

Abstract
Balanced and apparently balanced chromosome abnormalities (BCAs) have long been known to generate disease through position effects, either by altering local networks of gene regulation or positioning genes in architecturally different chromosome domains. Despite these observations, identification of distally affected genes by BCAs is oftentimes neglected, especially when predicted gene disruptions are found elsewhere in the genome. In this unit, we provide detailed instructions on how to run a computational pipeline that identifies relevant candidates of non-coding BCA position effects. This methodology facilitates quick identification of genes potentially involved in disease by non-coding BCAs and other types of rearrangements, and expands on the importance of considering the long-range consequences of genomic lesions.
Keywords: position effects, chromosome abnormality, cytogenetics, human genetics
Introduction
The compromised integrity of chromosome structure through translocations, inversions, insertions, deletion and duplications is associated with human disease through a variety of mechanisms (Kleinjan et al. 2001; Kleinjan and van Heyningen 2005; Zhang and Lupski 2015). Among the best characterized is the disruption of regulatory interactions between gene promoters and distal control elements such as enhancers, locus control regions, silencers, etc (Kleinjan et al. 2001; Kleinjan and van Heyningen 2005). In this context, even apparently balanced or balanced non-coding chromosome abnormalities (BCAs) may exert position effects associated with disease by inducing expression changes of neighboring genes without affecting their sequence. Well known examples of this phenomenon include translocations upstream and downstream of SOX9 linked to campomelic dysplasia (Velagaleti et al. 2005), and translocations downstream of PAX6, involved in congenital aniridia (Kleinjan et al. 2001; Velagaleti et al. 2005).
Because of the ever enlarging amount of sequencing information, the number of non-coding variants and rearrangements that need clinical interpretation has greatly increased (Ward and Kellis 2012; Zhang and Lupski 2015). As described in Ibn-Salem et al. 2014 and Zepeda-Mendoza et al. 2017 (Ibn-Salem et al. 2014; Zepeda-Mendoza et al. 2017), position effect analysis can in many cases enlighten the analysis of such variants. Zepeda-Mendoza et al. 2017 designed a computational pipeline that identifies and ranks potential position effect genes for chromosome rearrangements (either balanced or unbalanced) using publicly available enhancer, promoter and DNaseI hypersensitive sites (DHSs) datasets from the Encyclopedia of DNA Elements (ENCODE) project (2012), TAD boundaries (Dixon et al. 2012), predicted enhancer-promoter interactions (Thurman et al. 2012), as well as Human Phenotype Ontology (HPO) (Kohler et al. 2014) terms which tailors position effect gene ranking to those involved in a subject’s clinical phenotype. This unit provides a detailed method for running the position effect pipeline from Zepeda-Mendoza et al. 2017 to assess the contribution of distal genes in the generation of a clinical phenotype, and further allows for filtering of most relevant candidate genes. It offers examples and instructions that will provide clinical geneticists and researchers a quick and optimized way to identify additional genes in human disease.
Basic Protocol
The main purpose of this protocol is to identify potential position effect candidates related to particular clinical presentations in subjects with non-coding and apparently balanced chromosome rearrangements.
An example study case from Zepeda-Mendoza et al. 2017 is DGAP163 is a four-year-old boy with reported severe global developmental delay, absent speech, dysmorphic/distinctive facies, and hypospadias. As an infant he presented seizures, small left retinal coloboma, myopia, nystagmus, and he had a history of conductive hearing loss. He had normal fluorescence in situ hybridization (FISH) results for Smith Magenis syndrome (MIM#182290), DiGeorge syndrome (MIM#188400) and Velocardiofacial syndrome (MIM#192430), as well as a normal aCGH (1M Agilent array). Chromosome analysis revealed an apparently balanced translocation between chromosomes 2 and 14. His karyotype is 46,XY,t(2;14)(p23;q13)dn.arr(1–22)x2,(X,Y)x1 (Figure 1A). Genome sequencing revealed breakpoint locations to be chr2:39206240-39206242 and chr14:31717833-31717834.
Figure 1.
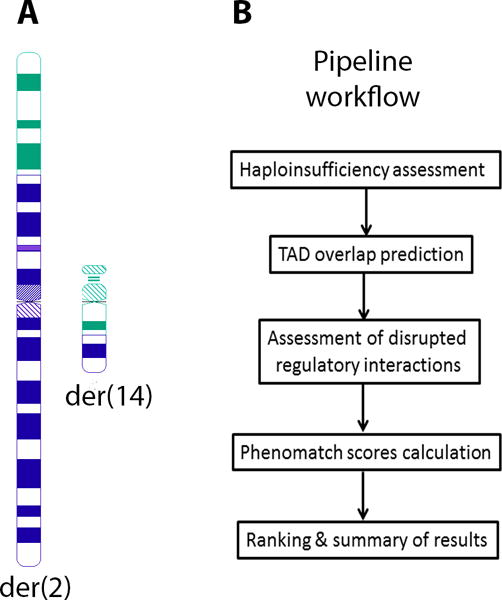
A) Ideogram of DGAP163’s t(2;14)(p23;q13). Chromosome 2 is depicted in blue while chromosome 14 is depicted in green. B) Analysis steps of the computational pipeline described in this protocol. A full analysis of chromatin interactions, haploinsufficiency and phenotypic relatedness is assembled and distal candidate genes are ranked based on their functional evidence.
