

SUMMARY
Cancer cachexia is characterized by extreme skeletal muscle loss that results in high morbidity and mortality. The incidence of cachexia varies among tumor types, being lowest in sarcomas, whereas 90% of pancreatic ductal adenocarcinoma (PDAC) patients experience severe weight loss. How these tumors trigger muscle depletion is still unfolding. Serendipitously, we found that overexpression of Twist1 in mouse muscle progenitor cells, either constitutively during development or inducibly in adult animals, caused severe muscle atrophy with features reminiscent of cachexia. Using several genetic mouse models of PDAC, we detected a marked increase in Twist1 expression in muscle undergoing cachexia. In cancer patients, elevated levels of Twist1 are associated with greater degrees of muscle wasting. Finally, both genetic and pharmacological inactivation of Twist1 in muscle progenitor cells afforded substantial protection against cancer-mediated cachexia, which translated into meaningful survival benefits, implicating Twist1 as a possible target for attenuating muscle cachexia in cancer patients.
Keywords: Cancer Cachexia, Muscle Cachexia, Muscle Progenitor Cells, Twist1, Pancreatic Ductal Adenocarcinoma, Activin/Myostatin Cytokines, Smad Signaling, Muscle Protein Breakdown
eTOC Blurb
Parajuli et al. show that both genetic and pharmacological inhibition of Twist1 preserves muscle mass and extends lifespan in several mouse models of cancer cachexia, highlighting Twist1 as an attractive therapeutic target for attenuating muscle cachexia and associated morbidity and mortality in advanced cancer patients.
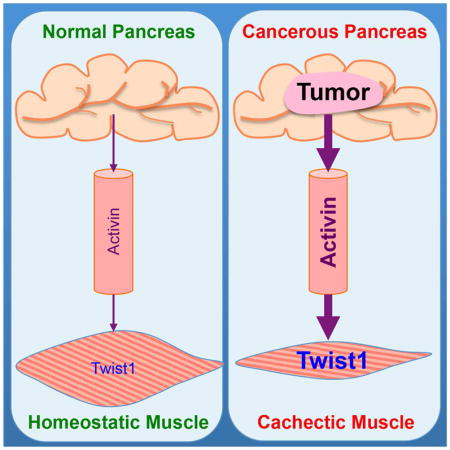
INTRODUCTION
Cancer cachexia is an insidious syndrome that has a dramatic impact on patient quality of life and is associated with poor responsiveness to chemotherapy and decreased survival (Cohen et al., 2015; Fearon et al., 2012; Tisdale, 2009). Cancer cachexia is characterized by the progressive depletion of skeletal muscle, which occurs in concert with the loss of body fat. Muscle cachexia arises through a combination of hypoanabolism together with increased catabolism of myofibrillar proteins. Several landmark studies documented the key role of the ubiquitin-proteasome pathway in myofibrillar protein degradation during cancer cachexia. In particular, two muscle-specific ubiquitin ligases, e.g., MuRF1 and Atrogin1/MAFbx, have been demonstrated to be essential for the degradation of muscle proteins, including myosin heavy chain (MHC) and eukaryotic initiation factor 3f (Elf-3f) (Clarke et al., 2007; Lagirand-Cantaloube et al., 2008).
The expression of MuRF1 and Atrogin1 is induced by a subset of secreted factors, most prominent among them is Myostatin (Mstn), whose deficiency culminates in muscle hypertrophy in both humans and animals (Fearon et al., 2012; Han et al., 2013; Sartori et al., 2014). Enforced expression of Mstn in mice triggers skeletal muscle atrophy, whereas its inactivation promotes muscle mass accrual (Lee and McPherron, 2001; McPherron et al., 1997). Moreover, genetic deletion of Mstn has been shown to protect against cancer-induced muscle cachexia (Gallot et al., 2014). Mstn is synthesized and secreted mainly from skeletal muscle cells, including muscle progenitor cells (satellite), and signals through a receptor complex composed of two serine/threonine kinases termed Activin type I (Alk) and type II (ActRIIB) receptors (Han et al., 2013; Sartori et al., 2014). Engagement of the ActRIIB-Alk receptor complex with Mstn leads to phosphorylation of Smad2 and Smad3 and their concomitant association with Smad4 and translocation into the nucleus, where they indirectly regulate expression of MuRF1 and Atrogin1 (Han et al., 2013; Sartori et al., 2014).
Besides Mstn, another transforming growth factor beta (TGF-β) superfamily member, Activin A (Act-A), also contributes to cachexia through similar mechanisms involving the same ActRIIB-Alk receptor complex and the same Smad downstream effectors (Cohen et al., 2015; Han et al., 2013; Sartori et al., 2014). Elevated levels of circulating Act-A have been noted in many cancer patients and are associated with severe cachexia and poor outcome (Fearon et al., 2012; Han et al., 2013; Loumaye et al., 2015; Sartori et al., 2014). Along these lines, an ActRIIB receptor-based trap (sActRIIB) was recently used to treat muscle wasting in several mouse models of cancer cachexia (Zhou et al., 2010). Intriguingly, sActRIIB was very potent at preventing further skeletal muscle loss as well as at restoring previous muscle mass. Mice treated with sActRIIB also experienced an improvement in survival despite persistent tumor growth, in line with the general notion that muscle cachexia represents a key determinant of cancer-related death (Tisdale, 2010; Zhou et al., 2010). At the molecular level, sActRIIB administration attenuated MuRF1 and Atrogin1 expression, thereby preventing degradation of myofibrillar proteins. Of particular importance, sActRIIB markedly promoted self-renewal of muscle progenitor cells and production of new muscle, implying that Act-A/Mstn signaling acts directly on muscle progenitor cells to trigger muscle cachexia (Zhou et al., 2010).
Twist1 is a transcription factor that fulfills important roles in the specification of mesenchymal stem cell fate during embryogenesis (Chen and Behringer, 1995; Qin et al., 2012). Twist1 inactivating mutations cause Saethre-Chotzen syndrome, an autosomal dominant inheritance disease characterized by a broad spectrum of malformations, including short stature (el Ghouzzi et al., 1997; Howard et al., 1997). Aberrant Twist1 overexpression is associated with progression of several human malignancies, including PDAC (Lee and Bar-Sagi, 2010; Qin et al., 2012; Yang et al., 2004). Mounting evidence suggests that Twist1 exerts multiple roles during carcinogenesis. For instance, Twist1 is able to override the failsafe cell senescence and apoptotic responses triggered by oncogenes and to facilitate metastasis by orchestrating the epithelial-to-mesenchymal transition (Lee and Bar-Sagi, 2010; Maestro et al., 1999; Valsesia-Wittmann et al., 2004; Yang et al., 2004). Noteworthy, recent studies reported that suppression of Twist1 recruitment to the transcriptional machinery by the small-molecule bromodomain inhibitor JQ1 was able to dampen mammary tumor growth in vivo (Shi et al., 2014), highlighting Twist1 as an attractive candidate target for anti-cancer therapy.
In this study, we combined several orthogonal approaches and models to demonstrate that Twist1 plays an essential role in muscle cachexia during cancer progression. From a mechanistic perspective, we provide evidence that tumor-derived Act-A acts on muscle to induce expression of Twist1, which in turn drives expression of MuRF1 and Atrogin1, leading to muscle protein degradation and attendant muscle cachexia. Genetic or pharmacological inactivation of Twist1 was sufficient to reverse muscle cachexia and improve survival in several genetic mouse models of cancer cachexia. Thus, our findings unveil an unusual function for the pro-malignant transcription factor Twist1 in orchestrating cancer-driven muscle cachexia and thus provide a paradigm for the design of dual chemotherapeutics to curb cancer progression and associated muscle cachexia simultaneously.
