

SUMMARY
A human variant in the BDNF gene (Val66Met; rs6265) is associated with impaired fear extinction. Using super-resolution imaging, we demonstrate that the BDNF Met prodomain disassembles dendritic spines and eliminates synapses in hippocampal neurons. In vivo, ventral CA1 (vCA1) hippocampal neurons undergo similar morphological changes dependent on their transient co-expression of a SorCS2/p75NTR receptor complex during peri-adolescence. BDNF Met prodomain infusion into the vCA1 during this developmental timeframe reduces dendritic spine density and prelimbic (PL) projections, impairing cued fear extinction. Adolescent BDNFMet/Met mice display similar spine and PL innervation deficits. Using fiber photometry, we found that in wild-type mice, vCA1 neurons projecting to the PL encode extinction by enhancing neural activity in threat anticipation and rapidly subsiding their response. This adaptation is absent in BDNFMet/Met mice. We conclude that the BDNF Met prodomain renders vCA1-PL projection neurons underdeveloped, preventing their capacity for subsequent circuit modulation necessary for fear extinction.
Keywords: BDNF, polymorphism, prodomain, synapse elimination, fear extinction, fiber photometry, ventral CA1, peri-adolescence, p75NTR, SorCS2
INTRODUCTION
Human carriers of a common human single nucleotide polymorphism (SNP) in the BDNF gene (Val66Met; rs6265) have impaired fear extinction learning (Soliman et al., 2010), decreased response to fear extinction-based therapies, and enhanced risk for fear related disorders such as anxiety or post-traumatic stress disorder (PTSD) (Felmingham et al., 2013; Zhang et al., 2014). The BDNF Val66Met mutation lies within the prodomain region that interacts with the Vps10 domain protein, sortilin, which has been shown to direct intracellular trafficking of proBDNF to the regulated secretory pathway (Chen et al., 2005). The variant BDNF exhibits decreased binding to sortilin, altered intracellular trafficking, and reduced activity-dependent secretion of mature BDNF (mBDNF) (Chen et al., 2005; Chen et al., 2006; Chen et al., 2004). Thus, the proposed mechanism by which the BDNF Met allele mediates fear extinction dysfunction has been conceptualized as a loss-of-function mutation due to decreased mBDNF bioavailability leading to reduced BDNF-TrkB-dependent signaling in fear-related circuitry. During the transition from childhood to adolescence, BDNF levels increase dramatically in the frontolimbic circuitry (Casey et al., 2015; Katoh-Semba et al., 1998; Kolbeck et al., 1999; Yang et al., 2009), which continues to develop during this time. Functional neuroimaging studies in human BDNF Met carriers indicate that there is altered fronto-amygdala activity reminiscent of similar findings in patients with anxiety disorders and depression when presented with an empty threat or aversive stimuli (e.g., fearful faces) (Liberzon et al., 1999; Rauch et al., 2000). However, the precise role of how BDNF may be involved in regulating the maturation of these circuits has not been determined.
Recently, we have shown that the cleaved BDNF prodomain can also be secreted in an activity-dependent manner from neurons (Anastasia et al., 2013). In addition, the presence of the Met amino acid induces structural changes within the prodomain involving a shift in transient secondary structure from β-sheet to a helical conformation in the residues surrounding the substitution. This shift confers bioactivity to the Met prodomain through differential interaction with a sortilin-related Vps10 domain sorting receptor 2 (SorCS2), leading to neuronal growth cone retraction in the presence of the p75 neurotrophin receptor (p75NTR) (Anastasia et al., 2013). However, this previous work only tested the effects of this newly discovered ligand on DIV3 neurons in vitro when the prodomain is barely detectable. In contrast, we sought to examine the effects of the Met prodomain at later developmental stages when its physiological levels peak in peri-adolescence (Anastasia et al., 2013). We hypothesized that key phenotypes that have been previously observed in human BDNF Met allele carriers as well as in BDNF Met knock-in mice may be due to the independent gain of function actions of the BDNF Met prodomain as opposed to reduced mBDNF bioavailability.
Here, we show that the BDNF Met prodomain serves as a potent ligand that triggers disassembly of the mature mushroom spines on hippocampal neurons and eliminates synapses by mobilizing multiple actin regulators through the SorCS2/p75NTR receptor complex. We identified a neuronal population in the ventral hippocampus projecting to the PL that is selectively affected by the BDNF Met prodomain due to their spatiotemporal expression of these receptors, which coincides with the peak in prodomain levels. We found that a single infusion of the BDNF Met prodomain into these neurons in peri-adolescent wild-type (WT) mice diminishes density of the projections to the PL and disrupts cued fear extinction. To bridge the defects found in spines of the ventral hippocampal neurons and the behavioral deficits, we used fiber photometry to observe in vivo circuit dynamics during the fear extinction learning. We demonstrate the anticipatory firing of the ventral CA1 (vCA1) neuronal projections recorded within the PL and their rapid return to baseline in the extinguished WT (BDNFVal/Val) mice. In contrast, the vCA1 neurons in knock-in mice with the BDNF SNP (BDNFMet/Met) fail to display this adaptation and exhibit calcium traces identical to those from the same animals during early extinction trials. These findings suggest that the reduced fear extinction learning in mice and humans with this BDNF SNP may stem from an underdevelopment of fear extinction circuitry in the presence of the BDNF Met prodomain.
RESULTS
BDNF Met prodomain disassembles spines and eliminates synapses
To test whether the BDNF prodomain affects mature neurons, we added recombinant Val or Met prodomain (10 ng/ml) to day in vitro (DIV)21 cultured hippocampal cells and analyzed their morphology. By 40 minutes of the Met prodomain treatment, there was a progressive transitioning from the mature mushroom spines into thin/filopodia-like protrusions, resulting in nearly 60% decrease in mushroom spines (Figures 1A-B). This effect was mirrored by an increase in thin spines with no changes in total spine density within one hour (Figures 1A, 1B, and S2A). Our analysis was conducted on cells stained with actin detecting phalloidin-Alexa 546 (Figure S1A) to examine non-manipulated cells imaged with high resolution 3D structured illumination microscopy (3D-SIM) (Nikon) (Gustafsson, 2005). This allowed us to significantly improve the visualization of these thin and/or filopodia-like spines by over 50% compared to the images of the same dendritic segments acquired with widefield microscopy (Figures S1B-C). Longer exposure to the BDNF Met prodomain (3 hours) led to nearly 30% reduction of total spines (Figures 1A, 1B, and S2B). In contrast to the neurons treated with the Met prodomain, there were no spine morphological or density changes in neurons treated with the Val prodomain for 1 or 3 hours (Figures 1A, 1B, and S2). To be certain that the Val prodomain has no effect on spines, neurons were exposed to higher concentrations of Val (50 ng/ml) for 1 hour, resulting in no morphological changes (Figure S2C). Since the mushroom spine structure is stabilized by the branched F-actin present in the spine head, we compared the F-actin levels in spines/filopodia following 1 hour Met prodomain treatment to a known actin depolymerizing agent Latrunculin A (Lat-A). We found that the 1 hour Met prodomain exposure resulted in over 50% decrease in F-actin fluorescence intensity normalized to underlying actin in the dendritic segment, an effect as potent as with a 3 hour 2.5 μM Lat-A exposure (Figures S1D-E).
Figure 1. BDNF Met prodomain eliminates synaptic connections in mature hippocampal neurons.
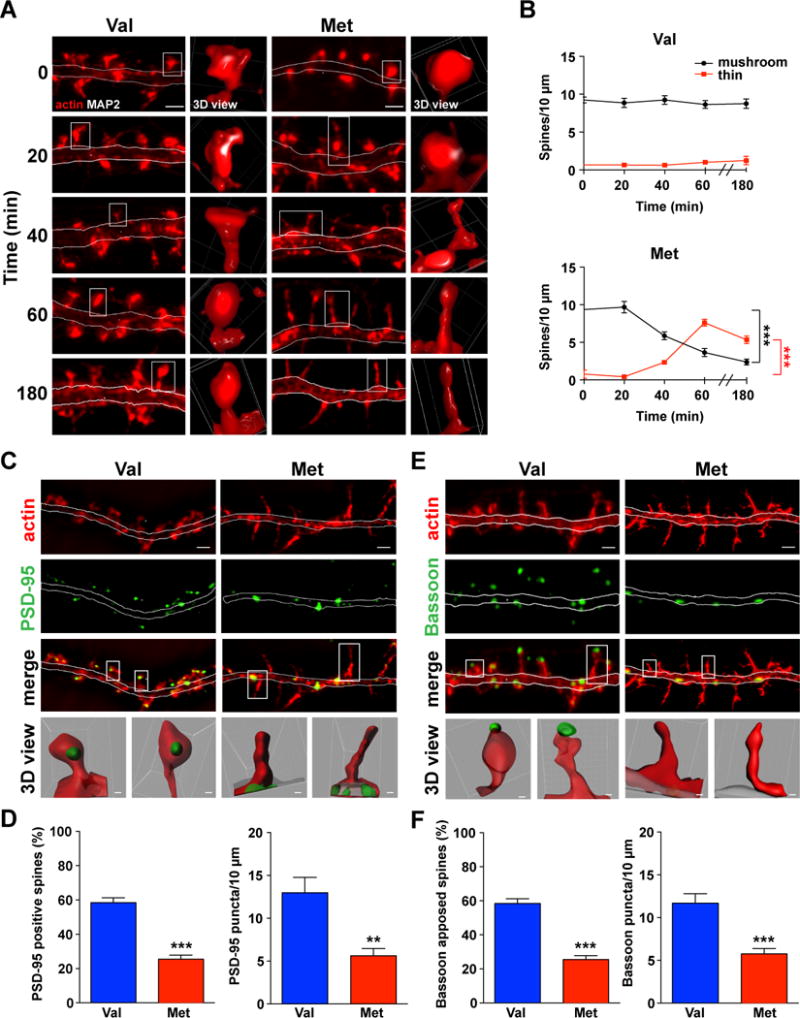
(A) 3D-SIM reconstructed Z-stack maximum intensity projection (MIP) images of actin-phalloidin-546 labeled neurons, treated with Val (left) or Met prodomain (right). Representative spines (white boxes) reconstructed in 3D (Nikon Elements). Scale bar 1 μm. See also Figure S1.