To establish whether the translocation may generate position effects on neighboring genes that may be responsible for all or part of DGAP163’s clinical phenotype, the position effect pipeline can be employed to determine the suitability of identified candidates (Figure 1B).
Necessary resources
Hardware
Any standard workstation running iOS, Windows, or any Linux distribution with a minimum of 4Gb RAM can run the position effect pipeline.
Software
The position effect prediction pipeline comprises a series of Perl, R and Java scripts that are run independently to categorize and rank different features of candidate genes. In order to run these scripts, it is recommended that users have access to a Linux-based operating system (i.e., iOS, Ubuntu, among others) or install the proper software additions. For more information on how to install Perl and R in Microsoft Windows refer to http://strawberryperl.com/, https://www.activestate.com/activeperl, and https://cran.r-project.org/bin/windows/base/rw-FAQ.html.
Files
All the necessary files can be downloaded from the following GitHub repository: https://github.com/ibn-salem/position_effect
Download and unzip the prediction effect pipeline scripts
-
1
The pipeline is available for download at https://github.com/ibn-salem/position_effect. The downloaded file, position_effect-master.zip, needs to be decompressed and stored in a user-defined folder. To unzip the file, write the following command in a terminal:
unzip position_effect-master.zip -d destination_folder
You will also need to unzip the data folder contained within the position_effect-master file. This folder contains the GRCh37 annotation files that will be used throughout the protocol, including happloinsufficiency scores, chromatin contacts, gene annotations, among others. To unzip the data folder, move to your destination folder and run in a terminal the following command:
unzip data/data_files.zip -d data/
Formatting of rearrangement breakpoint positions
-
2
The nucleotide positions of the BCAs or other rearrangements subjected to position effect analysis must be formatted in .bed style. In the case of DGAP163, translocation breakpoints were mapped at chr2(39206240-39206242) and chr14(31717833-31717834). Thus for this subject, open a text editor, create a new document, and write down the following lines using tabulator to separate column values:
| chr2 | 39206240 | 39206242 | DGAP163_A |
| chr14 | 31717833 | 31717834 | DGAP163_B |
These lines indicate the presence of two breakpoints in chromosomes 2 and 14, as well as their nucleotide positions and an identifier (breakpoint DGAP163_A and breakpoint DGAP163_B). Save this file in the analysis folder with the name DGAP163_positions.bed.
For additional bed formatting questions, the University of California Santa Cruz genome browser has a comprehensive guide that can be accessed at https://genome.ucsc.edu/FAQ/FAQformat.html
Selection of analysis window size
-
3
It is important to determine an analysis window size for the prediction of position effects. From our experience, the optimal window size for candidate gene discovery is +/− 1Mb upstream and downstream of the rearrangement breakpoints, as determined from analyzing 57 cases with known pathogenic and likely-pathogenic position effect variants. Results contained within the +/− 1Mb window are automatically included in the final candidate report (Step 9 of this protocol). Users are free to select smaller or larger analysis windows to suit their needs. The maximum recommended distance for predicting position effects is +/− 3Mb (i.e., 6Mb window size), according to reported distances of architectural loops within TADs (Krijger and de Laat 2016). For the rest of this protocol we will use a +/− 3Mb window to exemplify how many candidates could be obtained at the recommended maximal range of analysis.
