the ribose ring and nicotinamide. We took advantage of a pre-existing enzymatic synthesis of NAD+31 and coupled it with enzyme catalyzed “base exchange” and degradation for the production of isotopically labeled NRs. The easy access to NR isotopomers should facilitate the probing of NR metabolic pathways. Using these isotopically labeled NRs, we demonstrate that in mammalian cells NR can be converted to NAD+ independent of decomposition. We also establish the importance of NRK-dependent pathway in maintaining NAD+ biosynthetic capacity when salvage pathway is compromised, further highlighting the homeostatic mechanism in NAD+ biosynthesis.
RESULTS AND DISCUSSION
Design of the chemo-enzymatic synthesis
The biological importance of NAD+ in the hub of interconnected cellular events is increasingly recognized. More recently, NR has been identified as a precursor of NAD+. It contributes to the cellular NAD+ homeostasis through NRK-mediated pathway.10,12,17 NR is considered one of the most potent NAD+ boosting agents with no apparent toxicity. The study of NR pathway is still in its infancy, partly because in-depth functional interrogation of the pathway enzymes is rare, and these studies were further complicated by the competing biosynthetic and degradative pathways. The use of isotopically labeled NRs can allow the quantitative measurement of NR metabolites at extremely low levels. Stable isotope or radioisotope labeling proved to be an attractive strategy for monitoring sequential enzymatic conversions in complicated matrices including cell extracts and intact cells. However, these methods can be limited by the lack of available NR isotopomers. Reports of the synthesis of isotopically labeled NR are rather scarce.19 On the other hand, the enzymatic synthesis of isotopically labeled NAD+ is well established.31 We envision that the NAD+ isotopomers can be broken down via enzymatic degradations, leading to the formation of NR isotopomers. This should enable the incorporation of atomic labels in the ribose ring as well as in the NAM moiety.
The generation of NAD+ isotopomer contains two steps. The first step is a one-pot ten-enzyme coupled reaction in which isotopically labeled glucose (3H, 14C or 13C) can be converted to NaAD (Scheme 2A).31 Most of the enzymes involved in this step are commercially available, and the expression and purification of the others are well documented.31,32,33 In the second step, purified NaAD can be transformed to NAD+ by NAD+ synthetase (Scheme 2B). This method has been widely used for the creation of isotopically labeled NAD+s for kinetic isotope effect analysis,33,34,35,36 and was applied to the generation of poly(ADP-ribose) (PARP) with multiple labels.37 Atomic labels can be incorporated in the ribose ring using this method.
Scheme 2.

Enzyme coupled synthesis of isotopically labeled NAD+. A. Ten-enzyme coupled, one-pot synthesis of isotopically labeled NaAD; B. Conversion of NaAD to NAD+ catalyzed by NAD+ synthetase.
The study of NR metabolism, especially the degradation pathway, would greatly benefit from NRs with isotopic labels in the NAM moiety as well. This can be achieved through chemical synthesis of isotopically labeled NAM. Subsequently, NAM can be included into NAD+ by enzyme catalyzed “base exchange” reaction, leading to the formation of NAD+ with labels in both ribose and NAM (Scheme 3A). Ultimately, the NAD+ isotopomer can be degraded enzymatically (Scheme 3B). NR with multiple labels can thus be generated.
Scheme 3.

Enzymatic synthesis of isotopically labeled NR. A. ADP-ribosylcyclase catalyzed “base exchange” leads to the formation of 13C,18O-NAD+; B. 13C,18O-NR can be generated by phosphodiesterase I and alkaline phosphatase catalyzed NAD+ degradation.
Generation of isotopically labeled NRs
13C-NAD+ was created with the above-mentioned two-step protocol as described in “Methods and Materials” (Scheme 2). U-13C6-glucose, ATP, PEP, NADP+, α-ketoglutarate (αKG) and NA were used as the building blocks. The NaAD biosynthetic pathway (Scheme 2A) requires the participation of 10 enzymes among which pyruvate kinase, glutamate dehydrogenase and myokinase serve as the recycling enzymes. They were used for the regeneration of ATP and NADP+. Hexokinase, pyruvate kinase, glucose-6-phosphate dehydrogenase, glutamate dehydrogenase, 6-phosphogluconate dehydrogenase, phosphoriboisomerase, and myokinase were purchased from commercial vendors. 5-Phosphoribosyl-1-pyrophosphate synthetase and nicotinate phosphoribosyltransferase were expressed and purified as described before.32,33 Based on our previous experience with the protocol, nicotinic acid mononucleotide adenyltransferase (NMNAT) was cloned into pet28a vector, expressed and purified as described in “Methods and Materials” (Figure S1).