(B) Met prodomain begins to induce spine disassembly at 40 minutes (bottom line graph, black), when number of mushroom spines is reduced with a concomitant increase in thin spines (bottom line graph, red). At 3 hours of Met prodomain exposure, thin spines begin to be eliminated (n = 5 Met 0 min, n = 5 Met 20 min, n = 5 Met 40 min, n = 12 Met 60 min, n = 14 Met 180 min, n = 9 Val 0 min, n = 15 Val 20 min, n = 13 Val 40 min, n = 15 Val 60 min, n = 11 Val 180 min; one-way ANOVA with Bonferroni’s multiple comparisons test, ***p < 0.001; data are represented as mean ± SEM). See also Figure S2.
(C) PSD-95 in neurons exposed to Val or Met prodomain for 1 hour. Scale bar 1 μm. Representative spines (white box) were reconstructed in 3D using Imaris transparency feature to show PSD-95 inside the spine heads. Scale bars for Val-treated spines 0.1 μm, Met-treated spines 0.3 μm.
(D) There was a significant decrease in percentage of PSD-95 positive spines and in PSD-95 puncta density in Met prodomain treated neurons (n = 10 Val, n = 10 Met for PSD-95 positive spines; n = 7 Val, n = 7 Met for PSD-95 puncta density; two-tailed, unpaired Student t test, ***p < 0.001, **p < 0.01; data are represented as mean ± SEM).
(E) Bassoon in neurons exposed to Val and Met prodomain for 1 hour. Scale bar 1 μm. Representative spines (white box) reconstructed in Imaris showing presynaptic Bassoon apposed to the spines. Scale bars for Val-treated spines 0.1 μm, Met-treated spines 0.3 μm.
(F) There was a significant decrease in the percentage of spines apposed to the Bassoon and overall Bassoon density with 1 hour Met prodomain treatment (n = 7 Val, n = 12 Met for Bassoon positive spines; n = 8 Val, n = 9 Met for Bassoon puncta density; two-tailed, unpaired Student t test, ***p < 0.001; data are represented as mean ± SEM).
Given that the physical changes to the mushroom spine structure may disrupt synapse integrity (Lin and Koleske, 2010), we evaluated the presence of the pre- and postsynaptic markers following Met-induced morphological changes. Within 1 hour of the Met prodomain application, the percentage of spines containing postsynaptic density 95 (PSD-95) in the spine head and the overall PSD-95 density were both reduced by nearly 50% (Figures 1C-D) as opposed to cells exposed to the Val prodomain. The relocation of PSD-95 from spines to the dendritic shaft was concomitant with a significant decline in the number of the apposed presynaptic sites (~50%), labeled with anti-Bassoon antibody (Figures 1E-F). These results indicate that the BDNF Met prodomain disassembles spines and eliminates synapses.
SorCS2 and p75NTR are required for BDNF Met prodomain-induced synapse elimination
To define the mechanism by which the BDNF Met prodomain induces mature spine disassembly and synapse elimination, we examined the SorCS2/p75NTR receptor complex, previously shown to mediate growth cone collapse in immature neurons induced by proneurotrophins, including proBDNF, proNGF, as well as the BDNF Met prodomain (Anastasia et al., 2013; Deinhardt et al., 2011; Glerup et al., 2014; Sun et al., 2012). We found that in our hippocampal cultures, nearly 80% of the spines contained SorCS2 and p75NTR (Figures 2A-C). The specificity of the rabbit anti-p75NTR antibody was confirmed in p75NTR knockout (p75−/−) neurons (Figure S3A). In order to determine whether both receptors are necessary for the spine collapse effect, we prepared hippocampal cultures from p75NTR knockout (Lee et al., 1992) and SorCS2 knockout (SorCS2−/−) mice. We found that under basal conditions, total spine densities in SorCS2−/− (Figures 2D-E) and p75−/− neurons (Figure 2F-G) were elevated by nearly 20%, consistent with previously published hippocampal spine density analyses of p75−/− mice (Zagrebelsky et al., 2005). The addition of the Met prodomain had no effect on the number of mushroom or thin spines in neurons missing either one of the receptors (Figures 2D-G), indicating that p75NTR and SorCS2 are both necessary and required for the BDNF Met prodomain mediated processes.
Figure 2. BDNF Met prodomain induces acute spine changes by interacting with SorCS2 and p75NTR receptors.

(A-B) Both p75NTR and SorCS2 receptors are located inside the spine head in WT neurons. Scale bar 1 μm. Representative spines (white boxes) reconstructed in 3D using Imaris to visualize SorCS2 and p75NTR inside the spine. Scale bar 0.1 μm.
(C) About 80% of mature hippocampal spines contain SorCS2 and p75NTR. See also Figure S3.
(D-E) There was an increase in total and mushroom spine density in SorCS2−/− neurons (top, middle bar graphs) compared to WT neurons. There were no significant differences in thin spines (bottom bar graph). Met prodomain treatment for 1 hour did not induce any spine changes. Scale bar 1 μm (n = 10 control, n = 10 control’, n = 9 Val, n = 9 Met; one-way ANOVA with Bonferroni’s multiple comparisons test, **p < 0.01; data are represented as mean ± SEM).
(F-G) There was an increase in total and mushroom spine density in p75−/−neurons (top, middle bar graphs) compared to WT neurons. There were no differences in thin spines (bottom bar graph). 1 hour Met prodomain treatment did not induce any spine changes. Scale bar 1 μm (n = 7 control, n = 9 control’, n = 10 Val, n = 12 Met; one-way ANOVA with Bonferroni’s multiple comparisons test, **p < 0.01; data are represented as mean ± SEM).
BDNF Met prodomain induces acute spine remodeling by signaling to actin regulators
Since the spine shape is regulated by the organization of the actin cytoskeleton with the enrichment of the spine head with filamentous actin and Met prodomain reduces the levels of F-actin in a manner comparable to actin depolymerizing agent Lat-A (Figure S1D-E), we hypothesized that the Met prodomain signals through the SorCS2/p75NTR complex to actin modifying proteins. Previously, proNGF was shown to utilize this complex to signal through the guanine exchange factor (GEF) Trio to regulate growth cone retraction in immature cells (Anastasia et al., 2013; Deinhardt et al., 2011). Therefore, we examined the possible link between the BDNF Met prodomain and Trio in mature hippocampal spines after 1 hour when all the spines are still present (Figure S2A). Because the spines of neurons treated with the Val prodomain were indistinguishable from the untreated cells even at higher concentrations of Val (Figure S2C), we compared the effects of the BDNF Met to the Val prodomain on the spine localization of Trio. Met exposure led to significantly fewer Trio positive spines (Figures 3A-B). To determine whether this localized loss of Trio in dendritic spines affected Rac1 activity, we used an immunolabeling assay to detect active GTP-bound Rac1 using PAK1-GST probe (Harrington et al., 2011). To validate our assay, we added mBDNF as a positive control, as it is known to induce Rac1 activation (Miyamoto et al., 2006). As expected, mBDNF treatment led to a robust increase in the Rac1-GTP puncta density compared to the untreated control and Val-treated cells (Figures 3C-D). Conversely, the Met prodomain reduced Rac1-GTP puncta by nearly half of the basal levels (Figures 3C-D). Since downstream effectors of Rac1 are involved in spine maintenance by regulating actin stabilization (Nakayama et al., 2000), we examined the actin severing protein cofilin. Decreases in phospho-cofilin are linked with destabilization of mushroom spines and their transformation into thin ones (Pontrello et al., 2012; Shi et al., 2009; Zhou et al., 2004). Within 1 hour of the Met prodomain treatment, there was a significant reduction in phospho-cofilin puncta density (Figures 3E-F), suggesting that the effect of the Met prodomain on mature spines may be achieved through cofilin-mediated actin depolymerization. Another known parallel mechanism of actin destabilization involves actin bundling protein fascin. In growth cones treated with proNGF, fascin is phosphorylated by PKC and disassociates from actin, thus destabilizing actin filaments (Deinhardt et al., 2011). In mature spines, phospho-fascin levels were significantly increased following 1 hour of the Met prodomain application compared to the Val prodomain (Figures 3H-I). These findings suggest two parallel actin-regulating pathways triggered by the BDNF Met prodomain to promote the disassembly of spine heads: cofilin-mediated actin filament severing and phospho-fascin-mediated prevention of actin re-bundling.
Figure 3. BDNF Met prodomain remodels spines through actin regulators.

(A-B) There are fewer Trio positive spines in neurons treated for 1 hour with Met prodomain. Scale bar 1 μm (n = 8 Val, n = 14 Met; two-tailed, unpaired Student t test, ***p < 0.001; data are represented as mean ± SEM).
(C-D) Active Rac1-GTP levels in neurons treated with Met, but not Val, prodomain are significantly reduced. Mature BDNF condition was added as a positive control for Rac1 activation, showing an expected increase in Rac1-GTP puncta. Scale bar 1μm (n = 9 control, n = 6 Val, n = 6 Met, n = 6 BDNF; one-way ANOVA with Bonferroni’s multiple comparisons test, **p < 0.01, ***p < 0.001; data are represented as mean ± SEM).
(E-F) Neurons treated with Met prodomain for 1 hour showed marked reduction in the levels of inactive (phosphorylated) actin severing molecule cofilin. Scale bar 1μm (n = 7 Val, n = 8 Met; two-tailed, unpaired Student t test, **p < 0.01).
(G-H) Fascin (G) and p-fascin (H) in neurons treated with Val or Met prodomain for 1 hour. Scale bar 1 μm.
(I) With 1 hour Met prodomain treatment, there was significantly more (phosphorylated) form of actin p-fascin, preventing actin rebundling (n = 9 Val, n = 9 Met; two-tailed, unpaired Student t test, **p < 0.01; data are represented as mean ± SEM).