Analysis of neighboring genes haploinsufficiency and TAD overlaps
-
4
Gene happloinsufficiency is one of the main pathogenicity criteria used in the position effect prediction of this pipeline. Moreover, inclusion of a known or predicted haploinsufficient gene within the same TAD as the rearrangement breakpoints additionally strengthens its value as a candidate. This assessment is derived from the hypothesis that the rearrangement disrupts the native regulatory environment of the TAD; such disruption could lead to haploinsufficient gene misregulation and disease. The haploinsufficiency analysis and assessment of breakpoint-mediated TAD disruption is performed by running the dgap_features_check.pl script, which utilizes the following parameters:
perl dgap_features_check.pl \ -f GENOMIC_REGIONS \ -i HI_SCORES_FILE \ -g ENSEMBL_GENE_FILE_SUMMARY \ -c HIC_DOMAINS \ -n WINDOW_BP_SIZE \ -o OUTPUT_FOLDER_PATH
The GENOMIC_REGIONS file is the .bed file generated in Step 2 of this protocol. The HI_SCORES_FILE is the genomic haploinsufficiency score list published by Huang et al. 2010 (Huang et al. 2010), the ENSEMBL_GENE_FILE_SUMMARY is a gene annotation file derived from Ensembl BioMart (output columns selected include: Ensembl Gene ID, Chromosome Name, Gene Start (bp), Gene End (bp), HGNC ID(s), HGNC symbol, Gene type, WikiGene Description, Description, Phenotype description),and the HiC domains file is a processed bed file containing the TAD positions reported by (Dixon et al. 2012). All of these files are included in the data folder unzipped in Step 1 of this protocol WINDOW_BP_SIZE is the analysis window size selected in Step 3 of this protocol, and finally OUTPUT_FOLDER_PATH is the folder where the output analysis files will be saved. Included in the example data folder downloaded from the pipeline GitHub repository, the following files are used to run the script:
perl perl/dgap_features_check.pl \ -f DGAP163_positions.bed \ -i data/HI_Predictions_Version3.bed \ -g data/GRCh37.p13_ensembl_genes.txt \ -c data/hESC_hg37_domains.bed \ -n 3000000 \ -o.
This script generates the files HiC_list_DGAP.txt and HI_list_DGAP.txt. The first file contains a list of overlapped Hi-C domains by the rearrangement breakpoints, while the second file is a summary of breakpoint-overlapped Hi-C domains and genes with happloinsufficiency information within the selected analysis window. The output column structure for HI_list_DGAP.txt is shown in Table 1. For DGAP163, the output HiC_list_DGAP.txt should have two rows while HI_list_DGAP.txt has 46 rows of results. You can compare your output files to those included in the Supplementary Materials for this protocol.
Table 1.
HI_list_DGAP.txt column descriptions from Step 4 of this protocol.
| Column name | Description |
|---|---|
| Breakpoint_ID | Identification number of the studied rearrangement breakpoints |
| chr | Rearrangement breakpoint chromosome location |
| start | Rearrangement breakpoint start position |
| end | Rearrangement breakpoint end position |
| win_start | Analysis window start position |
| win_end | Analysis window end position |
| Hi_domain | TAD coordinates that contain the rearrangement breakpoint |
| HiC_chr | TAD chromosome location |
| HiC_start | TAD start position |
| HiC_end | TAD end position |
| HI_gene_chr | Chromosome location for gene contained within the analysis window |
| HI_gene_start | Position start for gene contained within the analysis window |
| HI_gene_end | Position end for gene contained within the analysis window |
| HI_gene_name | HUGO name for gene contained within the analysis window |
| LOD | Log-odds score derived from Huang et al., 2010 for gene haploinsufficiency |
| HI_prob | Calculated probability of a gene being haploinsufficient |
| HI_ensembl_gene_ID | Ensembl ID of gene contained within the analysis window |
| HI_chr | Ensembl chromosome location of gene contained within the analysis window |
| Gene_start | Ensembl reported start position of gene contained within the analysis window |
| gene_end | Ensembl reported end position of gene contained within the analysis window |
| HGNC_ID | HUGO Gene Nomenclature Committee identifier for gene contained within the analysis window |
| HGNC_symbol | HUGO Gene Nomenclature Committee symbol for gene contained within the analysis window |
| Gene_type | Ensembl reported type of gene contained within the analysis window |
| WikiGene_Description | WikiGene description of gene contained within the analysis window |
| Description | Gene description |
| Phenotype_description | Ensembl reported phenotypic associations of gene contained within the analysis window |
The files HI_SCORES_FILE, ENSEMBL_GENE_FILE_SUMMARY, and HIC_DOMAINS can be replaced with more recent versions or other custom files as long as the column structure is maintained.
Analysis of predicted enhancer/promoter interactions disrupted by the rearrangement breakpoints
-
5
The analysis pipeline predicts position effects based on haploinsufficiency information as well as disruption of chromosome organization. The alteration of enhancer-promoter interactions combines both criteria and further refines the list of candidate neighboring genes contributing to a clinical phenotype. The script enh_promoter_disruption_checker_DGAP.pl assesses the number of disrupted predicted DHS enhancer/promoter interactions (Thurman et al. 2012) within the analysis window. The script uses the following parameters:
perl enh_promoter_disruption_checker_DGAP.pl \ -f GENOMIC_REGIONS \ -d DHS_ENH_PROMOTER_FILE \ -a WINDOW_BP_SIZE \ -o OUTPUT_FILENAME
To run the analysis on DGAP163, use the DGAP163_positions.bed file from Step 2 of this protocol, the WINDOW_BP_SIZE selected in Step 3, as well as the DHS_ENH_PROMOTER_FILE from the data folder downloaded from the pipeline GitHub repository:
perl perl/enh_promoter_disruption_checker_DGAP.pl \ -f DGAP163_positions.bed \ -d data/genomewideCorrs_above0.7_promoterPlusMinus500kb_withGeneNames_32celltypeCategories.bed8 \ -a 3000000 \ -o DHS_promoter_broken_DGAP163.txt
This script will output the number and location of the predicted DHS enhancer/promoter interactions affected by the rearrangement breakpoints. The file used in this analysis was obtained from (Thurman et al. 2012), but additional regulatory interaction datasets may be used provided their column structures follow the one used in the DHS_ENH_PROMOTER_FILE.