All the building blocks, MgCl2, and NH4Cl were combined with 10 enzymes in reaction buffer and incubated at 37°C. The formation of NaAD was monitored with HPLC (Figure S2). The production of NaAD plateaued after 6 h of incubation. LC-MS analysis revealed an increase of 5 amu of 13C-NaAD compared to unlabeled NaAD (Figure S3). Isotopically labeled NaAD was then isolated from the reaction mixture using preparative HPLC. This compound can be degraded by phosphodiesterase to AMP and 13C-NaMN (Figure S4), further confirming the identity of this molecule.
Subsequently, the purified 13C-NaAD was subjected to the enzymatic conversion catalyzed by NAD+ synthetase. S. pneumoniae NAD+ synthetase (Figure S5) is a NH3-dependent enzyme capable of converting carboxylate in NaAD to amide in NAD+.38 The reaction was supplemented with ATP, MgCl2, NH4Cl and KCl. The formation of 13C-NAD+ was observed immediately as evidenced by the appearance of a new peak in the reaction mixture with the same retention time as the authentic NAD+ standard (Figure S6). Similar to NaAD, NAD+ was also purified by HPLC and concentrated by lyophilization. 13C-NAD+ can be readily converted to 13C-NMN and AMP by phosphodiesterase I (Figure S7).
To incorporate an atomic label into NAM, 18O-NAM was synthesized as reported before.39 The resulting product showed 2 amu increase by LC-MS analysis, and the incorporation of 18O label was determined to be 96.5% (Figure S8) by MALDI-TOF. 18O-NAM was then included in NAD+ structure by ADP-ribosylcyclase. ADP-ribosylcyclase catalyzes the intramolecular cyclization of NAD+ to form cyclic ADP-ribose (cADPR) and NAM.40 This transformation involves an enzyme-bound intermediate. The subsequent nucleophilic capture of the intermediate results in the formation of product or the regeneration of NAD+.41,42 The latter reaction is commonly referred as “base exchange”. With exogenously added NAM isotopomer, atomic labels can be introduced into NAD+. 13C-NAD+ was incubated with excess amount of 18O-NAM in the presence of ADP-ribosylcyclase. After 2 h of incubation, 13C,18O-NAD+ was isolated from the reaction mixture. The isotope incorporation was confirmed with MS (Figure S9).
The final step of the synthesis was the enzymatic degradation of 13C,18O-NAD+ with commercially available phosphodiesterase I and alkaline phosphatase. The digestive method led to the quantitative production of 13C,18O-NR (Figure S10). The identity of this compound was further confirmed with 1H NMR, 13C NMR and HRMS (Figure S11–S13).
The versatility of the synthetic approach was further demonstrated in the generation of 14C-NR. 14C-NAD+ was synthesized using unlabeled NAD+ and 14C-NAM via ADP-ribosylcyclase-mediated “base exchange”. The subsequent enzymatic degradation gave rise to radioisotopically labeled NR. 14C-NR was further characterized by two separate enzymatic transformations. In the first reaction 14C-NR was incubated with ATP and recombinant human NRK1 in 100 mM phosphate buffer pH 7.5. The reaction mixture was resolved on HPLC. Fractions were collected from HPLC and counted in a scintillation counter. The formation of 14C-NMN was determined by the co-elution of radioactivity with chemical standard of NMN (Figure S14A). In the second reaction, 14C-NR was incubated with human PNP. The phosphorolysis of NR was evidenced by the formation of 14C-NAM which has the same retention time as the chemical standard on the HPLC chromatogram (Figure S14B).
NR serves as a precursor of NAD+ biosynthesis
The synthesis of isotopically labeled NRs enabled the investigation of the mammalian NR metabolism. Neuro2a cells were cultured with 14C-NR for 2 or 6 h, after which cells were lysed and NR metabolites were resolved on HPLC (Figure 1A). Co-injection of unlabeled NR, NMN, NAM and NAD+ standards allowed the radiolabeled metabolites to be collected based on their retention times. The incorporation of 14C label was assayed by scintillation counting (Figure 1B). After 2 h of incubation, the accumulation of 14C-NR and the formation of 14C-NMN and 14C-NAD+ can be easily detected (Figure S15). The incorporation of 14C increased with longer incubation time. At the 6 h time point, 14C-NAD+ predominated (Figure 1B and S15). This is consistent with the previous report that NR serves as a precursor for NAD+ biosynthesis.18 The observation of 14C-NAM at both time points, although at low levels, suggested the presence of decomposition pathways for NR, NMN or NAD+. It is important to note that NAM readily diffuses across the cell membranes. So, the observed 14C-NAM level is most likely an underestimation of the actual 14C-NAM concentration.
Figure 1.

The bioconversion of NR can be monitored using radioisotope labeled NR. A. HPLC chromatogram showing the separation of NMN, NR, NAM and NAD+; B. The incorporation of 14C label into different metabolites can be quantified by scintillation counting.