RESULTS
Twist1 Overexpression in Muscle Progenitor Cells Induces Muscle Atrophy
The Twist1 gene is overexpressed or amplified in a number of mesenchymal stem cell-derived malignancies, including rhabdomyosarcoma, liposarcoma, and osteosarcoma (Maestro et al., 1999; Piccinin et al., 2012; Qin et al., 2012). However, given the embryonic-lethal phenotype of Twist1-null mice and the lack of an autochthonous system to study the impact of overexpressing Twist1 in vivo (Chen and Behringer, 1995), the contribution of Twist1 to the pathogenesis of these mesenchymal malignancies remains speculative. To address this issue rigorously, we set out to devise genetic experiments using mice carrying a Cre-activable Twist1 transgene, CAG-Loxp-Cat-Loxp-Myc-Twist1 (LCL-Twist1), in which the floxed chloramphenicol acetyltransferase (Cat) gene serves as both reporter and silencer, preventing Twist1 expression in the absence of Cre recombinase (Connerney et al., 2006). We crossed these mice with Prrx1.Cre mice, which express Cre in uncommitted mesenchymal stem cells that give rise to bone, muscle, and both white and brown adipose tissues (Logan et al., 2002). Intriguingly, LCL-Twist1;Prrx1.Cre mice exhibited a very lean phenotype that was mainly characterized by severe hypotrophy of skeletal muscle (Figure S1A). In support of this observation, we detected a marked decrease in weight of several skeletal muscles, including gastrocnemius (GC), extensor digitalis longus (EDL), soleus (Sol), and tibialis anterior (TA) (Figure S1B). Moreover, analysis of GC muscle histology by staining with hematoxylin and eosin (H&E) or immunostaining with anti-dystrophin antibody revealed a significant decrease in myofiber diameters in LCL-Twist1;Prrx1.Cre mice compared to control mice (Figure S1C). To corroborate these findings, we generated mice overexpressing Twist1 in muscle progenitor cells (satellite) using the Pax7.Cre strain (hereafter termed as mTwist1 mice). Congruent with our earlier observation, mTwist1 mice showed severe skeletal muscle hypotrophy, which again affects all types of hindlimb muscles analyzed, including GC, EDL, Sol, and TA (Figures 1A and 1B). Muscle histology demonstrated a marked decrease in myofiber diameters in mTwist1 mice relative to control mice (Figures 1C and S1D), providing further evidence that Twist1 overexpression can affect muscle mass.
Figure 1. Twist1 Overexpression Drives Muscle Atrophy.

(A) Representative pictures of two-month-old mTwist1 and control mice.
(B) The weights of GC, EDL, Sol, and TA muscles from two-month-old mTwist1 and control mice were normalized by body weight (BW) (n= 6).
(C) Analysis of GC muscle cross-sectional areas in two-month-old mTwist1 and control mice by H&E staining or dystrophin immunostaining. Scale bars: 50 μM.
(D–H) Three-month-old mTwist1ER or control mice were injected with vehicle or Tam as indicated (n= 12). Body weight was measured every other week (D). Grip strength (E), GC muscle weight per BW (F), and GC muscle cross-sectional areas (G) were analyzed 12 weeks following treatment. Physical activity was monitored by CLAMS beginning at 12 weeks following treatment (H). Scale bars: 50 μM.
Data in B, D, E, F, and H are expressed as mean ± SEM. **p < 0.01; ***p< 0.001; ns, not significant. See also Figure S1.
To investigate whether enforced expression of Twist1 in satellite cells could similarly compromise muscle homeostasis in healthy adult animals, we generated mice with Tamoxifen-controlled Twist1 expression in satellite cells using the Pax7.CreERT2 strain (referred to hereafter as mTwist1ER). Interestingly, inducing Twist1 expression in 3-month-old adult mTwist1ER mice resulted in severe muscle atrophy that became manifest following 4 weeks of treatment, as evidenced by the significant decrease in body weight, grip strength, muscle mass, and myofiber diameters (Figures 1D–G and S1E). EchoMRI analysis showed that Twist1 expression reduced lean mass, but had no effect on fat mass (Figures S1F and S1G). Comprehensive Laboratory Animal Monitoring System (CLAMS, known as metabolic cage) showed no changes in food consumption or respiratory exchange ratio (Figures S1H and S1I). In contrast, we noticed a progressive decline in physical activity in Tam-treated mTwist1ER mice (Figure 1H), further attesting to the ability of Twist1 to compromise muscle mass and function when overexpressed in adult muscle satellite cells.
Twist1 Drives Myostatin Synthesis in Satellite Cells
Perhaps surprisingly, Tam administration to 3-month-old adult mTwist1ER mice not only induced exogenous Twist1 expression in satellite cells, but also led to accumulation of endogenous Twist1 in cytoplasm and peripheral nuclei within myofibers, as gauged by immunoblotting and immunofluorescence (Figures 2A and 2B). As such, this observation raises the intriguing possibility that Twist1 overexpression in satellite cells might lead to production of a paracrine factor that in turn triggers endogenous Twist1 expression, at least in neighboring myofibers. To better understand this phenomenon, we generated mice with ubiquitous Tam-inducible Twist1 overexpression (gTwist1ER) using the CAG.CreERT2 strain, and assessed for the presence of endogenous Twist1 in other tissues besides skeletal muscle. Interestingly, inducing global transgenic Twist1 expression drove endogenous Twist1 expression exclusively in muscle (Figure 2A), supporting the hypothesis that Twist1 induces expression of a muscle intrinsic paracrine factor that triggers Twist1 expression in myofibers. To identify such a factor, we turned our attention on Myostatin (Mstn), a muscle atrophy-inducing cytokine that is expressed in satellite cells (McCroskery et al., 2003). Using either isolated Pax7-expressing satellite cells or whole muscle from mTwist1ER mice treated with Tam, we detected a significant increase in Mstn expression, concurring with increased Twist1 expression (Figures 2C, 2D, S2A, S2B). A similar conclusion could be drawn when satellite cells from mTwist1ER mice were treated with Tam in vitro (Figure S2C). We also detected a marked increase in Smad2 phosphorylation in muscle from Tam-treated mTwist1ER mice (Figure 2D), evocative of Mstn pathway activation. Taking advantage of the presence of two conserved Twist1 binding sites (E-boxes EMS1 and EMS2) within the Mstn promoter (Ma et al., 2001), we conducted ChIP experiments using satellite cell chromatin, and detected a significant increase in binding of Twist1 to both E-boxes in the Mstn promoter upon stimulation of mTwist1ER mice with Tam (Figure 2E). This binding is functional and specific, as expression of Twist1 induced gene expression from a luciferase reporter driven by the wild-type Mstn promoter, and mutating the two E-boxes abrogated this effect (Figure S2D). Finally, culturing wild-type myofibers in conditioned media of satellite cells from Tam-treated mTwist1ER mice resulted in increased Twist1 expression, and this effect was almost completely blocked by treatment with a Mstn neutralizing antibody (Figure 2F), indicating an ability of Mstn secreted by Tam-treated mTwist1ER satellite cells to induce Twist1 expression in myofibers. To demonstrate this directly, we used a lentiviral-based CRISPR/CAS9 strategy to inactivate Mstn in mTwist1ER mice. We found that although Mstn ablation did not affect transgenic Twist1 expression, it did suppress endogenous Twist1 expression (Figure 2G). Noteworthy, Mstn ablation was also effective at reversing muscle atrophy in mTwist1ER mice (Figure 2H). Taken together, these findings support the existence of a muscle intrinsic program in which Twist1 functions as a transcription factor in satellite cells to drive synthesis and secretion of Mstn, which in turn acts on myofiber to trigger Twist1 expression.
Figure 2. Twist1 Induces Muscle Atrophy.