Conversion of clinical phenotype to HPO terms
-
6
To further refine the selection of candidate position effect genes, a “phenomatch score” calculation is performed to compare the similarity of the subject’s clinical presentation and the potential neighboring position effect genes. Before calculating phenomatch scores (see next step in the protocol), users need to convert the clinical descriptions of their cases to Human Phenotype Ontology (HPO) terms (Kohler et al. 2014). The HPO project has curated over 11,000 terms, with the goal of facilitating the large scale computational analysis of human phenotypic annotations. Users can convert their clinical descriptions to HPO terms using Phenomizer (http://human-phenotype-ontology.github.io/tools.html), Phenotips (https://phenotips.org/), and the HPO Browser (http://human-phenotype-ontology.github.io/).
The file structure of the phenotype description for DGAP163 is as follows:
| ID | HPO |
| DGAP163 | HP:0000047 |
| DGAP163 | HP:0000480 |
| DGAP163 | HP:0000545 |
| DGAP163 | HP:0000639 |
| DGAP163 | HP:0001250 |
| DGAP163 | HP:0001270 |
| DGAP163 | HP:0001344 |
| DGAP163 | HP:0001763 |
| DGAP163 | HP:0008598 |
| DGAP163 | HP:0011344 |
Save this information in a file named DGAP163_phenotype.txt. Notice how all of the HPO IDs correspond to the clinical description provided in the beginning of the protocol (i.e., HP:0000047 is hypospadias, HP:0000480 is retinal coloboma, etc.) The phenomatch score calculation script uses a combined file with “chr”, “start”, “end”, “Breakpoint_ID”, “HPO” column. This file can be easily made by running the script combine_breakpoints_and_phenotypes.R. The parameters used are as follows:
Rscript combine_breakpoints_and_phenotypes.R \ SUBJECT_ PHENOTYPES \ GENOMIC_REGIONS \ WINDOW_SIZE \ OUTPUT_FILE
The SUBJECT_ PHENOTYPES file contains the case ID and the associated HPO terms, just as constructed for DGAP163_phenotype.txt. GENOMIC_REGIONS is the DGAP163_positions.bed file built in Step 2 of this protocol. The WINDOW_SIZE for this program will be the selected window size selected in Step 3, but multiplied by 2 (i.e., if 500Kb was selected for Step 3 of the protocol, 1Mb (1,000,000bp) will be used to run this script.) For DGAP163 run the script in the following manner:
Rscript R/combine_breakpoints_and_phenotypes.R DGAP163_phenotype.txt DGAP163_positions.bed 6000000 \ DGAP163_breakpoint_window_with_HPO.6MB_win.bed
The combine_breakpoints_and_phenotypes.R script requires the stringr and readr packages. For more information about how to install packages in R, visit https://www.r-bloggers.com/installing-r-packages/
Phenomatch scores calculation
-
7
The phenomatch score metric was created by (Ibn-Salem et al. 2014), and provides a quantitative measure of phenotypic similarity between two datasets of HPO terms. Comparing the similarity of the subject’s clinical presentation and the neighboring position effect genes is one of the most important pieces of information to rank position effect candidates, as genes without pathogenic consequences related to those of the cases we analyze are excluded. Phenomatch scores are calculated by running the phenomatch.jar script with the following parameters:
java -jar phenomatch.jar \ -i INPUT_FILE \ -g GENES \ -O PHENOTYPE_ONTOLOGY -a ANNOTATION_FILE \ -o OUTPUT_FILE
The INPUT_FILE is the file generated in Step 6, GENES is a list of annotated genes with a format of chr, start, end, entrez_id, strand; PHENOTYPE_ONTOLOGY is the obo file derived from HPO which contains all the ontological relationships between terms; ANNOTATION_FILE contains all annotated terms to each gene in the human genome, and is also obtained from the HPO webpage; finally, OUTPUT_FILE specifies the name of the file where the phenomatch scores of all breakpoint neighboring genes within the analysis window will be written.