NR conversion to NAD+ is independent of NAM
The initial findings not only indicated that it is possible to use radiolabeled NR to monitor NAD+ biosynthesis, but also implied possible competing degradative pathway in NR metabolism. NAM can be generated through the decomposition of NR by PNP (Scheme 1).28 NAM, in turn, can be harvested for NAD+ synthesis via the salvage pathway. The recycling pathway relies on the rate-limiting enzyme, NAMPT, which avidly reacts NAM with PRPP to form NMN.43 In order to establish that NR can be directly converted to NAD+ independently of the breakdown of NR to NAM and NAM recycling, the cells were treated with 20 μM 14C-NR in the presence or absence of 5 mM unlabeled NAM. If 14C-NAM is formed as an intermediate in the NR pathway, it will be rapidly diluted by the unlabeled NAM and is unlikely to be recycled. The addition of 5 mM unlabeled NAM only caused slight decrease of 14C incorporation into NAD+, suggesting that NR conversion to NAD+ is largely independent of NAM formation (Figure 2A).
Figure 2.


NR conversion to NAD+ is independent of NAM formation. A. 14C-NR increased 14C-NAD+ production in cells. Co-treatment of 14C-NR and unlabeled NAM (5 mM) caused slight reduction of 14C label incorporation into NAD+; B. FK866 treatment caused depletion of intracellular NAD+ level. This can be partially rescued by the addition of NAM at millimolar concentration. NR significantly stimulated cellular NAD+ production even in the presence of FK866. The NAD+ concentrations were determined using the cycling assay; C. The incorporation of 14C-NR into NAD+ was not affected by 20 nM FK866. The data represents the average of at least three independent experiments ± S.D. Statistical significance was determined with a Student’s t-test: *p < 0.01 vs control; #p < 0.01 vs FK866 treatment alone.
To corroborate these results, a known inhibitor of NAMPT was used to block the NAM salvage pathway. FK866 has been shown to significantly reduce intracellular NAD+ level through the inhibition of NAM recycling.44,45,46 Indeed, 20 nM FK866 led to 75% reduction of the intracellular NAD+ level after 4 h of incubation (Figure 2B). The addition of NAM in millimolar concentrations partially rescued this depletion. Strikingly, 1 mM unlabeled NR can increase cellular NAD+ content substantially (~160% increase). This NAD+ enhancing ability was decreased by only 15% in the FK866-NR co-treatment samples. Similarly, 14C-NR incorporation into NAD+ had a modest reduction in the presence of 20 nM FK866 (Figure 2C), further supporting the notion that breakdown of NR to NAM is not the major contributor of bioconversion of NR to NAD+.
NR can be degraded to NAM in cell extracts
With data suggesting that the majority of NR is integrated into NAD+ from a non-degradative mechanism, we sought to re-investigate the decomposition of NR by cell extracts, as first reported by Kornberg et al in 1951.27 14C-NR was incubated with freshly prepared Neuro2a cell extract for 1 h, after which 14C incorporation into different NR metabolites was assessed as described above. About 50% of the NR was degraded to NAM with a small amount of 14C-NAD+ (<5%) formation (Figure 3). PNP has been suggested as the key player in NR degradation.28 Consistent with this hypothesis, NR degradation can be effectively inhibited with the addition of immucillin H (ImmH),47 a potent inhibitor of mammalian PNP (Figure 3). At either 1 or 10 μM, ImmH substantially decreased NR degradation in the cell extract. Surprisingly, no increase in NAD+ formation was observed with ImmH treatment. We have confirmed that recombinant human PNP degrades NR efficiently (Figure S16). The Km and kcat of this degradation were determined to be 689 ± 18 μM and 9.3 ± 0.3 s−1, respectively. Collectively, these data suggested that degradation of NR is the dominant activity in cell extracts and PNP is most likely the enzyme responsible for this activity. The inability of NR to stimulate NAD+ synthesis under the above-mentioned condition can be attributed to the lack of NRK activity in cell extracts, although detailed mechanistic study is still needed.
Figure 3.

NR can be converted to NAM in cell extracts. 14C-NR (100,000 cpm) was incubated with freshly prepared Neuro2a cell extract at 37°C. Significant decomposition of 14C-NR to 14C-NAM was observed after 1 h of incubation. This degradation can be effectively inhibited with the addition of ImmH. Error bars represent S.D. of at least three replicates. Statistical significance was determined with a Student’s t-test: *p < 0.01 vs negative control; #p < 0.01 vs 14C-NR and ImmH co-treatment.
PNP is not a major metabolic factor for the conversion of NR to NAD+ in cells
Whether phosphorolysis of NR is critical for NR conversion to NAD+ in the cellular context was further explored. The total cellular NAD+ concentration was determined using the cycling assay.48 The addition of 10 μM ImmH caused negligible change to intracellular NAD+ level (Figure 4A). The presence of 1 mM unlabeled NR almost doubled the NAD+ concentration after a mere 4 h of incubation. And ImmH had very little effect on this NAD+ elevation effect. Furthermore, the incorporation of radioactivity into different NR metabolites was also examined. Cells were cultured with 14C-NR with or without ImmH (Figure 4B). The formation of 14C-NAD+ was not affected significantly by ImmH presence, suggesting that PNP activity is not essential for the bioconversion of NR to NAD+ in cells.