(A and B) Three-month-old mTwist1ER, gTwist1ER, or control mice were treated with Tam and analyzed for Twist1 expression 12 weeks following treatment. Twist1 expression in hindlimb muscles, skin, or testis was analyzed by immunoblotting using anti-Twist1 antibody (A). GC muscle sections from mTwist1ER or control mice were immunostained with anti-Twist1 or anti-Myc antibody to detect endogenous Twist1 or exogenous 6xMyc-Twist1 (6M-Twist1), respectively (B). Scale bars: 50 μM.
(C) Three-month-old mTwist1ER or control mice were treated with vehicle or Tam and satellite cells were isolated 12 weeks following treatment (n= 6). Expression of Mstn, Pax7, and Twist1 in isolated satellite cells was analyzed by immunoblotting.
(D and E) Three-month-old mTwist1ER or control mice were treated with vehicle or Tam and muscles were analyzed 12 weeks following treatment. Mstn expression and Smad2 phosphorylation were assessed by immunoblotting (D). Twist1 bound to the Mstn promoter was analyzed by ChIP and agarose gel (E).
(F) Myofibers from 3-month-old wild-type mice were cultured in conditioned media of satellite cells from 3-month-old Tam-treated mTwist1ER or control mice in the presence or absence of a neutralizing Mstn antibody. Expression of Twist1 was analyzed by immunoblotting.
(G and H) Muscle of 3-month-old mTwist1ER or control mice were injected with lentiviruses encoding control or gRNA targeting Mstn and then treated with Tam (n= 6). Expression of muscle Twist1 and Mstn was analyzed by immunoblotting 12 weeks following treatment. The weight of TA muscle was normalized by BW (H).
(I–L) Three-month-old mTwist1ER or control mice were treated with vehicle or Tam and muscles were analyzed 12 weeks following treatment (n= 8). The protein/DNA ratio was determined by quantifying total protein and DNA (I). Global protein ubiquitination was analyzed by immunoblotting using an anti-ubiquitin antibody (J). Expression of muscle wasting markers was analyzed by immunoblotting (K). Twist1 bound to the MuRF1 or Atrogin1 promoter was analyzed by ChIP and agarose gel (L).
Data in E, H, I, and L are expressed as mean ± SEM. **p < 0.01; ***p< 0.001; ns, not significant. See also Figure S2.
Twist1 Orchestrates Muscle Protein Degradation
The finding presented so far prompted us to extend our analysis beyond satellite cells to encompass general mechanisms that could account for the global muscle atrophy in mTwist1ER mice, including within myofibers. We found that treatment of mTwist1ER mice with Tam did not affect expression of master regulators of myoblast differentiation, including MyoD, Myf5, myogenin, muscle creatine kinase (MCK), myosin heavy chain (MyHC), and Pax7 (Figure S2E). Congruently, we detected a small fraction of myofibers in TA muscle that express exogenous 6xMyc-Twist1 (Figure S2F), supporting the inability of Twist1 to block myoblast differentiation, and further confirming previous studies that adult satellite cells can differentiate into myofibers under homeostatic conditions (Keefe et al., 2015; Pawlikowski et al., 2015). In an alternative experimental approach, we crossed LCL-Twist1 mice with MCK-Cre mice, and found that overexpressing Twist1 in myofibers did not affect muscle mass or integrity (Figures S2G and S2H).
Besides having little or no effect on myogenesis, we also found that Twist1 overexpression did not affect cell proliferation or apoptosis, as assessed by BrdU incorporation and capsase-3 cleavage, respectively (Figure S2I). In contrast, we detected a marked decrease in total muscle protein content, concurring with a global increase in protein ubiquitination (Figures 2I and 2J). In line, inducing Twist1 expression resulted in increased MuRF1 and Atrogin1 mRNA and protein expression, and this was further reflected in decreased abundance of their respective substrates, MHC and elF3-f (Figures 2K and S2B).
Because Twist1 overexpression in satellite cells led to accumulation of endogenous Twist1 within myofibers, we set out to investigate whether Twist1 could directly regulate expression of MuRF1 and Atrogin1. An in silico search identified three E-boxes in the MuRF1 promoter (EM1, EM2, EM3) and one E-box (EA) in the Atrogin1 promoter. To investigate whether Twist1 could bind to the MuRF1 and Atrogin1 promoters, we performed ChIP assays using chromatin from GC muscle of mTwist1ER mice. As shown in Figure 2L, Twist1 exhibited robust binding to EM2 and EM3 in the MuRF1 promoter and to EA in the Atrogin1 promoter in Tam-treated mTwist1ER mice as compared to untreated or control mice. To confirm these findings, we assessed the ability of Twist1 to activate luciferase reporters driven by wild-type or mutated (E-box) variants of the MuRF1 (MuRF1-Luc) and Atrogin1 (Atrogin1-Luc) promoters. Using C2C12 cells, we found that ectopic expression of Twist1 induced luciferase expression exclusively from the wild-type MuRF1-Luc and Atrogin1-Luc reporters (Figure S2D), validating the presence of Twist1 responsive elements within the MuRF1 and Atrogin1 promoters. Collectively, these findings provide strong evidence that Twist1 functions as a transcription factor for MuRF1 and Atrogin-1, thereby supporting a model in which Twist1 functions in myofibers to orchestrate muscle protein breakdown and muscle atrophy.
Increased Twist1 Expression in Muscle During Cancer Cachexia
Muscle cachexia is a prevalent syndrome among PDAC patients, often predicting treatment intolerance and severe morbidity and mortality (Fearon et al., 2012; Han et al., 2013). To investigate a possible role of Twist1 in cancer cachexia, we utilized a mouse model of PDAC that faithfully mimics the human disease (Hingorani et al., 2003). This model relies on the Pdx1.Cre strain to generate both pancreas-specific expression of an endogenous oncogenic Kras allele (KrasG12D) and deletion of one allele of the Trp53 tumor suppressor gene. For simplicity, KrasG12D;Trp53fl/+;Pdx1.Cre mice will be referred to hereafter as KP53 mice. Following PDAC formation (~12 weeks), all KP53 mice manifested a lethal wasting syndrome characterized by progressive weight loss and severe muscle cachexia (Figure 3A). In particular, the weight of GC muscle in KP53 mice decreased by more than 30% below levels in healthy animals, and this decrease was associated with reduced grip strength and myofiber diameters (Figures 3B–D). As such, this mouse model provides a valuable tool for exploring the mechanistic underpinnings of muscle cachexia under autochthonous PDAC conditions.
Figure 3. PDAC Triggers Muscle Twist1 Expression.

(A) Body weight of KP53 or control mice was measured every 4 weeks up to 32 weeks of age (n= 26 for control; n= 22 for KP53).
(B–D) Grip strength (B), GC muscle weight per BW (C), and GC muscle cross-sectional areas (D) of KP53 and control mice were analyzed at 24 weeks of age (n= 6). Scale bars: 25 μM.
(E) Muscles from KP53 or control mice were analyzed for Twist1 expression by immunoblotting and RT-PCR (n= 6). Immunblot shows expression of Twist1 in 3 representative mice.
(F) Satellite cells were isolated from KP53 and control mice and analyzed for Twist1 expression by immunoblotting or RT-PCR (n= 3).
(G) GC muscle sections from KP53 or control mice were analyzed for Twist1 expression by immunofluorescence (n= 6). Scale bars: 50 μM.
Data in A, B, C, E, and F are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p< 0.001. See also Figure S3.