For the DGAP163 example:
java -jar bin/phenomatch.jar \ -i DGAP163_breakpoint_window_with_HPO.6MB_win.bed \ -g data/knownGene.txt.entrez_id.tab.unique \ -O data/hp.obo \ -a data/ALL_SOURCES_TYPICAL_FEATURES_genes_to_phenotype.txt \ -o DGAP163_breakpoint_window_with_HPO.6MB_win.bed.phenomatch
More recent versions of HPO and gene phenotype annotations can be downloaded from: http://human-phenotype-ontology.github.io/downloads.html
Ranking of phenomatch scores
-
8
Once all phenomatch scores of breakpoint neighboring genes are calculated, a ranking system will prioritize the most likely candidates over those with no apparent contribution. This is performed by sorting the genes with the get_percentiles_DGAP_all.r script, which calculates the percentile rank for the phenomatch and max_phenomatch scores calculated in Step 7 of this protocol. The program is run using the following input files:
Rscript --vanilla get_percentiles_DGAP_all.r PHENO_FILE > OUTPUT_FILENAME
For DGAP163:
Rscript --vanilla R/get_percentiles_DGAP_all.r \ DGAP163_breakpoint_window_with_HPO.6MB_win.bed.phenomatch \ > percentiles_6Mb_pheno_maxpheno.txt
Summarizing results
-
9
9. The last step in the position effect analysis is putting together all of the results for a final ranking of the candidates. The script dgap_final_table_maker.pl will pull together all data and is run using the parameters:
perl dgap_final_table_maker.pl \ -f TAD_ANALYSIS_FEATURES_FILE \ -c HIC_DOMAINS\ -d DHS_ENH_PROMOTER_DISRUPTED_CONTACTS_FILE \ -h CLINGEN_HAPLO_FILE \ -t CLINGEN_TRIPLO_FILE \ -m PHENOMATCH_PERCENTILES_FILE \ -o OUTPUT_FILENAME
The TAD_ANALYSIS_FEATURES_FILE is the HI_list_DGAP.txt file that was obtained in Step 1. The file HIC_DOMAINS is the TAD information file used in Step 1. The DHS_ENH_PROMOTER_DISRUPTED_CONTACTS_FILE is the output file of Step 2 that is not the summary. CLINGEN_HAPLO_FILE and CLINGEN_TRIPLO_FILE are haploinsufficiency and triplosensitivity files derived from ClinGen Dosage Sensitivity Map (Rehm et al. 2015). Both of these are in bed format and contain the chr, start, end, gene HUGO name, dosage score. PHENOMATCH_PERCENTILES_FILE is the processed file obtained from Step 8. OUTPUT_FILENAME is the name of the file where the summary will be written.
Running this script will produce an additional section from the features analyzed in Step 1, and analyzes the genes’ inclusion in the initially selected size windows (Step 3), the +/− 1Mb (2Mb) window, the gene inclusion within the breakpoint TAD, whether or not predicted enhancer/promoter contacts were disrupted for the gene, and finally a summary of the genes phenomatch and max_phenomatch scores with their corresponding percentile values within the analyzed dataset (output of Step 4). Values of 0 and 1 indicate inclusion within the analyzed regions (Step 3 Mb windows, 2Mb window, TADs) or presence of disrupted contacts (enhancer/promoter).
For DGAP163:
perl perl/dgap_final_table_maker.pl \ -f HI_list_DGAP.txt \ -c data/hESC_hg37_domains.bed \ -d DHS_promoter_broken_DGAP163.txt \ -h data/ClinGen_haploinsufficiency_gene.bed \ -t data/ClinGen_triplosensitivity_gene.bed \ -m percentiles_6Mb_pheno_maxpheno.txt \ -o DGAP163_table_summary.txt
The output file has columns as described in Table 2. ClinGen haplo/triplo-sensitivity scores are indicated with ranges from 0 (no evidence) to 30 (known). Users need to establish their cut-off criteria for the selection of follow-up candidates. For example, to consider only genes with emerging evidence suggesting dosage sensitivity associated with the clinical phenotype, select ClinGen haplo/triplo-sensitivity scores from 2 and above and give those regions a final table value of 1. This can be done in the ClinGen file by substituting the desired scores with 1 and making everything else a 0, or by adding an extra column to the excel file and making this change with the IF selection formula. The same applies to the phenomatch and max_phenomatch percentiles (i.e., to include only the top quartile, assign a 1 to everything with >=0.75 percentile value).
Table 2.
Column descriptions for the summary file generated in Step 9 of this protocol.