SorCS2 and p75NTR receptors are selectively and transiently co-expressed in vCA1 hippocampal neurons during peri-adolescence
Having identified the molecular mechanism by which the BDNF Met prodomain disassembles dendritic spines, we expanded our study to the brain circuitry affected by the Met prodomain. Given that the prodomain is highly expressed in mice during peri-adolescence at P30 (Anastasia et al., 2013), we tested the expression of SorCS2 and p75NTR before, during, and after this time period. Given that humans with the Met allele and BDNFMet/Met mice have impaired fear extinction learning (Felmingham et al., 2013; Pattwell et al., 2012; Soliman et al., 2010), we focused on the fear extinction circuitry. Recent evidence points to a critical role of the vCA1 neurons that project selectively to the PL, gating the fear expression incoming from the amygdala during extinction by activating inhibitory interneurons in the PL (Sotres-Bayon et al., 2012). Due to discrepancies in the previous reports of the p75NTR expression, we utilized p75NTR-EGFP BAC reporter mice from the GENSAT Project (Schmidt et al., 2013). In these mice, the EGFP gene is under the p75NTR promoter, revealing neurons that possess the ability to express p75NTR. We immunolabeled p75NTR and SorCS2 in the hippocampus of pre-adolescent P23, peri-adolescent P30, and adult P60 mice. We found that the neurons that express both receptors are present in vCA1 at P23 (Figure 4A) and their levels rise at P30 (Figure 4B). However, by adulthood at P60, p75NTR is no longer present (Figure 4C). Figure S4 shows lower magnification images and the schematic pointing to vCA1 region represented in Figure 4. On the contrary, other brain regions related to fear extinction circuitry show the lack of p75NTR and SorCS2 co-expression. In the dorsal hippocampus across development, there are only incoming p75NTR positive fibers from the basal forebrain (Figure S3B). Additionally, there is no p75NTR present in the BLA and conversely no SorCS2 in the PL (Figure S3C-D). These results point to a unique population of vCA1 neurons in fear extinction circuitry that is predisposed to the effects of the Met prodomain through its temporal co-expression of the two necessary prodomain receptors.
Figure 4. SorCS2 and p75NTR are transiently and selectively co-expressed in vCA1 hippocampal neurons during peri-adolescence.

(A-C) Representative sections of p75-EGFP vCA1 of the hippocampus (Bregma −3.38 mm) with EGFP demarcating neurons with ability to express p75NTR at pre-adolescence P23 (A), peri-adolescence P30 (B), and adulthood P60 (C). These sections were stained with anti-p75NTR and anti-SorCS2 antibodies. Both p75NTR and SorCS2 levels peak at P30, with p75NTR expression diminishing by P60. Scale bars 40 μm. See also Figure S3 and Figure S4.
BDNF Met prodomain delivery into the vCA1 remodels hippocampal dendritic spines in vivo, changes projections to the PL, and alters fear extinction
Having identified a hippocampal subregion and developmental time point at which the SorCS2 and p75NTR receptors are co-expressed, we wanted to find out whether the delivery of exogenous Met prodomain at this particular time and location in vivo will modify the vCA1 neurons and consequently their projections to the PL. For this purpose, we injected a single dose of the Met or Val prodomain (400 ng/μl) into the vCA1 of P30 and P60 mice. Using Golgi staining, we examined the changes in dendritic spine morphology after 3 hours, which allowed for tissue recovery from the injection. In P30 mice, there were approximately 20% fewer spines after the Met prodomain injection, in contrast to the Val prodomain (Figure 5A-B). As predicted, there were no spine density changes between Met and Val prodomain injections in the P60 mice (Figure 5A-B). Considering that the exogenous Met prodomain was introduced into WT mice that normally produce the Val prodomain, we analyzed the effects of the Met prodomain in the presence of the Val prodomain in vitro. We found that the effect of the combined prodomains at 1:1 ratio on the neuronal spine morphology is intermediate compared to the Met and Val alone (Figure S5A B), suggesting that the smaller spine density reduction observed in vivo could be due to the combined effect of the two prodomains. To determine whether the acute reduction in spine density with exogenous BDNF Met prodomain in the vCA1 affected the fear extinction circuitry connectivity, we injected the Met or Val prodomain (400 ng/μl) into the vCA1 along with an anterograde tracer Phaseolus vulgaris leucoagglutinin (PHA-L), to label axonal projections from the vCA1 to the PL (Figure 5C). PHA-L positive fiber density analyses were conducted 20 days after the P25 injection at P45. In mice injected with the Met prodomain, there was a significant 25% decrease in fiber density of the vCA1 neurons projecting to the PL area at P45 compared to Val or saline-injected animals (Figures 5D-E). This effect was specific to the PL, as there was no difference in PHA-L positive fibers in the IL among all groups (Figure 5D-E). Given deficits in the spines and projections of the vCA1 neurons following single injection of the Met prodomain, we wanted to assess whether this discrete impairment could alter fear extinction learning. We used a well-established fear extinction paradigm, in which extinction consists of presenting the cue alone after the initial fear conditioning (day 1, Figure S6A). Comparing early extinction (days 2-3, Figure S6A) with late extinction trials (days 4-5, Figure S6A) (Pattwell et al., 2012a; Pattwell et al., 2012b) allows for differentiation between heightened anxiety and actual learning. We bilaterally injected the Met or Val prodomain (400 ng/μl) into the vCA1 of WT mice at P25 and assessed cued fear extinction behavior at P45. The Met prodomain-injected mice demonstrated significantly higher levels of freezing in late cued fear extinction trials, compared to the Val prodomain-injected mice (Figure 6A). There was no difference in fear extinction between Val prodomain-injected and saline-injected mice (Figure S6B). These results suggest that the Met prodomain infusion into the vCA1 does not result in a generalized dysregulation of the hippocampal-dependent cognitive processes; instead, it leads to a specific alteration in circuitry regulating cued fear extinction learning. In order to show spatiotemporal specificity of the Met prodomain’s effect on fear extinction circuitry, we performed this experiment in adult P60 WT mice, when p75NTR is no longer expressed, testing their extinction learning at P80. As predicted, we saw no differences in fear extinction behavior between Val and Met prodomain-injected adult mice (Figure 6B). When the BDNF Met and Val prodomains were injected into the PL at P25, there was also no difference in fear extinction (Figure 6C). Lastly, there was no difference in contextual fear acquisition or retrieval between Val and Met-injected mice during peri-adolescence (Figure 6D), indicating that the fear learning mechanism is unaffected by the BDNF Met prodomain and its effects are specific to cued fear extinction.
Figure 5. BDNF Met prodomain injection into vCA1 during peri-adolescence reduces total spine density and vCA1 projections to the PL.

(A) Golgi-stained neurons 3 hours after in vivo injection of Val or Met prodomain (400 ng/l) into the vCA1 at P30 and at P60. Scale bar 5 m.
(B) There was a significant reduction in total spine density after 3 hours of Met prodomain injected in vivo into the vCA1 at P30 (n = 20 Val at each age, n = 20 Met at each age; two-tailed, unpaired Student t test for each age, ***p < 0.001; data are represented as mean ± SEM). See also Figure S5.
(C) Schematic diagram showing Phaseolus vulgaris leucoagglutinin (PHA-L) anterograde tracer co-injected with either Val or Met prodomain (saline for control) into the vCA1 (Bregma −3.38 mm), labeling fibers in the PL and IL of the neurons simultaneously exposed to the prodomain.
(D) Representative images of the vPFC (left column, Bregma 1.545 mm) after saline, Val, or Met prodomain injections into vCA1. Scale bar 200 μm. Detailed view of the PHA-L positive fibers in the PL (middle column) and IL (right column). Scale bars 20 μm.
(E) There was reduced PHA-L fiber density in the PL with Met prodomain injections into vCA1, suggesting fewer connections projecting from vCA1 to PL as opposed to the IL, where no differences were found. (n = 4 saline, n = 4 Val, n = 4 Met; one-way ANOVA with Bonferroni’s multiple comparisons test, ***p < 0.001; data are represented as mean ± SEM)
Figure 6. Injecting BDNF Met prodomain into vCA1 at P25 reduces cued fear extinction.

(A) Mice with Met prodomain injections into vCA1 (Bregma −3.0 mm) during adolescence at P25 showed significantly higher levels of freezing in the late fear extinction trials when tested as adults at P45 (n = 8 early Val, n = 8 early Met, n = 6 late Val, n = 6 late Met; two-way ANOVA with Bonferroni’s multiple comparisons test, *p < 0.05; data are represented as mean ± SEM). See also Figure S6.
(B) There was no difference in freezing behavior during late cued fear extinction tests between mice that received Met or Val prodomain injections into the vCA1 during adulthood at P60 (n = 7 early Val, n = 8 early Met, n = 7 late Val, n = 8 late Met; two-way ANOVA, p > 0.05; data are represented as mean ± SEM)
(C) There was no difference in freezing behavior in mice that received Met or Val prodomain injections into the PL of vPFC (Bregma 1.7 mm) at P25 (n = 6 early Val, n = 7 early Met, n = 6 late Val, n = 6 late Met; two-way ANOVA, p > 0.05; data are represented as mean ± SEM)
(D) There was no difference in contextual fear retrieval between Val and Met prodomain injected mice at P25 and P60 (n = 6 Val P30, n = 6 Met P30, n = 8 Val P30, n = 8 Met P60; two-way ANOVA, p > 0.05; data are represented as mean ± SEM).
Fear extinction circuitry is altered in BDNFMet/Met mice
Recently, it has been shown that in developing WT mice, there is a significant surge in connectivity between vCA1 neurons projecting to the PL that peaks at P30 and decreases by P45 (Pattwell et al., 2016). To investigate the connectivity between the vCA1 and PL in mice carrying the BDNF Met allele, we injected a retrograde tracer, Fluoro-Gold (FG), into the PL of P23, P30, and P60 WT and BDNFMet/Met mice and assessed the cell body staining in the vCA1 projection neurons (Figure 7A). Similarly to the previously described findings, we found increased number of FG-labeled vCA1 neurons that projected to the PL between P23 and P30 in WT animals (Figures 7B-D). In contrast, this surge did not occur in BDNFMet/Met mice (Figures 7B-D) and the diminished connectivity persisted into adulthood (Figures 7B-D). To distinguish between the novel gain of function of the Met prodomain and the effect of the reduced BDNF levels, we also examined this developmental surge in heterozygote BDNF (BDNF+/−) mice with half the normal levels of mBDNF. We found that the connectivity surge across development in BDNF+/− mice was comparable to that of the WT animals (Figures 7B-D), indicating that reduced BDNF levels do not affect the development of this circuitry. In addition, BDNF+/− mice also do not demonstrate any differences in the fear extinction behavior (Figure S7A). Since the BDNFMet/Met showed reduction in projections from the vCA1 to the PL, we also examined whether dendritic spines of BDNFMet/Met neurons are affected. We prepared cultures from BDNFMet/Met, BDNFVal/Met and BDNFVal/Val mice and analyzed their spines at DIV21. The mature DIV21 BDNFMet/Met neurons had reduced spine head diameter and spine density while the BDNFVal/Met neurons demonstrated an intermediate phenotype between BDNFMet/Met and BDNFVal/Val neurons (Figure S7B-D).