Figure 4.

Intracellular conversion of NR to NAD+ was PNP-independent. A. The NAD+ elevating effect of NR was not affected by ImmH; B. The incorporation of 14C-NR to 14C-NAD+ in the cellular context was not changed by ImmH. The data represents the average of at least three independent experiments ± S.D. Statistical significance was determined with a Student’s t-test: *p < 0.01 vs ImmH treatment alone.
NR is converted to NAD+ as an intact molecule
To gain more insights on whether NR is incorporated into NAD+ in its entirety, NR with stable isotope labels in both ribose and NAM was employed. Cells were cultured with either unlabeled NR or 13C,18O-NR for 4 h before being harvested. The cellular NAD+ content was measured with MALDI-TOF. The relative intensity of unlabeled NAD+ was normalized (Figure 5A). The expected NAD+ isotopomers and their interconversions are illustrated in Figure 5B. The majority of the 13C,18O-NR was converted, as an intact molecule, to 13C,18O-NAD+ as evidenced by the species with m/z of 671. Interestingly, a significant amount of 18O-NAD+ was also detected. Two pathways could contribute to the production of this isotopomer. One is through NR degradation and NAM salvage, however, previous studies have indicated that this is not the major fate of NR in cells. The other possibility is the intracellular “base exchange”. Some NAD+-utilizing enzymes are known to catalyze the exchange of nucleobases. For example, human SIRT2 demonstrated robust nicotinamide exchange activity with a Km of 38 μM.49 The formation of 18O-NAD+ is highly likely due to this exchange pathway under physiological NAM concentration, which is normally in the mid-micromolar range.50,51 This hypothesis was further confirmed with the observation of 13C-NAD+ (m/z = 669). This isotopomer was only detected in the 13C,18O-NR treated samples. In addition to 18O-NAM, the phosphorolysis of 13C,18O-NR will also lead to the formation of 13C-ribose-1-phosphate, which is not a precursor of NAD+ biosynthesis. So the creation of 13C-NAD+ is probably a result of “base exchange” between 13C,18O-NAD+ and cellular unlabeled NAM (Figure 5B). Overall, our data clearly supports the notion that NR is incorporated into NAD+ in its intact molecular form in the cellular environment.
Figure 5.

NR was incorporated into NAD+ in its intact form. A. Relative intensity of different NAD+ isotopomers. Cells were cultured with 13C,18O-NR or unlabeled NR. The cellular metabolites were resolved with HPLC. NAD+ component was collected and subjected to MALDI-TOF analysis. The relative intensity of unlabeled NAD+ was normalized; B. The metabolic fate of NR and the interconversions between different NAD+ isotopomers.
CONCLUSIONS
Isotopically labeled NRs have been synthesized using a combination of chemical and enzymatic transformations. NAD+ biosynthesis, “base exchange” and enzymatic degradations were integrated for the production of 13C,18O-NR from commercially available 13C labeled glucose. This highly efficient synthesis was further extended to the generation of radioisotope labeled NR. When combined with unlabeled NAD+, 14C-NAM was conveniently incorporated into NAD+ structure enzymatically. The subsequent decomposition led to the formation of 14C-NR in excellent yield. The synthesis of stable or radioisotope labeled NRs will significantly facilitate the investigation of NR and NAD+ metabolism.
Given the central role of NAD+ in energy metabolism, it is not surprising that more than one biosynthetic pathway is used to produce NAD+ and some of these pathways are highly conserved across evolution.52,53 For example, the newly discovered NR pathway has been identified in different organisms. In yeast and fungus, NR contributes to cellular NAD+ homeostasis through two pathways: Nrk-mediated phosphorylation to NMN, or Pnp1/Urh1/Meu1-mediated degradation followed by NAM recycling.11,16,54 NMN generated from both pathways can be readily converted to NAD+ by NMN adenylyltransferases. Similarly, the mammalian NR pathway also contains phosphorylation component catalyzed by NR kinases NRK1/NRK2.16,19 What has been controversial is whether degradation is a key metabolic fate of NR in mammals. In the current study, we demonstrated that the bioconversion of NR to NAD+ can be monitored by using NRs with isotope labels. NR, indeed, demonstrated robust NAD+-elevating effect. This effect is mediated primarily by NRK-dependent pathway but is independent of NR degradation by PNP in the cellular context. The lack of PNP pathway might be due to the compartmentalization of PNP that makes NR inaccessible to the degradation enzyme, or more efficient NRK activity over PNP activity in mammalian cells. Further studies are expected to better answer this question. Moreover, NR can rescue NAD+ depletion caused by pharmacological inhibition of the salvage pathway. Maintaining cellular NAD+ homeostasis has been suggested as a promising therapeutic strategy for the treatment of metabolic disorders and age-related diseases. NR, as a potent NAD+-boosting agent, holds great therapeutic potential.