Remarkably, we found that Twist1 was highly expressed in muscles from KP53 mice, but barely detectable in muscles from healthy young animals (Figure 3E). A similar increase was also observed in isolated satellite cells from KP53 mice (Figure 3F). Immunofluorescence staining confirmed increased expression of Twist1 in both satellite cells and myofibers (Figure 3G). To substantiate this finding, we generated two PDAC cell lines from KP53 mice (termed KP1 and KP2 cells) that retained their capability to form PDAC tumors in syngeneic mice. Transplantation of these cells to 3-month-old wild-type mice resulted in muscle cachexia that was associated with a significant increase in muscle Twist1 expression (Figures S3A–D). This finding is not restricted to PDAC, as inoculation of CT-26 colon adenocarcinoma, B16 melanoma, or LLC lung carcinoma cells to 3-month-old syngeneic mice also caused muscle atrophy that was associated with a 6- to 9-fold increase in Twist1 expression, depending on the cell types (Figure S3E). Lastly, since muscle Twist1 expression vanishes when mice reach maturity and reappears in aged animals (Figure S3F), we extended our experiments to analyze the cachexia phenotype in old mice, which mimics more closely the chronological development of human PDAC. We found that inoculating KP1 cells to 15-month-old mice induced a ~1.9 fold increase in Twist1 expression, and this was associated with a significant decrease in body weight and muscle mass (Figures S3F–H), similar to what observed in 3-month-old mice. Collectively, these findings unveil that Twist1 expression is de-repressed in muscle undergoing cancer cachexia.
Twist1 Expression in Satellite Cells Mediates Cancer-Driven Muscle Cachexia
To investigate whether Twist1 activity in satellite cells contributes to muscle cachexia, we generated mice in which Twist1 can be conditionally deleted in a Tam-dependent fashion (hereafter termed as mTwist1KO;ER) using the Pax7.CreERT2 strain, as mice with constitutive muscle-specific Twist1 knockout (driven by Pax7.Cre) did not survive beyond 2 to 3 weeks due to hypertrophy of head muscles, which probably interfered with their feeding activity during lactation (Figure S4A). Tam administration to 3-month-old adult mTwist1KO;ER mice had minimal impacts on animal weight or health (Figures 4A–C), suggesting that Twist1 is dispensable for muscle maintenance in mature animal. Of note, Tam administration completely blunted endogenous Twist1 accumulation elicited by KP1 and KP2 cells without affecting the levels of the Pax7 protein that is coexpressed with Twist1 (Figure S4B), confirming the efficient Twist1 deletion in these mice. As anticipated, inoculating KP1 and KP2 cells into vehicle-treated mTwist1KO;ER mice induced muscle atrophy, as manifested by a significant reduction in body weight, muscle mass, and myofiber diameters (Figures 4A–C and S4C). Noteworthy, there were no significant changes in Pax7 or MyoD mRNA expression (Figure S4D). In contrast, deleting Twist1 almost completely rescued the muscle wasting phenotype (Figures 4A–C and S4C). Deletion of Twist1 also blunted expression of MuRF1, Atrogin1, and Mstn as well as Smad2 phosphorylation (Figures S4B and S4E), indicating that Twist1 deletion in satellite cells is sufficient to confer resistance to cancer cachexia. This scenario was further reflected in survival, as all vehicle-treated mTwist1KO;ER mice died within 46 days, whereas more than 85% of Tam-treated mTwist1KO;ER mice survived to 60 days, at which time point they were scarified because of tumor burden (Figure 4D). A similar protection against muscle cachexia and associated lethality was observed in Tam-treated mTwist1KO;ER mice inoculated with B16 melanoma cells, which also retained their capability to form tumors in syngeneic mice (Figures 4E–H, S4F, S4G). Thus, selective blockade of Twist1 in satellite cells suppresses muscle cachexia and prolongs survival in different mouse models of cancer cachexia.
Figure 4. Twist1 Mediates Cancer-Induced Muscle Cachexia.

(A–D) Three-month-old mTwist1KO;ER mice were treated with vehicle or Tam and then inoculated with KP1 or KP2 cells. Carcass weight (calculated by subtracting tumor weight from the total weight) (A), GC muscle weight per BW (B), and GC muscle cross-sectional areas (C) were measured 30 day following cell inoculation (n= 6). Kaplan Meier survival analysis was conducted using 12 mice for each group. p< 0.005 by log-rank test for significance (D). Scale bars: 25 μM.
(E–H) Three-month-old mTwist1KO;ER mice were treated with vehicle or Tam and then inoculated with B16 cells (n= 6). Carcass weight (E), GC weight per BW (F), and GC muscle cross-sectional areas (G) were examined 21 days following cell inoculation. Kaplan-Meier survival analysis was conducted using 12 mice for each group. p< 0.005 by log-rank test for significance (H). Scale bars: 25 μM.
Data in A, B, E, and F are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ns, not significant. See also Figure S4.
Activin A Induces Muscle Twist1 Expression During Cachexia
We and others have shown that elevated levels of circulating Act-A in cancer patients are associated with severe cachexia and poor outcome (Fearon et al., 2012; Han et al., 2013; Loumaye et al., 2015; Sartori et al., 2014). In keeping with these findings, we detected a significant increase in circulating Act-A levels in several cachectic mouse models, including transgenic KP53 mice and wild-type mice bearing KP1, KP2, or B16 tumors (Figures 5A, S5A, S5D). Depletion of Act-A in KP1 and KP2 cells blunted the increase in muscle Twist1 expression, and this was accompanied by decreased expression of Mstn and Smad2 phosphorylation (Figure 5B). Most importantly, knockdown of Act-A suppressed muscle cachexia without affecting tumor growth (Figures 5C, 5D, S5B, S5C). Similar results were obtained using B16 cells deleted of Act-A by CRISPR/CAS9 (Figures S5D–H), lending further support to the notion that tumor-derived Act-A triggers muscle Twist1 expression during muscle cachexia.
Figure 5. Tumor-Derived Act-A Induces Muscle Twist1 Expression.

(A) Circulating Act-A concentrations in KP53 and control mice were assessed by ELISA (n= 22 to 26).
(B–D) Three-month-old wild-type mice were inoculated with KP1 or KP2 cells stably expressing control (sh.NS) or shRNA targeting Act-A (sh.Act-A) (n= 6). Muscle Twist1 and Mstn expression and Smad2 phosphorylation (B), GC muscle weight per BW (C), and GC muscle cross-sectional areas (D) were examined 30 days following cell inoculation. Scale bars: 25 μM.
(E) Primary satellite cells from Smad4fl/fl (Smad4KO) or wild-type mice were transduced with Ad.Cre (n= 6) and then treated with or without Act-A for 48 hr. Cells were then analyzed for Twist1 expression by immunoblotting and RT-PCR.
(F) Skeletal muscle microbiopsies from non-cachectic (n= 19) or cachectic (n= 16) cancer patients were analyzed for TWIST1 mRNA expression by RT-PCR.
(G–I) Three-month-old SCID mice were inoculated with MiaPaca2 or Suit2 cells (n= 6). Carcass weight (G), GC muscle weight per BW (H), and expression of muscle wasting markers were examined 21 days following cell inoculation.
Data in A, B, C, E, F, G, H, and I are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p< 0.001; ns, not significant. See also Figure S5.
To investigate how Act-A induces muscle Twist1 expression, we focused our attention on Smad signaling, which has been shown to play an essential role in muscle cachexia (Cohen et al., 2015; Han et al., 2013; Sartori et al., 2014). To probe this possibility, primary satellite cells from hindlimb muscles of mice harboring a conditional Smad4 allele were transduced with Ad.Cre before being challenged with Act-A. We observed that Act-A treatment induced a marked increase in Twist1 protein and mRNA expression, and this effect was suppressed by deletion of Smad4 (Figure 5E). Similar results were obtained for Mstn (Figure S5I), which signals through the same Smad signaling pathway as Act-A. A ChIp experiment using control and Smad4-deleted satellite cells demonstrated that both Act-A and Mstn induced binding of Smad4 to an Activin-responsive element (ARE) present within the Twist1 promoter (Figure S5J), emphasizing Twist1 as a Smad target gene. When taken together with our earlier observations that Twist1 activates expression of MuRF1 and Atrogin1, these findings suggest that Twist1 acts downstream of Smad signaling to mediate muscle protein breakdown in response to Act-A/Mstn signaling.