Figure 7. Fear extinction circuitry is altered in the mice carrying the BDNF Met allele.

(A) Schematic diagram showing Fluoro-Gold (FG) retrograde tracer injected into the PL, labeling cell bodies of vCA1 neurons that projected to the PL.
(B) Representative images of Fluoro-Gold labeled cells in vCA1 (Bregma −3.28 mm) at ages P23, P30, and P60 in BDNFVal/Val, BDNF+/−, and BDNFMet/Met mice. Scale bar 50 μm.
(C) There was a significant increase in connectivity between vCA1 and vPFC from ages P23 to P30 in WT BDNFVal/Val and BDNF+/− mice, but not in BDNFMet/Met mice. The connectivity between vCA1 and vPFC decreased significantly in BDNFMet/Met mice at P60 (n = 5 for each condition, one-way ANOVA with Bonferroni’s multiple comparisons test, ***p < 0.001, *p < 0.05; data are represented as mean ± SEM). See also Figure S7.
(D) Although there was no difference in connectivity from vCA1 to vPFC at P23 across genotypes, there were significantly fewer connections between vCA and vPFC in BDNFMet/Met mice at P30 compared to BDNFVal/Val and BDNF+/− mice. The lack of the surge of connectivity was sustained through P60 (n = 5 for each condition; one-way ANOVA with Bonferroni’s multiple comparisons test, ***p < 0.001, *p < 0.05; data are represented as mean ± SEM).
Fiber photometry reveals adaptation deficits of the vCA1 projections to the PL of BDNFMet/Met mice with repeated extinction trials
In order to bridge the BDNF Met prodomain-induced dendritic spine changes and the behavioral fear extinction deficits, we utilized a live imaging technique, fiber photometry, which allows for a measurement of the real-time activity of neuronal populations within distinct circuit elements during specific behaviors. Importantly, through our use of fiber photometry recordings, we were able to establish the activity of vCA1 neuron terminals in PL with great temporal precision, i.e., time-locked to the onset of the fear cue. We injected an adeno-associated virus (AAV-Syn) carrying GCaMP6s unilaterally into vCA1 at P45 (Figure 8A). The GCaMP6s virus encodes a modified calmodulin and GFP fusion construct that serves as a proxy for neural activity (Akerboom et al., 2012). We obtained comparable numbers of neurons infected by the virus in vCA1 determined by the number of GFP labeled cell bodies (Figure 8B, Figure 8C left bar graph, and Figure S8A). In addition, an optical fiber implant was placed in the PL (Figures 8A-B and Figure S8B). Analysis of the distal fibers projecting from the vCA1 into the PL revealed a reduced number of fibers despite comparable numbers of cell bodies within vCA1 (Figure 8C). There was no detectable difference in the IL projecting fibers (Figure S8C-D). These results are consistent with our findings demonstrating reduced fiber numbers following the injection of the BDNF Met prodomain into the vCA1 and analysis of the projections labeled with anterograde tracer in the PL and IL (Figure 5D-E). Three weeks following surgery, the mice underwent cued fear conditioning and extinction procedures, and the GCaMP6s signal in PL-projecting vCA1 neurons was measured during extinction sessions by recording directly from distal fibers through the optical fiber implanted within the PL. There were no genotype differences in activity during early trials (trials 1-5); that is, all mice exhibited similarly high levels of freezing during early extinction trials, accompanied by an increase in neural activity immediately following onset of the fear cue (Figure 8D and Figures S8E-F). Consistent with our previously published results, the BDNFMet/Met mice demonstrated significantly higher levels of freezing than BDNFVal/Val mice in the late cued fear extinction trials (trials 16-20; Figure S8E). These differences were paralleled by two genotypic differences in the activity that emerged during late trials. First, there was an increase in neural activity in PL-projecting vCA1 terminals in the BDNFVal/Val, but not in BDNFMet/Met mice, during the period preceding the tone (Figure 8E and Figure S8G). These responses may indicate anticipatory adaptation emerging during late extinction trials that facilitates the priming of an appropriate behavioral response (i.e., less freezing). Secondly, tone-locked activity of the PL-projecting vCA1 terminals returned rapidly to baseline within ~2s of tone onset in the BDNFVal/Val mice (Figure 8E and Figure S8G). Again, this was not observed in BDNFMet/Met mice, which showed a sustained increase in tone-locked activity similar to activity patterns in the early trials, when fear has not been extinguished (Figure 8D and Figure S8F). Thus, during late cued fear extinction, the change in ΔF/F signal over time during the pre-tone period (−5.0 to 0.0 seconds) showed a significant increase in BDNFVal/Val mice relative to BDNFMet/Met mice (Figure 8G), while the change in ΔF/F signal over time during the initial five seconds of the tone (0.0 to 5.0 seconds) showed a significant decrease in BDNFVal/Val mice relative to BDNFMet/Met mice (Figure 8G). This genotypic difference is not observed during early trials (Figure 8F). This suggests that the response in BDNFMet/Met mice was not modulated despite repeated extinction trials. The rapid return to baseline observed in BDNFVal/Val neurons may reflect a properly functioning circuit and attenuated response to the fearful stimulus. Because the vCA1 has been shown to regulate fear expression through innervation of the PL (Jin and Maren, 2015; Sotres-Bayon et al., 2004; Wang et al., 2016), these data suggest that the lack of response preceding the tone and sustained activity of this projection in response to a fear cue may underlie the fear extinction deficits in BDNFMet/Met mice.
Figure 8. Fiber photometry shows adaptation of vCA1 projections to the PL during late fear extinction in BDNFVal/Val mice, but not BDNFMet/Met mice.

(A) Schematic diagram showing GCaMP6 virus infected cell bodies of the vCA1 neurons with projection fibers in the PL and optic fiber implanted in the PL. See also Figure S8.
(B) GCaMP6 AAV virus expression in the vCA1, injected at P45 (top row). Scale bar 40 μm. The tip of the optical fiber implant was centered above the PL (middle row). Scale bar 40 μm. Higher magnification views of the GCaMP6 positive fibers in the PL near the fiber implant are shown (bottom row). Scale bar 20 μm.
(C) There were fewer GCaMP6 positive fibers in the PL in BDNFMet/Met mice compared to BDNFVal/Val mice, despite no differences in GCaMP6 positive cells in the vCA1 (n = 8 BDNFVal/Val, n = 6 BDNFMet/Met; two-tailed, unpaired Student t test, ***p < 0.001; data are represented as mean ± SEM).
(D-E) Average fluorescent signal (ΔF/F) from mouse cohort across the first five presentations of the tone during early cued fear extinction (D) and during late cued fear extinction (E). ΔF/F is shown for the 5 seconds preceding the tone and the subsequent 5 seconds after tone onset (n = 12 BDNFVal/Val, n = 12 BDNFMet/Met; data are represented as mean ± SEM). There was no genotypic difference in signaling during the early trials. During late trials, BDNFVal/Val neurons showed increased activity in prediction of the tone, which subsided shortly after tone onset. Conversely, the BDNFMet/Met neurons did not show this adaptation with increased activity in prediction of the tone or return to baseline level of activity after tone onset.
(F) Neither the change in ΔF/F signal over time during the pre-tone period (−5.0 to 0.0 seconds) nor the change in ΔF/F signal over time during the initial five seconds of the tone (0.0 to 5.0 seconds) differed between BDNFVal/Val mice and BDNFMet/Met mice during early cued fear extinction (n = 12 BDNFVal/Val, n = 12 BDNFMet/Met; two-tailed, unpaired Student t test; p > 0.05; data are represented as mean ± SEM).
(G) The change in ΔF/F signal over time during the pre-tone period (−5.0 to 0.0 seconds) shows a significant increase in BDNFVal/Val mice relative to BDNFMet/Met mice. The change in ΔF/F signal over time during the initial five seconds of the tone (0.0 to 5.0 seconds) shows a significant decrease in BDNFVal/Val mice relative to BDNFMet/Met mice during late cued fear extinction. (n = 12 BDNFVal/Val, n = 12 BDNFMet/Met; two-tailed, unpaired Student t test; *p < 0.05; data are represented as mean ± SEM)
DISCUSSION
To our knowledge, this is the first report of a bioactive ligand inducing disassembly of mature dendritic spines and elimination of synapses in vitro and in vivo. The majority of synapse elimination studies in the mature CNS focuses on the removal of the molecules that regulate synapse maintenance or cell adhesion, such as neurexins, neuroligins or synaptic cell adhesion molecules (synCAMs). Using 3D-SIM, we show that the BDNF Met prodomain acts locally at the spine to mobilize key actin modifiers to eliminate synapses and disassembles spines that express the SorCS2/p75NTR receptor complex by driving Rac1 regulating molecule Trio out of the spines (Figure 3A-B). Recently, it has been demonstrated that Trio is present in dendritic spines of CA1 hippocampal neurons and functions as a critical and potent regulator of synaptic structure (Herring and Nicoll, 2016). Trio was initially discovered as a molecule interacting with leukocyte antigen-related (LAR) molecules known for transducing extracellular signals to regulate cytoskeletal rearrangements (Bateman and Van Vactor, 2001). While another member of the Trio family, Kalirin-7, has been more extensively studied in the context of spine remodeling due to its exclusive expression in the CNS, a recent analysis of Trio−/− knockout mice suggests its involvement in hippocampal learning (Zong et al., 2015). The relocation of Trio from spines to dendrites, triggered by the BDNF Met prodomain, could lead to the observed decrease in active Rac1 in spine heads (Figure 3C-D). The ability of Rac1 to maintain the spine morphology depends on keeping LIMK1 activated in order to phosphorylate and inactivate actin-severing protein cofilin (Nakayama et al., 2000). With lowered active Rac1 in the spine, the levels of active cofilin may rise. A recent model of the actin disassembly described in non-neuronal cells suggests that for the cofilin severing process to be efficient, the bundling protein fascin needs to dissociate from actin (Elkhatib et al., 2014). Previous EM studies analyzing mature dendritic spines did not detect fascin (Korobova and Svitkina, 2010); however, we observed fascin in spines with 3D-SIM (Figure 3G) in addition to increased phospho-fascin levels detected with another antibody following the Met treatment. (Figures 3H-I). Another study showed that an injection of cytochalasin D, an inhibitor of actin polymerization, into the CA1 inhibits the return of fear response after re-conditioning during the last extinction session (Motanis and Maroun, 2012). By driving up actin severing and preventing its bundling, the Met prodomain may maintain vCA1 spines in the collapsed state. Furthermore, as the Met prodomain disassembles spines, it may impose a physical constraint forcing PSD-95 to exit and further destabilize the dendritic spine suggested by a decrease in PSD-95 positive spines. Additionally, overall reduction in PSD-95 levels implies a degradation of PSD-95 (Figure 1C-D). Previous synapse elimination studies have shown that cell adhesion molecules, such as protocadherin-10 (Pcdh10) and ephrin-B3, also regulate PSD-95. The expression of the Pcdh10 is regulated by the MEF2 transcription factor and the Fragile X mental retardation protein (FMRP) (Tsai et al., 2012). As the MEF2 activates the ligase murine double minute-2 (Mdm2) that ubiquitinates PSD-95, it binds to Pcdh10 and is delivered to the proteasome for degradation. Similarly, ephrin-B3 stabilizes PSD-95 at synapses in its unphosphorylated state, but activity induced phosphorylation of ephrin-B3 leads to its detachment from PSD-95 and consequent destabilization and turnover of PSD-95 (Hruska et al., 2015). The reduction in spine number and spine head size of vCA1 neurons may reduce maturing vCA1 projections and render them incapable of efficiently innervating the PL neurons. To support this idea, strikingly, a single developmentally timed injection of the Met prodomain into the vCA1 of the WT mice phenocopies the connectivity and extinction deficits observed in BDNFMet/Met mice (Figure 6A, Figure 8 and Figure S8). Our detailed developmental analysis of the co-receptors pinpoints specific spatiotemporal window in which the Met prodomain can act on vCA1 neurons projecting into the PL during peri-adolescence.