METHODS AND MATERIALS
Reagents and instruments
All reagents were purchased from Aldrich or Fisher Scientific and were of the highest purity commercially available. UV spectra were obtained with a Varian Cary 300 Bio UV-visible spectrophotometer. HPLC was performed on a Dionex Ultimate 3000 HPLC system equipped with a diode array detector using a Macherey-Nagel C18 reverse-phase column. Radiolabeled samples were counted in a Beckman LS6500 scintillation counter. NMR spectra were acquired on a Bruker AVANCE III 500 MHz high-field NMR spectrometer and the data was processed using Topspin software. MS spectra were acquired with either a Q-Exactive Hybrid Quadrupole Orbitrap Mass Spectrometer (Thermo Scientific) coupled to an EASY-nLC 1000 Liquid Chromatography (Thermo Scientific), an LTQ Orbitrap Discovery (Thermo Scientific) Mass Spectrometer coupled to a Surveyor HPLC system (Thermo Scientific), or a Bruker Autoflex II MALDI-TOF mass spectrometer operated in positive ion reflectron mode.
Enzymes
Hexokinase, glucose-6-phosphate dehydrogenase, glutamate dehydrogenase, 6-phosphogluconate dehydrogenase, phosphoriboisomerase, myokinase, pyruvate kinase and ADP-ribosylcyclase were purchased from Sigma. Phosphodiesterase I was purchased from Worthington Biochemical. Alkaline phosphatase was purchased from New England Biolabs. Human PNP and NRK1 were purchased from Novus Biologicals. E. coli phosphoribosylpyrophosphate synthetase was expressed and purified using a published method.32 Nicotinate phosphoribosyltransferase was purified as described before.33
Expression and purification of yeast nicotinamide mononucleotide adenylyltransferase (NMNAT)
The gene NMA1 was cloned from Saccharomyces cerevisiae gDNA into the protein expression vector, Pet28a (Novagen). The PetNMA1 vector was transfected into BL21-CodonPlus(DE3)-RIPL competent cells (Agilent) and protein expression induced by 0.8 mM isopropyl-β-D-thiogalactopyranoside (IPTG) when the cells reached OD600 of 0.6~0.7 in LB media. The culture was grown for another 16 h at 37°C before the cells were pelleted and lysed with 3 freeze-thaw cycles. The protein was purified using a Ni-NTA resin (Thermo Fisher) affinity column and eluted with increasing concentrations of imidazole. The protein was aliquoted in 20% glycerol plus 2 mM DTT, flash frozen and stored at −80°C. Protein concentration was determined using Bradford assay. The protein was >95% pure as determined by SDS-PAGE gel electrophoresis.
Expression and purification of S. pneumoniae NAD+ synthetase
The gene for NAD+ Synthetase, NadE, was amplified from Streptococcus pneumoniae gDNA and cloned into the Pet28a vector (Novagen) at the NdeI-BamHI sites. The PetSPNadE vector was transfected into BL21-CodonPlus(DE3)-RIPL competent cells (Agilent) and protein expression induced by 0.8 mM IPTG when the cells reached OD600 of 0.6~0.7 in LB media. The culture was grown for another 16 h at 37°C before the cells were pelleted and lysed with 3 freeze-thaw cycles. The protein was purified using a Ni-NTA resin affinity column and eluted with increasing concentrations of imidazole. The protein was aliquoted in 20% glycerol plus 2 mM DTT, flash frozen and stored at −80°C. Protein concentration was determined using Bradford assay. The protein was >95% pure as determined by SDS-PAGE gel electrophoresis.
Synthesis of NR
NR was synthesized as described before.18
Synthesis of 18O-nicotinamide
This compound was synthesized as described previously.39,55 Briefly, 3-cyanopyridine (100 mg, 0.96 mmol) and 1,1′,3,3′-tetramethylguanidine (5.1 mg, 0.04 mmol) were dissolved in 200 μL 18O-water (97%, Cambridge Isotope Laboratories). The solution was heated at 100°C for 1 h before water and 1,1′,3,3′-tetramethylguanidine were removed by lyophilization. After crystallization from benzene, 18O-NAM was obtained (102 mg, 0.82 mmol, 85% yield). The incorporation of 18O was determined to be 96.5% by MALDI-TOF.