Severe cachexia with weight loss more than 10% results in general muscle weakness, leading to impairment of normal activities and eventually death due to respiratory failure (Cohen et al., 2015; Fearon et al., 2012; Tisdale, 2009). To attest to the clinical relevance of our findings, we assessed Twist1 expression in a cohort of cancer patients whose muscle mass and circulating Act-A levels were studied in the context of our previous clinical investigation of the etiology of muscle cachexia. In that previous clinical study, we have shown that higher circulating Act-A concentrations were associated with severe muscle loss and low quality of life (Loumaye et al., 2015). Using available skeletal muscle microbiopsies from 35 patients within this cohort, we observed a significant increase in muscle Twist1 expression in cachectic cancer patients as compared to non-cachectic cancer patients (Figure 5F). In efforts to substantiate this observation in vivo, we found that inoculating two human cancer cell lines derived from PDAC patients (i.e., MiaPaca2 and Suit2) to SCID mice induced muscle cachexia, and this was associated with increased expression of Twist1, MuRF1, Atrogin1, and Mstn (Figures 5G–I). These data, combined with the findings that Twist1 mediates muscle wasting in mouse models of PDAC, implicate a potential role of Twist1 in human cancer-associated cachexia.
Pharmacological Inhibition of Twist1 Suppresses Cachexia
Cancer-driven muscle cachexia reduces quality and length of life of cancer patients, and remains an unmet medical need. Therapeutic progress has been impeded by the marked heterogeneity of mediators and signaling pathways both within and between model systems, likely because most cachexia-oriented studies had relied heavily on artificial cancer systems involving inoculated cancer cells (Cohen et al., 2015; Fearon et al., 2012; Tisdale, 2009). The findings that KP53 mice develop severe muscle cachexia similar to human PDAC patients provided us with a unique opportunity to model the therapeutic efficacy of inhibiting Twist1 activity under autochthonous PDAC conditions. Accordingly, we sought to conduct experiments using JQ1, a small-molecule that interferes with Brd4-mediated recruitment of Twist1 to the transcriptional machinery (Shi et al., 2014). We initially performed specificity experiments using mTwist1ER mice, which display intrinsic muscle atrophy entirely provoked by Twist1 overexpression. Interestingly, administration of JQ1 to Tam-treated mTwist1ER mice was able to reverse the muscle wasting phenotype following 6 weeks of treatment, as evidenced by the normalization of body weight, grip strength, muscle mass, and myofiber size (Figures S6A–D). Remarkably, there were no effects of JQ1 on body weight or muscle physiology in the absence of Tam (Figures S6A–D), suggesting that JQ1 might block muscle atrophy through inhibition of the activity of Twist1 rather than the activity of another Brd4-associated transcription factor, such as c-Myc (Delmore et al., 2011). Of note, although JQ1 treatment did not alter muscle Twist1 expression, it did blunt Twist1 binding to the MuRF1, Atrogin1, and Mstn promoters (Figures S6E and S6F), further attesting to the ability of JQ1 to inhibit Twist1 activity in muscle.
Next, we found that JQ1 administration to cachectic KP53 mice prevented weight loss, and this was accompanied with profound improvement in grip strength, muscle mass, myofiber size, and survival (Figures 6A–E and S6G). Because of these protective effects of JQ1, we extended our analysis to assess possible inhibitory effects of JQ1 on PDAC growth. To facilitate this analysis, we generated KP53 mice harboring a Cre-inducible luciferase allele (Cheung et al., 2008) (KP53Luc) to monitor PDAC growth behaviors in live animals. When JQ1 completely reversed cachexia in KP53Luc mice following 3 weeks of treatment, there was no significant decrease in PDAC tumor volumes (Figures 6F and 6G), indicating that cachexia regression was not primarily due to tumor shrinkage. However, maintaining JQ1 treatment beyond the complete remission from cachexia prevented further tumor growth (Figures 6F and 6G). In light of previous data that Twist1 is required for KrasG12D-driven PDAC (Lee and Bar-Sagi, 2010), we surmised that prolonged JQ1 treatment might constrain PDAC growth by inhibiting Twist1 activity in the pancreatic epithelium. In fact, using KP53 mice with pancreas-specific Twist1 deletion (KP53T), we were not able to detect any signs of invasive PDAC or cachexia over a period of 30 weeks, time at which 80% of KP53 mice succumbed to PDAC (Figures S6H and S6I).
Figure 6. Pharmacological Inhibition of Twist1 Suppresses Muscle Cachexia.

(A–E) KP53 or control mice were treated with JQ1 for 6 weeks (n= 10 or 12). Body weight (A), grip strength (B), GC muscle weight per BW (C), and GC muscle cross-sectional areas (D) were analyzed up to 6 weeks of treatment. Kaplan Meier survival was analyzed using 12 mice per group. p< 0.005 by log-rank test for significance (E). Scale bars: 25 μM.
(F and G) KP53Luc mice were treated with JQ1 for 3 or 6 weeks (n= 6). Tumor volumes in live mice were analyzed by IVIS bioluminescence (F). PDAC tumor weights were measured following 3 or 6 weeks of treatment (G).
Data in A, B, and C are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p< 0.001; ns, not significant. See also Figure S6.
Because Twist1 has been shown to promote KrasG12D-driven PDAC by antagonizing p16Ink4a (Lee and Bar-Sagi, 2010), we sought to conduct pharmacological studies using KrasG12D mice deleted of Twist1 and p16Ink4a (hereafter termed KP16LucT) instead of Trp53 to firmly establish that JQ1 can actually reverse muscle cachexia without suppressing tumor growth. To achieve p16Ink4a deletion, we used p16Ink4a-Luc mice in which the endogenous p16Ink4a gene was replaced by luciferase, allowing for non-invasive assessment of tumor growth in live animals, as p16Ink4a is expressed only in tumors in young mice (Burd et al., 2013). As we anticipated, KP16LucT mice formed PDAC tumor with onset similar to that of KP16Luc mice (Figure S7A). Importantly, although treating KP16LucT mice with JQ1 for 6 weeks almost completely blocked muscle cachexia and prolonged survival, it was unable to dampen PDAC tumor growth (Figures 7A–F and S7B), validating our earlier observation that suppression of muscle cachexia by JQ1 did not occur as a result of tumor growth inhibition. Of note, although JQ1 stimulation did not alter Twist1 expression or circulating Act-A levels, it did suppress Twist1 binding to the MuRF1, Atrogin1, and Mstn promoters (Figures S7C–E). Collectively, these results demonstrate that pharmacological inhibition of Twist1 can afford substantial protection against PDAC-associated cachexia, ultimately preserving muscle mass and function to an extent that improves overall survival.
Figure 7. Pharmacological Inhibition of Twist1 Suppresses Muscle Cachexia Without Affecting Tumor Growth.

(A–D) KP53 or KP16LucT mice were treated with JQ1 for 6 weeks (n= 6 or 12). Body weight (A), GC muscle weight per BW (B), and GC muscle cross-sectional areas (C) were analyzed up to 6 weeks of treatment. Kaplan Meier survival was analyzed using 12 mice for each group (D). p< 0.005 by log-rank test for significance. Scale bars: 25 μM.
(E and F) KP53Luc or KP16LucT mice were treated with JQ1 for 6 weeks (n= 6). Tumor volumes in live animals were analyzed by IVIS bioluminescence (E) and PDAC tumor weights (F) were measured following 6 weeks of treatment.
Data in A and B are expressed mean ± SEM. *p< 0.05; **p< 0.01; ns, not significant. See also Figure S7.