Our use of fiber photometry to analyze in vivo neural ensemble activity at vCA1 neuron terminals is the first report linking neuronal activity differences in vCA1 neurons projecting to the PL with altered fear extinction learning, establishing that these differences emerge during extinction training. Our data suggest that as a result of the repeated extinction trials, in BDNFVal/Val mice, the vCA1 neurons adapt their response and raise their activity in anticipation of the tone. This activity rapidly returns to baseline following tone onset in contrast to BDNFMet/Met mice that exhibit impaired extinction and fail to raise the activity in prediction of the tone (2 seconds before), but also improperly maintain sustained activity following the tone onset for at least 5 seconds (Figure 8 and S8). Our observation that the vCA1 neurons of BDNFMet/Met mice are unable to properly adapt their response with repeated extinction trials suggests that this impairment could lead to failure in response of the PL neurons, thus resulting in persistent fear expression. It is conceivable that having neurons with thinner and fewer spines with reduced vCA1 fibers in the PL may contribute to extinction learning deficits by reducing the influence of vCA1 on PL and correct processing of the fear extinction information and incorporating it within the circuit. Although extinction learning has been shown to involve the IL, the PL directly influences the efficacy of extinction training by mediating fear expression (Vidal-Gonzalez et al., 2006). Thus, decreased fear following extinction is mediated by increasing the fear suppressing properties of the IL and by reducing the fear expressing properties of PL (Giustino and Maren, 2015; Sotres-Bayon et al., 2012). In this context, we did not observe the difference in the vCA1 projections to the IL (Figure 5 and Figure S8). This could be attributed to the distinct maturation and refinement of the vCA1 projections to the IL. In particular, it has been shown that in WT mice, unlike vCA1 projections to the PL, there is no developmental surge in connectivity of vCA1 neurons to the IL during peri-adolescence (Pattwell et al., 2016). Notably, there is evidence of feed-forward inhibition of PL by IL neurons (Ji and Neugebauer, 2012; van Aerde et al., 2008), which may facilitate extinction learning. Alterations in PL activity resulting from the genotypic differences we have highlighted in PL-projecting vCA1 neurons may prevent the normal microcircuit communication between PL and IL. Future work should consider the dynamics by which multiple afferent and efferent PL projections interact to regulate fear extinction behavior.
Our findings suggest that the Met prodomain has a novel gain of function with regards to fear extinction-related circuitry, rather than a loss of function as a result of reduced mBDNF bioavailability as previously reported (Chen et al., 2006). In particular, our experiments using BDNF+/− mice that have reduced mBDNF levels show no deficits in this developmental surge in connectivity between the vCA1 and the PL (Figure 7B-D) and no impairments in fear extinction (Figure S7A). As previously reported, developmental expression of the proBDNF in the CNS declines significantly after P15 (Yang et al., 2014). Furthermore, it is rapidly converted intracellularly into mBDNF or released in response to neuronal activity and converted extracellularly; therefore its levels are negligible, and our results cannot be attributed to proBDNF (Dieni et al., 2012; Matsumoto et al., 2008). Our in vivo studies of injection of the Met prodomain into the vCA1 show acute effects on circuitry and fear extinction behavior despite the mBDNF and the BDNF Val prodomain being present in vCA1 of the WT mice. An experiment mimicking this in vivo situation demonstrated that by adding both Met and Val prodomains to hippocampal neurons in vitro has an intermediate effect on spines even when the Val prodomain is added in excess (Figure S5), compensating for the fact that there are physiologically lower levels of the released Met prodomain. Finally, in cultured BDNFMet/Met hippocampal neurons, spine head diameter and spine density were reduced compared to the BDNFVal/Val neurons. The cultured BDNFVal/Met hippocampal neurons show an intermediate phenotype (Figure S7). These findings correlate with previous behavioral studies showing impaired fear extinction in BDNFMet/Met mice compared to the BDNFVal/Val mice (Soliman et al., 2010).
Together, our findings provide a mechanism by which the BDNF Met prodomain directly alters maturation of fear-related circuitry in a defined developmental “sensitive period”. In addition, it suggests the refinement of the neurotrophic hypothesis with regards to the divergent roles of the BDNF prodomain and mature BDNF in fear related behaviors.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| sheep anti-SorCS2 | R&D Systems | Cat# AF4237, RRID: AB_2192264 |
| rabbit anti-p75 9992 | New York University | Cat# MC0002, RRID: AB_2335792 |
| goat anti-Trio | Santa Cruz | Cat# sc-6061, RRID: AB_2209233 |
| rabbit anti-fascin | Abcam | Cat#: ab126772, RRID: AB_11143231 |
| rabbit anti-phospho S39 fascin | Abcam | Cat# ab90618, RRID: AB_2049642 |
| mouse anti-PSD-95 | Sigma | Cat# P246, RRID: AB_260911 |
| mouse anti-Bassoon | Enzo | Cat#: ADI-VAM-PS003, RRID: AB_10618753 |
| chicken anti-MAP2 | Abcam | Cat# ab92434, RRID: AB_2138147 |
| Alexa Fluor 546 phalloidin | Thermo Fisher Scientific | Cat# A-22283, RRID: AB_2632953 |
| Latrunculin A | Cayman Chemical | Cat#: 76343-93-6 |
| Fluoro-gold antibody | Fluorochrome | Fluoro-gold, RRID: AB_2314408 |
| PHA-L (Phaseolus vulgaris agglutinin) antibody | Vector Laboratories | Cat# 2300, RRID: AB_2315140 |
| Bacterial and Virus Strains | ||
| BL21(DE3)pLysS Chemically Competent Cells | Invitrogen | Cat#: C606003 |
| AAV1.Syn.GCaMP6s.WPRE.SV40 | Penn Vector Core | Cat#: AV-1-PV2824 |
| Biological Samples | ||
| Embryonic and postnatal brains from C57BL/6 and transgenic mice as described in Experimental | This paper | N/A |
| Model and Subject Details. | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant BDNF prodomain | Anastasia et al., 2013 | N/A |
| Critical Commercial Assays | ||
| Ni-NTA purification system | Invitrogen | Cat#: K95001 |
| Experimental Models: Cell Lines | ||
| Sorcs2tm1a(EUCOMM)Wtsi | EUCOMM | MGI Cat#: 1932289, NCBI: 81840 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BDNFMet/Met | Chen et al., 2006 | N/A |
| Mouse: BDNFtm1Jae/BDNF+ (BDNF+/−) | The Jackson Laboratory | IMSR Cat# JAX:002266, RRID: IMSR_JAX:002266 |
| Mouse: p75NTR-EGFP BAC | GENSAT | NINDS Contracts N01NS02331 & HHSN271200723701C to The Rockefeller University, New York, NY |
| Mouse: B6.129S4-Ngfrtm1Jae/J (p75−/−) | The Jackson Laboratory | IMSR Cat# JAX:002213, RRID: IMSR_JAX:002213 |
| Mouse: SorCS2−/− as described in Experimental Model and Subject Details. | This paper | N/A |
| Mouse: B6.FVB-TG(Ella-Cre)C5379mgd/J | The Jackson Laboratory | Cat# JAX:003724, RRID: IMSR JAX:003724 |
| Mouse: B6;SJL-Tg(ACTFLPe)9205Dym/J | The Jackson Laboratory | IMSR Cat# JAX:003800, RRID: IMSR JAX:003800 |
| Recombinant DNA | ||
| Plasmid: pET28 | Novagen | Cat# 69864-3 |
| Software and Algorithms | ||
| NIS-Elements software 4.20 with N-SIM | Nikon Instruments | https://www.nikoninstruments.com/Products/Software; RRID: SCR_014329 |
| Nikon Elements AR 4.2 | Nikon Instruments | https://www.nikoninstruments.com/Products/Software; RRID: SCR_014329 |
| Imaris 7.6.3 | Bitplane | http://www.bitplane.com/imaris/imaris; RRID: SCR_007370 |
| Neurolucida | MBF Bioscience | http://www.mbfbioscience.com/neurolucida; RRID: SCR_001775 |
| Stereo Investigator software | MBF Bioscience | http://www.mbfbioscience.com/stereo-investigator; RRID: SCR 002526 |
| Graphic State 4 software | Coulbourn Instruments | http://www.coulbourn.com/category_s/363.htm |
| MATLAB | Mathworks | http://www.mathworks.com/products/matlab/; RRID: SCR_001622 |
| Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_015807 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Francis S. Lee (fslee@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animal care was in accordance with Weill Medical College of Cornell University’s Institutional Animal Care and Use Committee. Mice were maintained with a 12-hour light/dark cycle at 18°C - 22°C and had ad libitum access to water and food. All mice were healthy with no obvious behavioral phenotypes, and none of the experimental mice were immune compromised. For primary hippocampal neuronal cultures, C57BL/6 pregnant mice were purchased from Charles River (Wilmington, MA, USA). For all mouse studies, mice of the male sex were used and were randomly allocated to experimental groups. The BDNFMet/Met mice were as reported previously (Chen et al., 2006). Briefly, a targeting vector was used to introduce the desired Val66Met substitution (G196A) in the BDNF sequence. This knock-in allele is regulated by endogenous BDNF promoters. WT (BDNFVal/Val) and BDNFMet/Met animals were derived from heterozygous BDNFVal/Met mating pairs. BDNF+/− mice (BDNFtm1Jae) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). p75-EGFP BAC mice expressing GFP in neurons that express endogenous p75, were obtained from the Gene Expression Nervous System Atlas (GENSAT) project (NINDS Contracts N01NS02331 & HHSN271200723701C to The Rockefeller University, New York, NY) (Schmidt et al., 2013). The p75−/− mice were purchased from The Jackson Laboratory (B6.129S4-Ngfrtm1Jae/J, Jackson Laboratories). The SorCS2−/− mice were generated by inserting a vector purchased from EUCOMM (the European Conditional Mouse Mutagenesis Program), which contained L1L2_Bact_P cassette composed of FRT site, lac Z gene, and loxP site followed by neomycin resistance gene under the control of the beta-actin promoter, second FRT and second loxP site flanking exon II of the SorCS2 gene and third loxP site downstream of the exon II. Following electroporation into 129 ES cells, positive clones were identified by neomycin resistance and resulting animals were screened by Southern Blotting detecting disrupted allele at 12.6kb as oppose to 7.5kb WT fragment resulting from HindIII restriction. The mice possessing disrupted SorCS2 allele were bred with flp deleter strain (B6;SJL-Tg(ACTFLPe)9205Dym/J, Jackson Laboratories) to remove the neo cassette flanked by FRT sites, followed by crossing with EIIa-Cre deleter strain (B6.FVB-TG(Ella-Cre)C5379mgd/J, Jackson Laboratories) to excise exon 2 of the SorCS2 gene surrounded by loxP sites. The mice were then backcrossed to C57Bl/6 for over eight generations.