Synthesis of 13C-NAD+
This is done in a two-step sequence as described before.31 D-Glucose (U-13C6, 99%) was converted to labeled NaAD via a ten enzymes coupled one-pot reaction (Scheme 2A). Briefly, a typical reaction was performed in 50 mM phosphate buffer pH 7.5 in a total volume of 1 mL. The reaction contained 1 mM U-13C6-glucose, 2 mM ATP, 3 mM MgCl2, 50 mM KCl, 5 mM DTT, 10 mM PEP, 10 mM α-ketoglutarate, 100 μM NADP, and 2 mM nicotinic acid. All the enzymes were added as follows: 0.1 U/mL hexokinase, 2 U/mL pyruvate kinase, 0.1 U/mL glucose 6-phosphare dehydrogenase, 0.5 U/mL glutamate dehydrogenase, 0.1 U/mL 6-phosphogluconate dehydrogenase, 6 U/mL phosphoriboisomerase, 0.5 U/mL myokinase, 0.02 U/mL nicotinate phosphoribosyltransferase, 0.2 U/mL phosphoribosylpyrophosphate synthetase, 0.25 U/mL nicotinamide mononucleotide adenylyltransferase. The reaction was initiated by the addition of hexokinase. The reaction was incubated at 37°C for 4 h before being heated at 120°C for 1.5 min. The sample was centrifuged at 14,000 × g for 5 min before it was purified with HPLC. The sample was injected on an HPLC fitted to an XTerra C18 preparative column. NaAD was separated from other metabolites using a gradient of 0 to 20% methanol in 20 mM ammonium acetate with a flow rate of 20 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing NaAD were collected and lyophilized to dryness. The yields of purified 13C-NaAD were in the range of 75%~91%.
After HPLC purification, 13C-NaAD was dissolved in 50 mM phosphate buffer pH 7.5 to a final concentration of 2 mM. To this solution were then added 50 mM KCl, 3 mM MgCl2, 5 mM DTT, 4 mM ATP and 5 mM NH4Cl. The reaction was initiated by the addition of 0.2 U/mL S. pneumoniae NAD+ synthetase and incubated at 37°C for 2 h. The reaction mixture was injected on an HPLC fitted to an XTerra C18 preparative column. 13C-NAD+ was separated from other metabolites using a gradient of 0 to 20% methanol in 20 mM ammonium acetate with a flow rate of 20 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing NAD+ were collected and lyophilized to dryness. The yields of purified 13C-NAD+ were in the range of 81%~89%.
Synthesis of 13C,18O-NAD+
A typical reaction contained 1 mM 13C-NAD+ and 5 mM 18O-NAM in 100 mM phosphate buffer pH 7.5. The reaction was initiated by the addition of ADP-ribosylcyclase. The reaction was incubated at 37°C for 2 h before being quenched by 10% TFA. 13C,18O-NAD+ can then be purified using HPLC as described above. The yields of purified 13C,18O-NAD+ were in the range of 65%~68%.
Synthesis of 13C,18O-NR
A typical reaction contained 400 μM 13C,18O-NAD+ in 100 mM phosphate buffer pH 7.5. The reaction was initiated by the addition of phosphodiesterase I and alkaline phosphatase. The reaction was incubated at 37°C for 2 h. The reaction mixture was injected on an HPLC fitted to a Aquasil C18 column. 13C,18O-NR was separated using a gradient of 0 to 20% methanol in 20 mM ammonium acetate with a flow rate of 1 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing NR were collected and lyophilized to dryness. The yield of this transformation was almost quantitative.
Synthesis of 14C-NR
A typical reaction contained 1 mM unlabeled NAD+, 5 mM [carbonyl-14C]-nicotinamide (14C-NAM, American Radiolabeled Chemicals Inc.) in 100 mM phosphate buffer pH 7.5. The reaction was initiated by the addition of ADP-ribosylcyclase. The reaction was incubated at 37°C for 2 h before being quenched by 10% TFA. 14C-NAD+ can then be purified using HPLC as described above. Subsequently, 14C-NAD+ was re-suspended in 100 mM phosphate buffer pH 7.5 to a final concentration of 400 μM. The reaction was initiated by the addition of phosphodiesterase I and alkaline phosphatase. The reaction was incubated at 37°C for 2 h. 14C-NR was purified as described above. The overall yields of purified 14C-NR were in the range of 54%~67% based on the starting radioactivity.
Cell culture
Neuro2a cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were maintained in a humidified 37°C incubator with 5% CO2.
Radioisotope labeled NR treatment
Neuro2a cells were plated onto 6-well plates and grown to 70~80% confluence. Cells were then incubated with 200,000 to 450,000 cpm 14C-NR for 2 to 6 h. Cells were harvested, rinsed and re-suspended in 1 mL of fresh medium. Cell number was determined using a hemocytometer. The cell suspension was then re-pelleted for NAD+ metabolites measurements.
Unlabeled NR treatment
Cells were incubated with or without 1 mM NR for 4 h. Cells were harvested, rinsed and re-suspended in 1 mL of fresh medium. Cell number was determined using a hemocytometer. The cell suspension was then re-pelleted for NAD+ concentration determination.