DISCUSSION
Cancer cachexia occurs in more than 80% of patients with advanced stages of pancreatic, gastrointestinal, and lung cancers and accounts for nearly 30% of cancer-related death, yet there is currently no effective therapy (Cohen et al., 2015; Fearon et al., 2012; Sartori et al., 2014; Tisdale, 2009). Therefore, a deeper understanding of the underlying mechanisms that lead to the complex metabolic defects of cachexia, coupled with effective treatment options, would facilitate management of muscle mass deterioration and ultimately improve survival in cancer patients. Here, we show that Twist1 plays a key role in cancer-associated muscle cachexia. We serendipitously found that conditional overexpression of Twist1 in either mesenchymal stem cells or muscle progenitor cells affected muscle mass, albeit without altering expression of master myogenic markers. We went on to show that overexpression of Twist1 in muscle progenitor cells in adult mice was also able to trigger muscle atrophy that was characterized by constitutive muscle protein degradation, providing us with an outstanding platform for exploring a potential implication of Twist1 in muscle cachexia. In fact, we detected a significant increase in endogenous Twist1 expression in skeletal muscles from several mouse models of cancer cachexia, including PDAC, colon carcinoma, lung carcinoma, and melanoma. Remarkably, conditional deletion of Twist1 in satellite cells was sufficient to suppress cancer-induced muscle wasting, and this protective effect was mirrored by a striking improvement in overall survival. Likewise, pharmacological inhibition of Twist1 also afforded substantial protection from muscle cachexia and associated death, highlighting Twist1 as an attractive drug candidate for treatment of cancer-associated muscle cachexia. Thus, elaboration of potent strategies to inactivate Twist1 should be an important therapeutic goal, not just to restore muscle mass needed to maintain normal activity and good quality of life, but also to prolong the lives of cancer patients. It would also be interesting to determine whether Twist1 drives muscle cachexia associated with other human diseases, such as AIDS, chronic kidney disease, or congestive heart failure.
In light of our findings, one would speculate that this muscle wasting mechanism might function under homeostatic conditions to fine-tune the rates of muscle mass accrual and breakdown depending on the phase of embryonic development or postnatal growth, physical activity, or aging. Alternatively, it is conceivable that Twist1 might function exclusively to integrate the muscle wasting processes under pathological conditions, such as occurs in cancer cachexia. Thus, the molecular framework that we propose gives rise to a number of areas for future studies to foster our understanding of the physiological and molecular mechanisms that impact muscle mass under health and disease conditions. Previous in vitro studies have shown that overexpression of Twist1 was able to inhibit MyoD activity and suppress myoblast differentiation in immortalized myoblastic cell systems (Hamamori et al., 1997; Spicer et al., 1996). However, subsequent studies revealed that Twist1 and MyoD are not mutually exclusive (Maestro et al., 1999), creating a conundrum as to whether indeed Twist1 represses MyoD expression. Interestingly, MyoD knockout mice did not display any discernable muscle phenotype (Rudnicki et al., 1992), further challenging the possibility that Twist1 inhibits myoblast differentiation through repression of MyoD. Consistent with this view, we found that conditional overexpression of Twist1 in satellite cells resulted in muscle atrophy without altering expression of MyoD or of any other master myogenic markers we tested. Although we cannot exclude the possibility that Twist1 may also block myoblast differentiation under specific circumstances, our gain- and loss-of function genetic approaches strongly suggest that Twist1 affects muscle mass at least by inducing muscle wasting, which proceeds through degradation of myofibrillar proteins.
Perhaps the most impressive finding in this study is the increase in Twist1 expression in muscle undergoing cachexia, which occurs irrespective of the cancer cachexia model interrogated. This scenario is clinically relevant, as high levels of Twist1 in skeletal muscle are associated with greater degrees of muscle cachexia in cancer patients. This finding raises the tantalizing possibility that Twist1 might serve as a marker to predict clinical outcome in patients with cancer cachexia. In our earlier clinical studies involving the same cohort of cancer patients (Loumaye et al., 2015), we found that elevated levels of circulating Act-A were strongly associated with severe muscle cachexia and high morbidity. Here, we extend this clinical finding and provide a mechanistic aspect by demonstrating that tumor-derived Act-A acts on satellite cells to induce expression of Twist1, which in turn drives synthesis of Mstn to trigger its own expression in myofibers, where it regulates expression of MuRF1 and Atrogin1 and thereby leads to global muscle atrophy. It is worth mentioning that our previous study showed slightly decreased levels of circulating Mstn in lung and colon cancer patients (Loumaye et al., 2015), but this observation does not appear to be in conflict with our new study showing increased Mstn expression specifically in muscle. We suggest that the increase in circulating Act-A observed in cancer patients might compensate for Mstn, since both factors activate the same signaling pathway leading to muscle loss. It would be very interesting to dissect in future studies the exact contribution of Mstn and Act-A to muscle cachexia in caner patients. Regardless of this provocative question, our findings suggest that Twist1 may not only function to integrate muscle wasting signaling in satellite cells, but also orchestrate a signaling program within myofibers to execute the muscle breakdown process once it has commenced. Beyond these mechanistic insights, the finding that Twist1 fulfills dual functions during muscle cachexia will likely leverage the design of bimodal therapeutic agents to prevent muscle loss if cancer is detected early enough and to restore normal muscle mass at the advanced stages of the disease.
Our study also provides the proof-of-principle that blocking Twist1 activity, either genetically or pharmacologically, can confer substantial protection against cancer-driven muscle cachexia and associated morbidity and mortality. Interestingly, both genetic and pharmacological approaches led to inhibition of PDAC growth in KP53 mice, supporting a role for Twist1 in PDAC progression in addition to mediating muscle cachexia. Nonetheless, although both genetic and pharmacological approaches were effective at reducing muscle wasting in KP16Luc mice, they failed to suppress PDAC formation and progression, suggesting that suppression of muscle cachexia through inhibition of Twist1 might occur independently of the shrinkage of the host tumor. Though these findings seem to confirm earlier observations that p16Ink4a functions downstream of Twist1 to promote PDAC formation and progression (Lee and Bar-Sagi, 2010), recent studies have revealed that Twist1 deletion was ineffective at suppressing PDAC driven by the combined expression of KrasG12D and gain-of-function mutant p53, and, therefore, questioning the contribution of Twist1 to PDAC development (Zheng et al., 2015). In our study, we found that Twist1 deletion in the pancreatic epithelium elicited either no effect or complete suppression of PDAC, depending on the genetic background (i.e., p16Ink4a versus Trp53). Thus, our genetic approaches seem to reconcile these previously incongruent observations, and further shed important insights into the complex roles of this pro-malignant transcription factor in PDAC, and perhaps in other human malignancies. Regardless of its involvement in PDAC, our study provides evidence that both genetic and pharmacological inhibition of Twist1 can confer resistance to muscle cachexia even without affecting PDAC growth, and thus dissociates the relative contributions of Twist1 to cancer progression and muscle cachexia.
Extensive epidemiological studies indicate that one in four of the general population will die from cancer, and cachexia affects the majority of patients with advanced malignancies. Sadly, few therapeutic options are currently available for preventing or delaying cancer cachexia (Cohen et al., 2015; Fearon et al., 2012; Sartori et al., 2014; Tisdale, 2009). Thus, by demonstrating that Twist1 plays a pivotal role in cancer-driven muscle cachexia and validating its clinical relevance, our findings open up new opportunities for effective therapeutic interventions to reverse muscle cachexia. Since pharmacological inhibition of Twist1 also dampens PDAC tumors with specific genetic alterations, targeting Twist1 could provide a dual strategy to curb both muscle cachexia and PDAC in a subset of patients. Finally, one could also envision combinatorial therapeutic strategies comprising Twist1 and Act-A/Mstn inhibitors, which could mitigate potential drug toxicity by lowering the dose needed for each medicine and combat the development of resistance. Of critical relevance, Act-A/Mstn receptor inhibitors and JQ1 or analogs are being tested in clinical trials for a variety of hematological malignancies, leveraging the concept that targeting the Act-A/Mstn/Twist1 axis holds promise for developing therapeutic strategies with immediate clinical applicability in fatal cancer cachexia.