Hippocampal neuronal cultures
DIV18-21 primary hippocampal neurons were prepared from C57BL/6 pregnant mice as previously described (Kaech and Banker, 2006) with some modifications. Briefly, hippocampi from E18 mice were collected and digested with papain (Worthington Biochemical Corporation, Lakewood, NJ, USA), in the presence of deoxyribonuclease I (Sigma), 1.5 mM CaCl2, and 0.75 mM EDTA solution in 37°C/5% CO2 incubator for 25 min. The tissue was triturated with fire-polished glass pipettes and cells were plated on nitric acid-treated, poly-L-lysine (Sigma) coated glass cover slips (Electron Microscopy Sciences, Hatfield, PA, USA). The cells were incubated for several hours in Neurobasal medium (Gibco, Grand Island, NY, USA), supplemented with 1 mM sodium pyruvate (Gibco), 6mM Glutamax (Gibco), 10% fetal bovine serum, 0.5% glucose, and 50 μg ml−1 primocin (Invivogen, San Diego, CA, USA). After allowing the cells to settle for a few hours, the media were switched to Neurobasal medium supplemented with B27 (Gibco), 1 mM pyruvate (Gibco), 2 mM glutamine (Gibco), 50 μg ml−1 primocin, and 4 μM cytosine-1-β-D-arabinofuranoside and maintained in culture for 18-21 days without any media changes/additions.
METHOD DETAILS
All culture and animal experiments were quantified blinded as to the experimental manipulation. Mice were randomly allocated to experimental groups and all data collected in the course of these studies were included in the individual analyses. No data were excluded and therefore there were no exclusion criteria. Due to technical limitations on sample collection no sample-size estimates were conducted. All attempts were made to use a maximal sample size in each experiment whenever possible.
Antibodies
The following primary antibodies were used: anti-SorCS2 (R&D systems, Minneapolis, MN, USA), anti-p75 9992 (a gift from Dr. Moses Chao; Huber and Chao, 1995), anti-Trio (Santa Cruz Biotechnology, Inc., Dallas, TX), anti-fascin (Abcam, Cambridge, MA USA), anti-phospho S39 fascin (Abcam), anti-MAP2 (Abcam), anti-PSD-95 (Sigma, St. Louis, MO, USA), and anti-Bassoon (Enzo Life Sciences, Farmingdale, NY, USA). Secondary antibodies were Alexa Fluor antibodies (Life Technologies, Norwalk, CT, USA), except for the 405nm fluorescent DyLight (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). For visualization of actin cytoskeleton, Alexa Fluor 546 phalloidin (Life Technologies, Thermo Fisher Scientific) was used. Latrunculin A was used for actin depolymerization experiments (Cayman Chemicals, Ann Arbor, MI).
Expression and purification of recombinant BDNF prodomain
Recombinant BDNF prodomain proteins were produced as previously described (Anastasia et al, 2013). Briefly, the plasmid pET28 (Novagen, Madison, WI, USA) containing the gene for the human BDNF prodomain (Val or Met) (amino acids 19–128 preceded by an N-terminal His-tag and SUMO) (Mossessova and Lima, 2000), was transformed in BL21(DE3) pLysS competent cells (Invitrogen, Grand Island, NY, USA). Prodomain samples were purified using a nickel resin column (Ni-NTA, Invitrogen). SUMO was cleaved by Ulp-1 (His6 tagged) proteolysis. Subsequently, the sample was negatively selected in a second nickel resin column where the untagged BDNF prodomain was obtained in the flow-through fraction. The prodomain was purified by reverse phase HPLC using a Vydac C18 column, with gradient solutions A: 0.1% formic acid, 5% acetonitrile, 94.9% H2O and B: 0.1% formic acid, 99.9% acetonitrile with prodomain elution at 67% B. Prodomain containing fractions were lyophilized and stored at −20°C for further use. The resulting protein was resuspended and dialyzed for further use into 50 mM NaH2PO4, 100 mM NaCl, pH 7.0. The sample purity was assessed by SDS–PAGE using Coomassie blue and silver staining. The prodomain concentrations were determined by UV using a calculated extinction coefficient of 0.418 O.D. at 280 nm for a 1 mg/ml solution and confirmed using the Bradford method.
Immunostaining
For immunostaining of cells, cultured primary hippocampal neurons were starved for 3-4 hours in Neurobasal medium supplemented with 0.5% glucose. Following starvation, the neurons were treated with 1nM (10 ng ml−1) of Met and Val prodomains at 37°C/5% CO 2. After the prodomain applications, the neurons were briefly washed with pre-warmed Hank’s Balanced Salt Solution (Gibco) and fixed with pre-warmed 4% paraformaldehyde/4% sucrose solution for 15 minutes at room temperature. Cells were permeabilized and blocked with 5% fetal bovine serum with 0.1% Triton X-100 in phosphate buffered saline for 30 minutes at room temperature. Primary antibodies were applied for 1 hour at room temperature, and subtype-specific Alexa fluorescent secondary antibodies were added for 15 minutes at room temperature. Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). For immunostaining of tissue sections, coronal sections (40 mm) of whole brain were cut by using a sliding microtome frozen by powdered dry ice. Free-floating serial sections were washed in TBS, incubated for 30 minutes in a blocking solution containing 4% normal horse serum (vol/vol), 1% BSA in TBS with 0.2% Triton X-100 (TBS-Tx) and incubated overnight at 4°C with primary antibodies diluted in blocking solution. Sections were then washed in TBS and incubated for 2 hours with subtype-specific Alexa fluorescent secondary antibodies at room temperature. After washing three times for 10 minutes, sections were mounted, cover-slipped with water soluble glycerol-based mounting medium, and sealed with nail polish.
3D Structured Illumination Microscopy (3D-SIM) imaging
For super resolution analysis of fixed culture samples, Nikon’s Structured Illumination Microscope (N-SIM) Nikon Eclipse Ti (Nikon Instruments Inc., Melville, NY, USA) was used. Images were acquired using a 100x Apo TIRF lens with 1.49 N.A. (Nikon Instruments Inc.) and Andor iXon3 DU-897E EMCCD camera (Andor Technology Ltd, Belfast, UK), set to 3D-SIM mode. Nikon’s TIRF 405 laser was used for imaging of fluor 405. Acquisition settings were as following: 10MHz, 14-bit, EM gain without binning, conversion gain 1x, 2.4x or 5x depending on the signal. All laser power settings were set at 50%, and exposure times were adjusted to keep histogram values approximately at 4000 maximum counts for each channel (green, red, far red). Acquisition settings were kept consistent across all conditions within a given experiment. For every Z-stack, 15 images were generated resulting from three angles and five phases. These images were subsequently reconstructed using NIS-Elements software 4.20 with N-SIM analysis plug-in (Nikon Elements) to generate super-resolution data image. Reconstruction parameters (Auto structured illumination contrast, 1.00 apodization filter parameter, 0.05 width of 3D-SIM filter) were optimized and kept consistent to generate the best Fourier transforms, allowing for image resolutions up to 85 to 100 nm (depending on the fluor used) with improved visualization of thin spines.
Tissue imaging and analysis
Serial coronal sections were observed under Nikon 80i fluorescent microscope with a DAPI/FITC/Rhodamine Tri-color Filter. Microphotographs were created using the MicroFire digital camera and the FireFrame software (Optronics, Princeton, NJ, USA). Stereological estimations of cell density were performed using the Stereo Investigator Software (MBF Bioscience, Williston, VT, USA). Contours of the basolateral nucleus (BLA) and ventral hippocampus were made with random sampling of the contours. Total volumes of the BLA and the ventral hippocampus were estimated by using Cavalieri estimation. Total cell numbers were estimated with the optical fractionator method and systemic random sampling, using counting frame size of 25 × 25 × 40 μm and sampling grid size of 100 × 100 μm.