NAD+ metabolites analysis
To the cell pellet was added 30 μL of ice-cold 7% perchloric acid. The sample was then vortexed for 30 s and sonicated on ice for 5 min. The vortex-sonication cycle was repeated three times. The sample was centrifuged at 14,000 x g for 3 min at RT. Clear supernatant was removed and neutralized to pH 7 with 3 M NaOH and 1 M phosphate buffer (pH 9). The sample was then injected on an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. To aid the identification of these species, unlabeled authentic standards of NAM, NR, NMN and NAD+ were co-injected with the cellular components. NMN, NR, NAM and NAD+ were resolved using a gradient of 0 to 20% methanol in 20 mM ammonium acetate with a flow rate of 1 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing each of the NAD+ metabolites were collected and radioactivity was determined by scintillation counting.
NAD+ concentration determination
The cellular NAD+ level was measured using NAD+ cycling assay as described previously.48
Cell lysate preparation
Cells were harvested and lysed with RIPA buffer (Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific).
FK866 or ImmH treatment
For cell culture experiments, FK866 (20 nM) or ImmH (1 or 10 μM) was added to the medium together with either unlabeled or 14C-labeled NR and incubated at 37°C. The cells were then harvested, rinsed and re-suspended in 1 mL of fresh medium. Cell number was determined using a hemocytometer. The cell suspension was then re-pelleted for either NAD+ metabolites measurements or NAD+ concentration determination.
For cell lysate experiments, the lysate was incubated with ImmH (1 or 10 μM) together with 100,000 cpm 14C-NR at 37°C before it was quenched by 10% TFA. The sample was centrifuged at 14,000 × g for 3 min at RT. Clear supernatant was removed and neutralized to pH 7 with 3 M NaOH and 1 M phosphate buffer (pH 9). The sample was then injected on an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. NMN, NR, NAM and NAD+ were resolved using a gradient of 0 to 20 methanol in 20 mM ammonium acetate with a flow rate of 1 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing each of the NAD+ metabolites were collected and radioactivity was determined by scintillation counting.
13C,18O-NR treatment
Cells were cultured with either unlabeled NR or 13C,18O-NR for 4 h. The cells were then harvested, rinsed and re-suspended in 1 mL of fresh medium. Cell number was determined using a hemocytometer. The cell suspension was then re-pelleted. To the cell pellet was added 30 μL of ice-cold 7% perchloric acid. The sample was then vortexed for 30 s and sonicated on ice for 5 min. The vortex-sonication cycle was repeated three times. The sample was centrifuged at 14,000 × g for 3 min at RT. Clear supernatant was removed and neutralized to pH 7 with 3 M NaOH and 1 M phosphate buffer (pH 9). The sample was then injected on an HPLC fitted to a Macherey-Nagel Nucleosil C18 column. NMN, NR, NAM and NAD+ were resolved using a gradient of 0 to 20% methanol in 20 mM ammonium acetate with a flow rate of 1 mL/min. Chromatograms were analyzed at 260 nm. Fractions containing NAD+ were collected and lyophilized to dryness. The sample was then subjected to MALDI-TOF analysis.
Supplementary Material
Acknowledgments
This work was supported in part by start-up funding from ACPHS (to Y.C.) and 1R15GM123393 from NIH/NIGMS (to Y.C.).
References
- 1.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krishnakumar R, Kraus WL. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sauve AA, Youn DY. Curr Opin Chem Biol. 2012;16:535–543. doi: 10.1016/j.cbpa.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Bai P, Canto C. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Chang HC, Guarente L. Trends Endocrinol Metab. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang T, Fu M, Pestell R, Sauve AA. Trends Endocrinol Metab. 2006;17:186–191. doi: 10.1016/j.tem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurnasov O, Goral V, Colabroy K, Gerdes S, Anantha S, Osterman A, Begley TP. Chem Biol. 2003;10:1195–1204. doi: 10.1016/j.chembiol.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 9.Belenky P, Bogan KL, Brenner C. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, Nedyalkova L, Yang T, Sauve AA, Park HW, Brenner C. PLoS Biol. 2007;5:e263. doi: 10.1371/journal.pbio.0050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Bogan KL, Brenner C. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich LS, Fuller L, Yero IL, Martinez L. J Biol Chem. 1966;241:188–191. [PubMed] [Google Scholar]
- 14.Preiss J, Handler P. J Biol Chem. 1958;233:488–492. [PubMed] [Google Scholar]