STAR METHODS
KEY RESOURCES TABLE
| REAGENTS or RESOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Atrogin1/Fbx32 | Abcam | ab168372 |
| BrdU | Cell Signaling | 52925 |
| Cleaved caspase 3 | Cell Signaling | 9661 |
| Cytokeratin 19 | Abcam | ab52625 |
| Dystrophin | Abcam | ab15277 |
| elF3-f | Abcam | ab64177 |
| MHC | Abcam | ab71808 |
| Myostatin | Abcam | ab71808 |
| Myostatin | R&D Systems | AF788 |
| MuRF1 | Cell Signaling | 4305 |
| Myc tag | Cell Signaling | 22765 |
| Smad4 | Santa Cruz | sc-7966 |
| Smad2 | Cell Signaling | 5339 |
| pSmad2 | Cell Signaling | 3108 |
| Twist1 | Santa Cruz | sc-81417 |
| Pax7 | Abcam | Ab92317 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Activin A | R&D Systems | 338-AC-050 |
| bFGF | Sigma-Aldrich | F3685 |
| Collagenase D | Sigma-Aldrich | 11088858001 |
| Collagenase type 1 | Sigma-Aldrich | SCR103 |
| Dispase II | Sigma-Aldrich | 4942078001 |
| D-luciferin | Perkin Elmer | 122799 |
| JQ1 | Sigma-Aldrich | SML-1524 |
| Myostatin | R&D Systems | 788-GB-010 |
| puromycin | Sigma-Aldrich | P9620 |
| Tamoxifen | Sigma-Aldrich | T5648 |
| Critical Commercial Assays | ||
| Activin A ELISA kit | R&D Systems | DAC00B |
| SURVEYOR Mutation Detection Kit | Integrated DNA Technologies | 706025 |
| Recombinant DNA | ||
| Adenovirus-CMV.Cre (Ad.Cre) | Baylor College | Ad5-CMV-Cre |
| Atrogin1-Luc | This study | N/A |
| Mstn-Luc | This study | N/A |
| MuRF1-Luc | This study | N/A |
| pBABE-puro-mTwist1 | Addgene | 1783 |
| pGIPZ-sh.Activin | Dharmacon | EG16323 |
| pGIPZ-sh.control | Dharmacon | RHS4346 |
| LentiCISPRv2 | Addgene | 52961 |
| pSpCas9(BB)-2A-Puro (PX459) | Addgene | 62988 |
| Experimental Models: Cell Lines | ||
| B16 | ATCC | CRL-6322 |
| Suit2 | ATCC | CRL-1687 |
| MiaPaca2 | ATCC | CRL-1420 |
| CT-26 | ATCC | CRL-2638 |
| KP2 | This study | N/A |
| KP1 | This study | N/A |
| LLC | ATCC | CRL-1642 |
| Experimental Models: Organisms/Strains | ||
| CAG-CreERT2 | Jackson Lab. | 004682 |
| CAG-Loxp-CAT-Loxp-Twist1 | Jackson Lab. | 018543 |
| CAG-Loxp-Stop-Loxp-Luciferase | NCI | 01XAC |
| Loxp-Stop-Loxp-KrasG12D | NCI | 01XJ6 |
| NOD/SCID | Jackson Lab. | 001303 |
| p16Ink4A-Luciferase | Dr. Sharpless | Burd et al., 2013 |
| Pax7.Cre | Jackson Lab. | 010530 |
| Pax7.CreERT2 | Jackson Lab. | 012476 |
| Pdx1.Cre | NCI | 01XL5 |
| Prrx1.Cre | Jackson Lab. | 005584 |
| MCK.Cre | Jackson Lab. | 006475 |
| Trp53.Loxp/Loxp | Jackson Lab. | 008462 |
| Twist1.Loxp/Loxp | Dr. Xu | Baylor College of Medicine |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact Azeddine Atfi, azeddiine.atfi@inserm.fr; aatfi@umc.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Mississippi Medical Center.
CAG-Loxp-CAT-Loxp-Twist1 (LCL-Twist1), CAG-Loxp-Stop-Loxp-Luciferase (LSL-Luc), Trp53.Loxp/Loxp (Trp53fl/fl), CAG.CreERT2, Pax7.Cre, Pax7.CreERT2 Prrx1.Cre, and MCK.Cre mice were obtained from Jackson Laboratory. Loxp-Stop-Loxp-KrasG12D (LSL.KrasG12D) and Pdx1.Cre were obtained from the NCI Mouse Repository. Twist1.Loxp/Loxp (Twist1fl/fl) and p16Ink4A-Luciferase (p16-Luc) were described in previous studies from other groups (Burd et al., 2013; Xu et al., 2013)
LCL-Twist1 mice harbor a floxed CAT (chloramphenicol acetyltransferase) gene that serves as silencer (STOP) in the absence of Cre recombinase (Connerney et al., 2006). To generate mice with conditional overexpression of Twist1 in mesenchymal stem cells, LCL-Twist1 mice were crossbred with Prrx1.Cre mice. To generate mice with conditional overexpression of Twist1 in muscle progenitor cells (referred in the manuscript as mTwist1), LCL-Twist1 mice were crossbred with Pax7.Cre mice. To generate mice with Tam-inducible expression of Twist1 in muscle progenitor cells (referred in the manuscript as mTwist1ER), LCL-Twist1 mice were crossbred with Pax7.CreERT2 mice. To generate mice with ubiquitous Tam-controlled Twist1 expression (referred in the manuscript as gTwist1ER), LCL-Twist1 mice were crossbred with CAG.CreERT2 mice. To generate mice with constitutive overexpression of Twist1 in myofiber, LCL-Twist1 mice were crossbred with MCK.Cre mice. To generate mice with Tam-inducible deletion of Twist1 in muscle progenitor cells (referred in the manuscript as mTwist1KO;ER), Twist1fl/fl mice were crossbred with Pax7.CreERT2. To generate mice with constitutive muscle-specific deletion of Twist1 in muscle progenitor cells (referred in the manuscript as mTwist1KO), Twist1fl/fl mice were crossbred with Pax7.Cre.
The PDAC mouse models were generated through successive crossbreeding of Pdx1.Cre, LSL-KrasG12D, Trp53fl/fl, LSL-Luc, Twist1fl/fl, and p16-Luc mice as appropriate. Their genotypes are as follow:
KP53: LSL-KrasG12D;Trp53fl/+;Pdx1.Cre
KP53Luc: LSL-KrasG12D;Trp53fl/+;LSL-Luc;Pdx1.Cre
KP53T: LSL-KrasG12D;Trp53fl/+;Twist1fl/fl;Pdx1.Cre
KP16Luc: LSL-KrasG12D;p16Ink4a-Luc;Pdx1.Cre
KP16LucT: LSL-KrasG12D;p16Ink4a-Luc;Twist1fl/fl;Pdx1.Cre
All mice were maintained on a mixed C57BL/6 and FVB/N genetic backgrounds, except mTwist1KO;ER mice, which were backcrossed to the C57BL/6 background for more than 8 generations. Mice were maintained in 12 hr light:dark cycles (6:00 am to 6:00 pm) at 22 °C and fed a standard rodent chow di et.