Stereotactic injections
Mice from different age groups were microinjected stereotactically with the Met or Val prodomain (400 ng/μl) for spine examination and behavioral studies. For fear extinction circuitry connectivity analyses, the mice were injected with a retrograde fluorescent tracer fluorogold (FG) (Fluorochrome, Denver, CO, USA) or the anterograde tracer Phaseolus vulgaris leucoagglutinin (PHA-L) (Vector Laboratories, Burlingame, CA). Each mouse was anesthetized with a ketamine and xylazine cocktail (100 mg and 10 mg ml−1) at dosage of 0.1 ml per 10 g of total body weight. The animal was mounted on a stereotactic frame (David Kopf Instruments, Tujunga, California, USA) prior to surgical exposure of the skull. For enhanced visualization of the skull sutures, a 3% of H2O2 solution was applied on the top of skull. Stereotactic coordination was performed by using an electrical drill mounted on a manipulator (David Kopf Instruments). The settings for each age group to target PL were the following: for P23 and P25, AP = 1.7 mm, ML = 0.4 mm, DV = 1.4 mm; for P29, AP = 1.95 mm, ML = 0.5 mm, DV = 1.95 mm; and for P45 and P60, AP = 1.98 mm, ML = 0.5 mm, and DV=1.98 mm. The settings to target vCA1 were AP=−3.0 mm, ML=2.5 mm, DV=2.3 mm. Microinjections were performed using a Nanoject II Auto-Nanoliter Injector (Drummond Scientific Company, Broomall, PA, USA) equipped with glass micropipettes (tip diameter 25 m). A total of 10 nl of 4% FG was injected into each animal 3 times over 2 minutes. 2.5% PHA-L 20nl and either prodomain (1 μl) were injected separated by the air bubble. To minimize leakage up the needle track after the injection, the needle was kept in place for an additional 10 minutes prior to withdrawal. In case of FG injection, 7 days later, the mice were deeply anesthetized with sodium pentobarbital. Using the Perfusion Two Automated Pressure Perfusion system (Leica Microsystems, Buffalo Grove, IL, USA), they were perfused transcardially with 30 mL of 0.9% saline, followed by 120 mL of 4% paraformaldehyde in 0.01 M sodium phosphate buffer (pH 7.4), at a flowing rate of 25 mL per min. The brains were then removed and post-fixed with 4% paraformaldehyde in 0.01 M sodium phosphate buffer (PB) at 4° C overnight and transferred to a sucrose solution (30% sucrose in 0.1 M PB) at 4° C for 48 hours. Coronal sections (40 m) were prepared using a freezing microtome. One in every three sections was immediately mounted, while the other 2 sections were set in an antifreeze solution (30% glycerol, 30% ethylene glycol and 40% 0.25 M PB) and stored at −20° C. The mounted sections were air dried for 3 hours before applying coverslips with a FG-enhancing solution (10% SiO2, 0.1 M Tris, pH 11). In case of PHA-L, tissue fixation and slicing were same as FG animals. Free-floating serial sections (take one every third) were washed (three times for 10 min each) in TBS, incubated for 30 min in a blocking solution containing 4% normal donkey serum and 1% BSA in Tris buffered saline (TBS) with 0.2% Triton X-100 (TBS–Tx). Sections then were transferred in rabbit anti PHA-L primary antibody (1:1000, Vector lab) diluted in the blocking solution mentioned above and incubated 24 hours at 4 °C with rotating. After washed in TBS and sections were incubated for 2 hours with Alexa Fluor-labelled donkey anti-rabbit-555 (1:500, Invitrogen) and washed. The sections were mounted on chromium/gelatin-coated slides and air-dried for at least 2 hours in dark. Slides were cover-slipped by water-soluble glycerol-based mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) and sealed with nail polish.
Golgi staining and imaging
Three hours after stereotactic prodomain injections into the vCA1, the mice were euthanized using euthasol (0.1 ml/10 g body weight). The brains were rapidly removed, submerged in modified Golgi-Cox solution containing 1% HgCl, 1% potassium dichromate and 0.8% potassium chromate and left at room temperature, in the dark for 10 days. They were subsequently transferred to 30% sucrose solution at 4°C for 3 days. The brains were cut into 120 μm coronal sections mounted on gelatin coated slides. Tissue on slides was allowed to dry and following washes was placed in blackening solution containing 0.4 % potassium hydroxide for 10-15min. Following this step, the tissue went through series of dehydration steps and placed in Histo-Clear (Sigma). Sections were covered by coverslips with DPX mounting medium, air dried for 3 days and imaged under the light microscope. The neurons for spine analysis were chosen from the areas surrounding the radius of 1000 μm of the injection but excluding 250 μm radius area of the injections site to minimize sampling of neurons from the lesion site. Neurons were analyzed by the experimenter blinded to the conditions. Dendritic spine density measurements were performed using Neurolucida program (MBF Bioscience, VT, USA). Spines chosen for analysis came from untruncated secondary dendritic branches longer than 30 μm of neurons isolated from neighboring impregnated cells. Spines were categorized using previously described criteria (Risher et al., 2014).
Cued fear conditioning and extinction
The task was conducted as described previously (Pattwell et al., 2012b). Briefly, the fear conditioning apparatus consisted of a mouse shock-chamber (Coulbourn Instruments, Whitehall, PA, USA) placed in a sound-attenuated box. On day 1 (fear acquisition), the mice were acclimated to the conditioning chamber (scented with 0.1% peppermint in 70% EtOH) for 2 minutes prior to the administration of the three tone-shock pairings. The tone-shock pairing consisted of a 30-second presentation of a 5 kHz, 70 dB tone (conditioned stimulus, CS) that ended with a 0.7mA foot shock (unconditioned stimulus, US) during the last 1.0 second of the tone. The intertrial interval (ITI) between the tone-shock pairings was set at 30 seconds. Mice remained in the conditioning chamber for 1 minute after the final tone-shock pairing before being returned to their home cages. On day 2 (24 hours after fear acquisition), the mice were exposed to 5 presentations of the CS tone in the absence of shock (fear extinction). These tones were presented in a novel context (white cylindrical arena scented with 0.1% lemon in 70% ETOH) in order to minimize confounding interactions of contextual fear. Tone presentations lasted for 30 seconds with an ITI of 30 seconds. After the final tone presentation, mice remained in the cylindrical chamber for 1 minute before being returned to their home cages. Fear extinction trials were repeated daily for a total of 4 days. Graphic State 4 software (Coulbourn Instruments) was used to control experiments, and all trials were videotaped for manual analysis. Freezing was defined as the absence of visible movement except that required for respiration (Fanselow, 1980). The percentage time spent freezing was calculated by dividing the amount of time spent freezing during the CS tone by the total duration of the tone. For mice included in the fiber photometry experiment, to minimize handling stress associated with patch cord attachment to each mouse’s optical fiber implant, extinction was limited to two sessions (15 trials per session of an un-shocked 30 second tone separated by a 30 second ITI). To maintain consistency with previous extinction protocols, fiber photometry recordings from trials 1-5 (“early”) and trials 16-20 (“late”) were selected for analysis.
Fiber photometry
Adult mice (P45) were prepped for stereotactic surgery as described above. Holes were drilled in the skull unilaterally over PL (AP = 1.98 mm, ML = 0.5 mm) and vCA1 (AP = −2.8 mm, ML = 2.5 mm). Microinjection of the viral vector AAV1.Syn.GCaMP6s.WPRE.SV40 (Penn Vector Core) was performed using a 10 l Nanofil syringe (World Precision Instruments) equipped with a 33-gauge beveled needle and connected to an infusion pump (UMP, Micro4; World Precision Instruments). A volume of 200nl of GCaMP6s virus was infused at a rate of 50nl/min into vCA1 at depth of 2.3 mm from brain surface. The needle was left in place for an additional 10 minutes after the infusion. A 400 μm diameter optical fiber (Doric) was implanted into PL at a depth of 1.6 mm from brain surface. Optical fibers were secured with Metabond (Parkell). Three weeks following surgery, mice underwent cued fear conditioning and extinction procedures, and GCaMP6s signal was recorded during extinction. The GCaMP6s virus encodes a modified calcium binding protein bound to a fluorescent indicator that serves as a proxy for neural activity. The fiber photometry rig was based on a design described in detail elsewhere (Gunaydin et al., 2014; Cui et al., 2013). A 470 nm LED (Thorlabs) modulated at a frequency of 521 Hz and passed through a filter (Semrock, FF02-472/30) was connected to the optical fiber implant via a 0.48 NA fiber optic patch cord (Doric). Light from the LED served to excite neurons such that those neurons expressing the GCaMP6s emitted an activity-dependent fluorescent signal (Akerboom et al., 2012; Gunaydin et al., 2014). The fluorescent signal was also collected by the fiber optic implant, separated from the excitation light using a dichroic (Semrock, FF495-Di03), passed through a single band filter (Semrock, FF01-535/50), focused on a photodetector (Newport, Model 2151), and recorded by a real-time processor (Tucker Davis Technologies). A pulse from the behavioral system (Graphic State 4) was also passed to the processor allowing fluorescent signal to be time-locked to stimulus presentations. After the completion of extinction procedures mice were perfused and brains were removed and sectioned as described above. Sections were then analyzed to verify expression of the GCaMP6s and placement of the optical fiber using Stereo Investigator software (MBF Bioscience) and a fluorescent microscope (Nikon eclipse).
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample sizes (n) indicated in figure legends 1, 2, 3, 5B correspond to the number of individual neurons analyzed. Sample sizes (n) indicated in figure legends 5E, 6, 7, 8 correspond to the number of mice used in the experiments. All statistical parameters are presented as means ±SEM (Standard Error of Mean). All data were analyzed with GraphPad Prism 6.0 software (San Diego, CA, USA). Statistical significances were calculated via unpaired Student t test (for two group comparisons), one-way ANOVA with Bonferroni post hoc tests to control for multiple comparisons (for three or more group comparisons), or two-way ANOVA with Bonferroni post hoc tests (to assess statistical significance between means), as indicated within individual figure legends. In figures, asterisks denote statistical significance marked by *p < 0.05, **p < 0.01, ***p < 0.001, and “n.s.” indicates no statistical significance.