| Column name | Description |
|---|---|
| Breakpoint_ID | Identification number of the studied rearrangement breakpoints |
| chr | Rearrangement breakpoint chromosome location |
| start | Rearrangement breakpoint start position |
| end | Rearrangement breakpoint end position |
| win_start | Analysis window start position |
| win_end | Analysis window end position |
| Hi_domain | TAD coordinates that contain the rearrangement breakpoint |
| HiC_chr | TAD chromosome location |
| HiC_start | TAD start position |
| HiC_end | TAD end position |
| HI_gene_chr | Chromosome location for gene contained within the analysis window |
| HI_gene_start | Position start for gene contained within the analysis window |
| HI_gene_end | Position end for gene contained within the analysis window |
| HI_gene_name | HUGO name for gene contained within the analysis window |
| LOD | Log-odds score derived from Huang et al., 2010 for gene haploinsufficiency |
| HI_prob | Calculated probability of a gene being haploinsufficient |
| HI_ensembl_gene_ID | Ensembl ID of gene contained within the analysis window |
| HI_chr | Ensembl chromosome location of gene contained within the analysis window |
| Gene_start | Ensembl reported start position of gene contained within the analysis window |
| gene_end | Ensembl reported end position of gene contained within the analysis window |
| HGNC_ID | HUGO Gene Nomenclature Committee identifier for gene contained within the analysis window |
| HGNC_symbol | HUGO Gene Nomenclature Committee symbol for gene contained within the analysis window |
| Gene_type | Ensembl reported type of gene contained within the analysis window |
| WikiGene_Description | WikiGene description of gene contained within the analysis window |
| Description | Functional gene description |
| Phenotype_description | Ensembl reported phenotypic associations of gene contained within the analysis window |
| SelectedWindowMb | Location of gene within the selected analysis window. 1 indicates presence, 0 is absence. By default, this column should always be 1 |
| 2Mb | Location of gene within a 2Mb analysis window. 1 indicates presence, 0 is absence |
| TAD | Location of gene within TAD analysis window. 1 indicates presence, 0 is absence |
| DHS_promoter | Presence of disrupted enhancer-promoter interactions. 1 indicates presence, 0 is absence |
| Haploinsufficiency_score | ClinGen reported haploinsufficiency score for the gene |
| Triplosensitivity_score | ClinGen reported triplosensitivity score for the gene |
| Phenomatch_score | Phenomatch score for the gene |
| MaxPhenoScore | Max Phenomatch score for the gene |
| Pheno_percentile | Percentile of phenomatch score for the gene within the analysis window |
| MaxPheno_percentile | Percentile of max phenomatch score for the gene within the analysis window |
For the final candidate selection and ranking, add the 0 and 1 values from different fields. Based on our experience, adding the following columns gives the best candidate ranking: PERC+DHS+TAD+HAPLO+TRIPLO or PERC+DHS+2Mb+HAPLO+TRIPLO. When sorting by these values, the first few rows of results should look similar to the ones in the table in Figure 3. From this table, we conclude that there are two important candidates which may explain part or all of DGAP163’s observed phenotypic characteristics. The first candidate is SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1, MIM#182530), whose potential misregulation could be involved DGAP163’s developmental delay and neurological problems. A quick look at SOS1 in chr2 reveals how the rearrangement disrupts several long-range interactions present in the region, potentially causing misregulation of this gene (Figure 2). The second candidate, cochlin (COCH, MIM#603196) could be related to the hearing loss present in DGAP163. It is important to remark that while these candidates represent an initial step in understanding DGAP163’s clinical features, functional experiments should be performed to unveil the pathogenic mechanisms by which they may contribute to the phenotype, especially since DGAP163 does not present with the full clinical spectrum of Noonan syndrome (MIM#610733).
Figure 3.
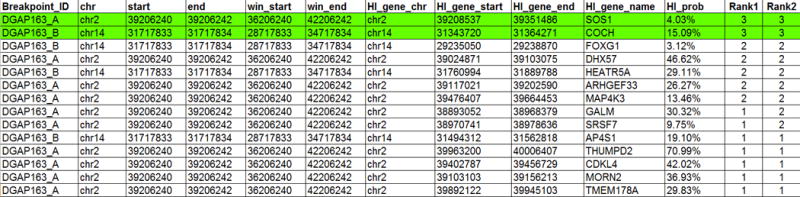
First 14 rows of the results file obtained in Step 9 of this protocol. Notice that several data columns were omitted in order to highlight the top candidate gene names and their ranking. The Rank1 column corresponds to the PERC+DHS+TAD+HAPLO+TRIPLO addition, while Rank2 represents the PERC+DHS+2Mb+HAPLO+TRIPLO sum. The best two candidates are highlighted in green. Notice how the top candidates have equal Rank1 and Rank2 values (3).
Figure 2.
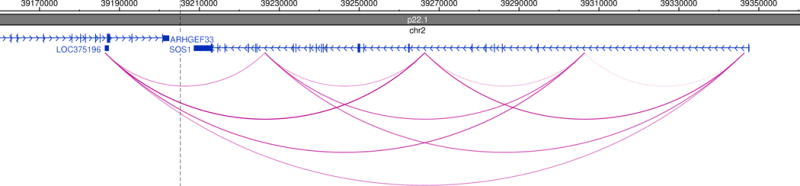
SOS1 region in chromosome 2. Genes present in the region are depicted in blue, including SOS1. The translocation breakpoint is indicated in a dashed black vertical line. Chromatin interactions in H1-hESC cells at 40Kb resolution are depicted in pink arcs for this region. This image was obtained through the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/browser/), an excellent resource for the quick visualization of chromatin interactions and regulatory element datasets (Zhou and Wang 2012).