- 15.Xu P, Sauve AA. Mech Ageing Dev. 2010;131:287–298. doi: 10.1016/j.mad.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Bieganowski P, Brenner C. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva Xavier G, Garten A, Elhassan YS, Redpath P, Migaud ME, Philp A, Brenner C, Canto C, Lavery GG. Mol Metab. 2017;6:819–832. doi: 10.1016/j.molmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang T, Chan NY, Sauve AA. J Med Chem. 2007;50:6458–6461. doi: 10.1021/jm701001c. [DOI] [PubMed] [Google Scholar]
- 19.Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C, Canto C. Nat Commun. 2016;7:13103. doi: 10.1038/ncomms13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DAG, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Canto C, Conley KE, Walter L, Lovering RM, Chin ER, Jasmin BJ, Marcinek DJ, Menzies KJ, Auwerx J. Sci Transl Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaur P, Brugg B, Mericskay M, Li Z, Schmidt MS, Vivien D, Orset C, Jacotot E, Brenner C, Duplus E. FASEB J. 2017;31:5440–5452. doi: 10.1096/fj.201700221RR. [DOI] [PubMed] [Google Scholar]
- 22.Wu LE, Sinclair DA. Cell Res. 2016;26:971–972. doi: 10.1038/cr.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canto C, Auwerx J. Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauve AA. J Pharmacol Exp Ther. 2008;324:883–893. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- 25.Hershberger KA, Martin AS, Hirschey MD. Nat Rev Nephrol. 2017;13:213–225. doi: 10.1038/nrneph.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canto C, Menzies KJ, Auwerx J. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowen JW, Kornberg A. J Biol Chem. 1951;193:497–507. [PubMed] [Google Scholar]
- 28.Wielgus-Kutrowska B, Kulikowska E, Wierzchowski J, Bzowska A, Shugar D. Eur J Biochem. 1997;243:408–414. doi: 10.1111/j.1432-1033.1997.0408a.x. [DOI] [PubMed] [Google Scholar]
- 29.Tanimori S, Ohta T, Kirihata M. Bioorg Med Chem Lett. 2002;12:1135–1137. doi: 10.1016/s0960-894x(02)00125-7. [DOI] [PubMed] [Google Scholar]
- 30.Mikhailopulo IA, Pricota TI, Timoshchuk VA, Akhrem AA. Synthesis. 1981;1981:388–389. [Google Scholar]
- 31.Rising KA, Schramm VL. J Am Chem Soc. 1994;116:6531–6536. [Google Scholar]
- 32.Tolbert TJ, Williamson JR. J Am Chem Soc. 1996;118:7929–7940. [Google Scholar]
- 33.Cen Y, Sauve AA. J Am Chem Soc. 2010;132:12286–12298. doi: 10.1021/ja910342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berti PJ, Blanke SR, Schramm VL. J Am Chem Soc. 1997;119:12079–12088. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheuring J, Berti PJ, Schramm VL. Biochemistry. 1998;37:2748–2758. doi: 10.1021/bi972594x. [DOI] [PubMed] [Google Scholar]
- 36.Rising KA, Schramm VL. J Am Chem Soc. 1997;119:27–37. [Google Scholar]
- 37.Schultheisz HL, Szymczyna BR, Williamson JR. J Am Chem Soc. 2009;131:14571–14578. doi: 10.1021/ja903155s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson MD, Echlin H, Dao TH, Rosch JW. Microbiology. 2015;161:2127–2136. doi: 10.1099/mic.0.000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang T, Sauve AA. AAPS J. 2006;8:E632–643. doi: 10.1208/aapsj080472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HC, Aarhus R. Cell Regul. 1991;2:203–209. doi: 10.1091/mbc.2.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauve AA, Munshi C, Lee HC, Schramm VL. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- 42.Sauve AA, Deng H, Angeletti RH, Schramm VL. J Am Chem Soc. 2000;122:7855–7859. [Google Scholar]
- 43.Revollo JR, Grimm AA, Imai S. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 44.Hasmann M, Schemainda I. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 45.Skoge RH, Dolle C, Ziegler M. DNA Repair (Amst) 2014;23:33–38. doi: 10.1016/j.dnarep.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Graham E, Rymarchyk S, Wood M, Cen Y. ACS Chem Biol. 2018;13:782–792. doi: 10.1021/acschembio.7b00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. Biochemistry. 1998;37:8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 48.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. J Biol Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 50.Smythe GA, Braga O, Brew BJ, Grant RS, Guillemin GJ, Kerr SJ, Walker DW. Anal Biochem. 2002;301:21–26. doi: 10.1006/abio.2001.5490. [DOI] [PubMed] [Google Scholar]
- 51.Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 52.Gossmann TI, Ziegler M, Puntervoll P, de Figueiredo LF, Schuster S, Heiland I. FEBS J. 2012;279:3355–3363. doi: 10.1111/j.1742-4658.2012.08559.x. [DOI] [PubMed] [Google Scholar]
- 53.Rongvaux A, Andris F, Van Gool F, Leo O. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 54.Ma B, Pan SJ, Zupancic ML, Cormack BP. Mol Microbiol. 2007;66:14–25. doi: 10.1111/j.1365-2958.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- 55.Kołodziejska-Huben M, Kamiński Z, Paneth P. J Labelled Comp Radiopharm. 2002;45:1005–1010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.