3D-SIM image analysis
For accurate image analysis, the background signals were subtracted from all images prior to creation of maximum intensity projections. Regions of interest (ROI) were carefully selected and appropriate thresholds for each channel were kept consistent across all conditions within a given experiment. Spines were manually categorized as mushroom (spine head/neck ≥ 1.5) or thin (head/neck ≤ 1.5, length > 1 μm). Puncta and colocalization analyses were performed on Nikon Elements AR software 4.2 (Nikon Instruments Inc.) using the software’s automated object count and measurement tools. Colocalized puncta were automatically determined by creating the intersection between the two thresholded channels. 3D visualizations of spines were reconstructed using both the volumetric view tool on Nikon Elements AR and the surface creation tool on Imaris 7.6.3 (Bitplane AG, Zurich, Switzerland). All quantifications were performed by the person blinded to the conditions and all experiments were done in triplicates and representative experiment was shown.
Fiber photometry data analysis
Data was analyzed in MATLAB (Mathworks) where the fluorescent signal from each mouse was normalized by calculating the ΔF/F. To do this, the median value of an 80 second window (40 seconds before and after a given data point) was calculated. This value was then subtracted from each data point across the 17.5-minute recording period, and each outcome was divided by the same median. The average ΔF/F across a ten-second window (five-seconds preceding the onset of the tone and the initial five-seconds following tone onset) was calculated for each mouse by collapsing across “early” or “late” trials (trials 1-5 and 16-20, respectively). Subsequently, the ΔF/F signal was averaged across one-second time bins (“Time Points”) across the ten-second window. The change in ΔF/F signal over time was calculated for each mouse during the pre-tone period by subtracting the average ΔF/F at −5.0 to −4.0 seconds from the average ΔF/F at −1.0 to 0.0 seconds. Similarly, the change in ΔF/F signal over time was calculated for each mouse during the initial five-seconds of the tone by subtracting the average ΔF/F at 0.0 to 1.0 seconds from the average ΔF/F at 4.0 to 5.0 seconds and dividing by the length of time (5 seconds). Statistical significances were calculated via two-tailed, unpaired Student t tests. In addition, the one-second time bins of ΔF/F signal were subjected to a repeated measure ANOVA, with Genotype as the between-subjects factor, and Time Point as the within-subjects factor. At time bins representing baseline (−5.0 to −4.0 seconds), pre-tone (−1.0 to 0.0 seconds), and tone (4.0 to 5.0 seconds), paired Student t tests relative to tone onset (0.0 to 1.0 seconds) were used to calculate statistical significance.
Supplementary Material
HIGHLIGHTS.
The BDNF Met prodomain eliminates spines in vCA1 neurons during peri-adolescence.
The BDNF Met prodomain alters maturation of the fear extinction circuitry.
BDNFMet/Met vCA1 neurons fail to adapt their activity during fear extinction trials.
Acknowledgments
We acknowledge the resources at the Bio-Imaging facility at Hunter College supported by a Research Centers in Minority Institutions Program grant from the National Institute on Minority Health and Health Disparities (MD007599) of the NIH and thank their staff, Dr. Diana Bratu, Dr. Lloyd Williams and Zhong Wang. We thank Dr. Moses Chao for providing us with the anti-p75NTR antibody. This work was supported in part by National Institutes of Health (NIH) grants NS052819 (BLH, FSL) and 5UL1TR000457 (FSL), a generous gift by the Mortimer D. Sackler, M.D. family, the Brain and Behavior Research Foundation NARSAD grant (JIG, DJ, FSL), the New York-Presbyterian Youth Anxiety Center (FSL), the Pritzker Neuropsychiatric Disorders Research Consortium (FSL), and the DeWitt-Wallace Fund of the New York Community Trust (FSL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes eight figures.
AUTHOR CONTRIBUTIONS
BLH, FSL, JIG and AA designed the study. JIG, DJ, HCM, HB, and ID conducted experiments. JIG, JK, HCM, CB, ID, DJ, CL and KL analyzed data and interpreted the results. JY generated and provided SorCS2−/− mice for the study. JIG and FSL wrote the manuscript. All authors reviewed and approved the final manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasia A, Deinhardt K, Chao MV, Will NE, Irmady K, Lee FS, Hempstead BL, Bracken C. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat Commun. 2013;4:2490. doi: 10.1038/ncomms3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J, Van Vactor D. The Trio family of guanine-nucleotide-exchange factors: regulators of axon guidance. J Cell Sci. 2001;114:1973–1980. doi: 10.1242/jcs.114.11.1973. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Glatt CE, Lee FS. Treating the Developing versus Developed Brain: Translating Preclinical Mouse and Human Studies. Neuron. 2015;86:1358–1368. doi: 10.1016/j.neuron.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG, Nykjaer A, Hempstead BL, Lee FS. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25:6156–6166. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–4411. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, Chao MV, Hempstead BL. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal. 2011;4:ra82. doi: 10.1126/scisignal.2002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. 2012;196:775–788. doi: 10.1083/jcb.201201038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkhatib N, Neu MB, Zensen C, Schmoller KM, Louvard D, Bausch AR, Betz T, Vignjevic DM. Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr Biol. 2014;24:1492–1499. doi: 10.1016/j.cub.2014.05.023. [DOI] [PubMed] [Google Scholar]
- Felmingham KL, Dobson-Stone C, Schofield PR, Quirk GJ, Bryant RA. The brain-derived neurotrophic factor Val66Met polymorphism predicts response to exposure therapy in posttraumatic stress disorder. Biol Psychiatry. 2013;73:1059–1063. doi: 10.1016/j.biopsych.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustino TF, Maren S. The Role of the Medial Prefrontal Cortex in the Conditioning and Extinction of Fear. Front Behav Neurosci. 2015;9:298. doi: 10.3389/fnbeh.2015.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glerup S, Olsen D, Vaegter CB, Gustafsen C, Sjoegaard SS, Hermey G, Kjolby M, Molgaard S, Ulrichsen M, Boggild S, et al. SorCS2 regulates dopaminergic wiring and is processed into an apoptotic two-chain receptor in peripheral glia. Neuron. 2014;82:1074–1087. doi: 10.1016/j.neuron.2014.04.022. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, St Hillaire C, Zweifel LS, Glebova NO, Philippidou P, Halegoua S, Ginty DD. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell. 2011;146:421–434. doi: 10.1016/j.cell.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Nicoll RA. Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc Natl Acad Sci U S A. 2016;113:2264–2269. doi: 10.1073/pnas.1600179113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska M, Henderson NT, Xia NL, Le Marchand SJ, Dalva MB. Anchoring and synaptic stability of PSD-95 is driven by ephrin-B3. Nat Neurosci. 2015;18:1594–1605. doi: 10.1038/nn.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain. 2012;5:36. doi: 10.1186/1756-6606-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Maren S. Fear renewal preferentially activates ventral hippocampal neurons projecting to both amygdala and prefrontal cortex in rats. Sci Rep. 2015;5:8388. doi: 10.1038/srep08388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh-Semba R, Semba R, Takeuchi IK, Kato K. Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence-accelerated mice: a comparison to those of nerve growth factor and neurotrophin-3. Neurosci Res. 1998;31:227–234. doi: 10.1016/s0168-0102(98)00040-6. [DOI] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. Journal of neurochemistry. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Amdur R, Jung TD, Chamberlain KR, Minoshima S, Koeppe RA, Fig LM. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999;45:817–826. doi: 10.1016/s0006-3223(98)00246-7. [DOI] [PubMed] [Google Scholar]
- Lin YC, Koleske AJ. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu Rev Neurosci. 2010;33:349–378. doi: 10.1146/annurev-neuro-060909-153204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamauchi J, Tanoue A, Wu C, Mobley WC. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci U S A. 2006;103:10444–10449. doi: 10.1073/pnas.0603914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motanis H, Maroun M. Differential involvement of protein synthesis and actin rearrangement in the reacquisition of contextual fear conditioning. Hippocampus. 2012;22:494–500. doi: 10.1002/hipo.20915. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci. 2012;32:2410–2421. doi: 10.1523/JNEUROSCI.5205-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattwell SS, Liston C, Jing D, Ninan I, Yang RR, Witztum J, Murdock MH, Dincheva I, Bath KG, Casey BJ, et al. Dynamic changes in neural circuitry during adolescence are associated with persistent attenuation of fear memories. Nat Commun. 2016;7:11475. doi: 10.1038/ncomms11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A. 2012;109:E442–451. doi: 10.1073/pnas.1118803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB, Orr SP, Pitman RK. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry. 2000;47:769–776. doi: 10.1016/s0006-3223(00)00828-3. [DOI] [PubMed] [Google Scholar]
- Schmidt EF, Kus L, Gong S, Heintz N. BAC transgenic mice and the GENSAT database of engineered mouse strains. Cold Spring Harb Protoc. 20132013 doi: 10.1101/pdb.top073692. [DOI] [PubMed] [Google Scholar]
- Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn Mem. 2004;11:525–535. doi: 10.1101/lm.79504. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–812. doi: 10.1016/j.neuron.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Lim Y, Li F, Liu S, Lu JJ, Haberberger R, Zhong JH, Zhou XF. ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PLoS One. 2012;7:e35883. doi: 10.1371/journal.pone.0035883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, Huber KM. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151:1581–1594. doi: 10.1016/j.cell.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aerde KI, Heistek TS, Mansvelder HD. Prelimbic and infralimbic prefrontal cortex interact during fast network oscillations. PLoS One. 2008;3:e2725. doi: 10.1371/journal.pone.0002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Jin J, Maren S. Renewal of extinguished fear activates ventral hippocampal neurons projecting to the prelimbic and infralimbic cortices in rats. Neurobiol Learn Mem. 2016;134(Pt A):38–43. doi: 10.1016/j.nlm.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, Jing D, Lafrancois JJ, Bath KG, Mark W, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014;7:796–806. doi: 10.1016/j.celrep.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, et al. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci. 2005;25:9989–9999. doi: 10.1523/JNEUROSCI.2492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Benedek DM, Fullerton CS, Forsten RD, Naifeh JA, Li XX, Hu XZ, Li H, Jia M, Xing GQ, et al. PTSD risk is associated with BDNF Val66Met and BDNF overexpression. Molecular psychiatry. 2014;19:8–10. doi: 10.1038/mp.2012.180. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zong W, Liu S, Wang X, Zhang J, Zhang T, Liu Z, Wang D, Zhang A, Zhu M, Gao J. Trio gene is required for mouse learning ability. Brain Res. 2015;1608:82–90. doi: 10.1016/j.brainres.2015.02.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.