The dosage sensitivity map can be downloaded from https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/
The full list of files generated throughout the protocol can be downloaded from https://drive.google.com/open?id=1PRkdQHxagzR6HpwnoHDf2de1p3QX0qzw
Commentary
Recent studies in diverse cell lines and tissues have revealed an extensive and intrinsic modular organization of the human genome into topologically associating domains (TADs) (Dekker and Heard 2015; Nora et al. 2012). TADs are defined as DNA segments, usually ~1Mb in size, that have a higher number of chromatin interactions contained within the region compared to neighboring segments. At the functional level, TADs have been associated with the establishment and regulation of cell developmental programs (Beagan et al. 2016; Gorkin et al. 2014; Phillips-Cremins et al. 2013) and genome replication (Thurman et al. 2012). The high degree of correlation between genomic function and chromatin organization makes evident the importance of studying potential clinical pathogenic consequences. Chromosome rearrangements, even when not directly disrupting a gene’s coding sequence, have been shown to generate disease by altering long-range associations between gene promoters and their regulatory elements (Giorgio et al. 2015; Groschel et al. 2014; Ibn-Salem et al. 2014; Lupianez et al. 2015; Roussos et al. 2014; Visser et al. 2012). Therefore, even apparently balanced or balanced non-coding chromosome abnormalities (BCAs) may exert position effects which could be further associated with disease, such as those seen in translocations nearby SOX9 linked to campomelic dysplasia (Velagaleti et al. 2005), and translocations downstream of PAX6, in congenital aniridia (Kleinjan et al. 2001).The computational method described in this protocol provides a streamlined pipeline that assesses the contribution of distal genes in the generation of a clinical phenotype. It is important to highlight that position effect analyses are not typically included in studies of balanced or unbalanced chromosomal abnormalities, thus biasing for negative results in the case of non-coding breakpoints.
With the availability of a computational method to automate such tasks, clinicians and researchers are now able to provide a quick overview of the region beyond the rearrangement location, and thus focus their functional characterizations towards relevant candidate genes.
All types of chromosome rearrangements are amenable to position effect studies (see paper by Ibn-Salem et al. 2014 regarding the study of position effects in copy number variants (CNVs) and Zepeda-Mendoza et al. 2017 for non-coding BCAs); however, careful attention must be paid in the interpretation of chromosome abnormalities that disrupt coding sequences. In such cases, the detection of position effects may complement the diagnosis or explain features not fully accounted for by the disrupted gene sequence(s).
Finally, we would like to mention that the accuracy of the position effect prediction method is directly dependent upon the curation of public databases. Major efforts are being carried out by projects such as ClinGen (Rehm et al. 2015) and DECIPHER (Firth et al. 2009) to advance the annotation of the clinical genome. The results of a position effect prediction analysis may differ between different annotations as additional information becomes available about gene function.
Troubleshooting
It is important to note that the position effect analysis requires the mapping of rearrangement breakpoints, either through next-generation sequencing and/or Sanger sequencing. Positions derived from standard or high resolution cytogenetic karyotypes or estimated regions by fluorescence in situ hybridization (FISH) probes comprise thousands of base pairs containing several regulatory regions and interactions, and are thus not specific or amenable to this analysis.
In addition to having base-pair resolution mapped breakpoints, because the protocol relies on comparing specific rearrangement breakpoint coordinates to annotated genes and regulatory element interactions it is essential to use the same reference genome. For example, hg19 was used throughout this protocol. Other reference genome versions can be used as long as regulatory genomic features are reported in said genome version. Some difficulty may arise when formatting new and updated files downloaded from public databases such as Ensembl (Yates et al. 2016) and UCSC (Kent et al. 2002). To address this problem, it is important to ensure that the downloaded file structure matches the ones described in this protocol. In this regard, Ensembl’s BioMart tool has the option to output desired data annotation columns, while UCSC has extensive documentation regarding file column descriptions. For UCSC files, the described position effect prediction protocol does not require further formatting.
Anticipated Results
The position effect analysis pipeline described in this protocol provides users with a ranked list of genes that may contribute in the phenotype of a subject with balanced or apparently balanced chromosome abnormalities. The number of highest ranking candidate genes depends on the specificity and availability of phenotypic information per subject, as well as the annotation of the neighboring genes in public clinical databases. Users should expect one or more high ranking candidates; however, certain cases do not reach a significance ratio. We recommend not pursuing functional validation of candidate genes with less than two different lines of evidence supporting their inclusion.
Time considerations
Analysis times usually take less than an hour for 20 breakpoints or less, using a computer with 4Gb RAM memory. For a higher number of breakpoints, consider partitioning your files for a faster analysis run.
Acknowledgments
C.C.M. is supported by a grant from the National Institutes of Health (GM061354).
Literature Cited
- An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagan JA, Gilgenast TG, Kim J, Plona Z, Norton HK, Hu G, Hsu SC, Shields EJ, Lyu X, Apostolou E, Hochedlinger K, Corces VG, Dekker J, Phillips-Cremins JE. Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell Stem Cell. 2016;18:611–24. doi: 10.1016/j.stem.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Heard E. Structural and functional diversity of Topologically Associating Domains. FEBS letters. 2015;589:2877–84. doi: 10.1016/j.febslet.2015.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. American journal of human genetics. 2009;84:524–33. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, Gasparini L, Ferrera D, Canale C, Guipponi M, Pennacchio LA, Antonarakis SE, Brussino A, Brusco A. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) Human molecular genetics. 2015;24:3143–54. doi: 10.1093/hmg/ddv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–75. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, van der Velden VHJ, Havermans M, Avellino R, van Lom K, Rombouts EJ, van Duin M, Dohner K, Beverloo HB, Bradner JE, Dohner H, Lowenberg B, Valk PJM, Bindels EMJ, de Laat W, Delwel R. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS genetics. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibn-Salem J, Kohler S, Love MI, Chung HR, Huang N, Hurles ME, Haendel M, Washington NL, Smedley D, Mungall CJ, Lewis SE, Ott CE, Bauer S, Schofield PN, Mundlos S, Spielmann M, Robinson PN. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014;15:423. doi: 10.1186/s13059-014-0423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome research. 2002;12:996–1006. doi: 10.1101/gr.229102. Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001;10:2049–59. doi: 10.1093/hmg/10.19.2049. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V. Long-Range Control of Gene Expression: Emerging Mechanisms and Disruption in Disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GC, Brown DL, Brudno M, Campbell J, FitzPatrick DR, Eppig JT, Jackson AP, Freson K, Girdea M, Helbig I, Hurst JA, Jahn J, Jackson LG, Kelly AM, Ledbetter DH, Mansour S, Martin CL, Moss C, Mumford A, Ouwehand WH, Park SM, Riggs ER, Scott RH, Sisodiya S, Van Vooren S, Wapner RJ, Wilkie AO, Wright CF, Vulto-van Silfhout AT, de Leeuw N, de Vries BB, Washingthon NL, Smith CL, Westerfield M, Schofield P, Ruef BJ, Gkoutos GV, Haendel M, Smedley D, Lewis SE, Robinson PN. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic acids research. 2014;42:D966–74. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nature reviews. Molecular cell biology. 2016;17:771–782. doi: 10.1038/nrm.2016.138. [DOI] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–5. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–95. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS. ClinGen–the Clinical Genome Resource. The New England journal of medicine. 2015;372:2235–42. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, Okada Y, Siminovitch KA, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Plenge RM, Raychaudhuri S, Fromer M, Purcell SM, Brennand KJ, Robakis NK, Schadt EE, Akbarian S, Sklar P. A role for noncoding variation in schizophrenia. Cell reports. 2014;9:1417–29. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velagaleti GV, Bien-Willner GA, Northup JK, Lockhart LH, Hawkins JC, Jalal SM, Withers M, Lupski JR, Stankiewicz P. Position effects due to chromosome breakpoints that map approximately 900 Kb upstream and approximately 1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am J Hum Genet. 2005;76:652–62. doi: 10.1086/429252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Kayser M, Palstra RJ. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome research. 2012;22:446–55. doi: 10.1101/gr.128652.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nature biotechnology. 2012;30:1095–106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Keenan S, Lavidas I, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Nuhn M, Parker A, Patricio M, Pignatelli M, Rahtz M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Birney E, Harrow J, Muffato M, Perry E, Ruffier M, Spudich G, Trevanion SJ, Cunningham F, Aken BL, Zerbino DR, Flicek P. Ensembl 2016. Nucleic acids research. 2016;44:D710–6. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda-Mendoza CJ, Ibn-Salem J, Kammin T, Harris DJ, Rita D, Gripp KW, MacKenzie JJ, Gropman A, Graham B, Shaheen R, Alkuraya FS, Brasington CK, Spence EJ, Masser-Frye D, Bird LM, Spiegel E, Sparkes RL, Ordulu Z, Talkowski ME, Andrade-Navarro MA, Robinson PN, Morton CC. Computational Prediction of Position Effects of Apparently Balanced Human Chromosomal Rearrangements. Am J Hum Genet. 2017;101:206–217. doi: 10.1016/j.ajhg.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24:R102–10. doi: 10.1093/hmg/ddv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang T. Using the Wash U Epigenome Browser to examine genome-wide sequencing data. Current protocols in bioinformatics. 2012 doi: 10.1002/0471250953.bi1010s40. Chapter 10: Unit10 10. [DOI] [PMC free article] [PubMed] [Google Scholar]