Clinical Samples
Patients were enrolled in ACTICA study, a cross-sectional study aimed at assessing cachexia in patients diagnosed with colorectal or lung cancer. The study was performed at the Cliniques Universitaires Saint-Luc, Brussels, Belgium from January 2012 to March 2014. The IRB protocol was approved by the local ethical committee of the Université Catholique de Louvain and written consent was given prior to entry into the study. The inclusion and exclusion criteria have been precisely described previously in the original paper (Loumaye et al., 2015). Cachexia was defined, according to the definition proposed by Fearon et al. (Fearon et al., 2011), as an involuntary weight loss >5 % over the past 6 months or weight loss >2% and BMI <20 kg/m2 or weight loss >2% and low muscularity. Among 152 patients enrolled in the study, a skeletal muscle microbiopsy was performed in 35 patients under general anesthesia, just before the surgery for cancer and before any other therapeutic intervention. The microbiopsies were taken from vastus lateralis of the quadriceps, with a 14 Gauge true-cut biopsy needle (Bard Magnum Biopsy gun; Bard, Inc.). The muscle samples were cleaned of gross blood contamination and fat or fibrous tissue prior to being frozen in liquid nitrogen and stored at −80°C until RNA extraction.
Below is a table that describes the patients enrolled in the study.
| Cancer Patients | Non-Cachectic | Cachectic |
|---|---|---|
| n | 19 | 16 |
| Age | 67 (44–83) | 69 (40–95) |
| Sex ratio F/M | 44/56 | 50/50 |
| Primary/Recurrent (%) | 78/22 | 88/12 |
| Weight loss (%) | 0 (0–3) | 7 (2–13) p<0.0001 |
Cell Lines
C2C12, HEK293T, MiaPaca2, Suit2, B16, CT-26, and LLC cells were obtained for the American Type Culture Collection (ATCC). They were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics.
Primary satellite cells were isolated from 3-month-old wild-type, mTwist1ER, or Smad4fl/fl mice using a standard procedure. GC muscle was dissected, washed with PBS, minced with sterile razor blades, and digested for 45 min at 37 °C in PBS containing 10mM CaCl2, 2.4U/ml Dispase II, and 1.5U/ml Collagenase D. Digested tissue was filtered through a 40-μM strainer, centrifuged at 600g for 5 min, and cultured in Ham’s F-10 nutrient mixture (Gibco) containing 20% FBS (Atlanta Biologicals) supplemented with 2.5ng/ml bFGF. Cells were allowed to recover for 72 hr before being treated with 50ng/ml Act-A or Mstn for 48 hr. To delete Smad4, cells were infected with Ad.Cre and allowed to recover for 5 days before being treated with 50ng/ml Act-A or Mstn for 48 hr. Satellite cells carrying mTwist1ER were treated with Tam at 10mM for 48 hr.
Single myofibers were isolated from GC muscles as previously described (Rosenblatt et al., 1995) with minor modifications. Briefly, muscles were gently dissected and incubated with rotation in isolation medium containing 0.2% Collagenase type 1 in DMEM for 2.5 hr at 37 °C in an atmosphere of 5% CO2. After digestion, single myofibers were isolated under a dissecting microscope by gentle trituration using a plastic pipette, washed three times in DMEM, and transferred into a matrigel-coated 24-well plate using a fire-polished Pasteur pipette. The isolated myofibers were grown on matrigel-coated in DMEM supplemented with 5% horse serum in an atmosphere of 5% CO at 37 °C. After 24 hr cultivation period, myofib ers were culture in conditioned media of satellite cells isolated from Tam-treated mTwist1KO;ER or control mice in the presence or absence of Mstn neutralizing antibody for 48 hr. Myofibers were then analyzed for Twist1 protein expression by immunoblotting.
Pancreatic cancer cell lines KP1 and KP2 were prepared from KP53 mice. Briefly, freshly isolated tumor specimens were minced, digested with Dispase II and Collagenase D for 1 hr at 37°C, washed, resuspended in RPMI containing 20% FCS, and seeded on fibronectin-coated plates. Cells were passaged by trypsinization. For stable depletion, KP1 and KP2 cells were infected with pGIPZ lentiviruses encoding control (sh.NS) or shRNA targeting Activin (sh.Act) and selected with puromycin. Resistant colonies were then pooled and expanded as single populations.
To generate B16 cells with Act-A inactivation, the gRNA/CAS9 plasmid (PX459) was transfected into cells using Lipofectamine 2000 (Invitrogen). Cells were then selected with 10μg/ml puromycin and individual clones were screened for deletion by mutation-specific PCR and SURVEYOR assay according to the manufacturer’s instructions (Integrated DNA Technologies). Several clones deleted for Act-A were pooled and used for inoculation experiments.
METHOD DETAILS
Plasmid Constructs
pBABE-puro-mTwist1 was obtained from Addgene. pGIPZ-sh.control (sh.NS, non silencing) and pGIPZ-sh.Activin (sh.Act) were purchased from Dharmacon. To generate the MuRF1-Luc, Atrogin1-Luc, and Mstn-Luc reporters, genomic fragments (600 bp for MuRF1, 712 bp for Atrogin1, 2158 bp for Mstn) upstream of the 5′ UTR of their genes were amplified by the Genomic-GC PCR amplification kit (BD Biosciences) using murine genomic DNA obtained from C2C12 cells. Unique KpnI and XhoI sites were incorporated at the 5′ and 3′ ends of the sequence, respectively, to simplify directional cloning into KpnI and XhoI sites in the reporter plasmid, pGL3-basic (Promega). Introduction of mutations into the three E-boxes of the MuRF1 promoter (EM1: −143 bp; EM2: −66 bp; E-M3: −44 bp), the unique E-box of the Atrogin1 promoter (EA: −79 bp), or the two E-boxes in the Mstn promoter (EMS1: −1,134 bp; EMS2: −1,755 bp) were generated by PCR using the QuickChange Site-Directed Mutagenesis kit according to the manufacturer’s instructions (Stratagene). All cloned DNA fragments and their corresponding mutants were checked by sequencing.
The following primers were used:
MuRF1-For 5′-GGGTACCGCAGCAAGCCCCTTTACCAC-3′
MurRF1-Rev 5′-GCTCGAGGCTGCACCTCTGTCACATGA-3′
Atrogin1-For 5′-GGGTACCCAGAGCGCGGACGCGACCGA-3′
Atrogin1-Rev 5′-GCTCGAGTGCACCCCTACCCGTCCCCA-3′
Mstn-For 5′-GGGTACCGTCAGAGCTGAGTGAATGGC-3′
Mstn-Rev 5′-GCTCGAGCGAGAGACAACCTGCCACGCCAGG-3′
For Act-A gene editing, the PX459 plasmid expressing the CRISPR/Cas9 system was used for cloning following recently published protocols (Ran et al., 2013). Specific gRNAs were designed using CRISPR Design. The following Act-A specific gRNA (PAM sequence underlined) was used in experiments: GGACTGCCCGTCCTGTGCGCTGG. For gRNA cloning, pSpCas9(BB)-2A-Puro vector was digested and ligated with annealed oligonucleotides with complementary fragments.
For Mstn genome editing, a specific gRNA was designed using CRISPR Design: AATCCTCAGTAAGCTGCGCCTGG (PAM sequence underlined). The gRNA and its complementary strand were annealed and then subcloned into LentiCRISPRv2 as described previously (Sanjana et al., 2014; Shalem et al., 2014).
Lentivirus Production
Lentiviruses were produced by co-transfection of HEK293T cells with lentiviral backbone constructs and the packaging vectors delta8.2 and VSV-G using X-tremeGENE9 (Roche). Supernatant was collected 72 hr post-transfection, concentrated by ultracentrifugation at 25,000 RPM for 90 minutes and resuspended in OptiMEM (Gibco).
Mice Treatment and Analysis
Mice were divided into treatment groups randomly while satisfying the criteria that the average body weight in each group would be about the same. Formation of PDAC in all cachectic mice enrolled in the study was confirmed using pancreatic tissue sections stained with hematoxylin and eosin (H&E) or immunostained with an antibody to the ductal marker Cytokeratin 19 (see blow). For intramuscular injection, tibialis anterior muscle of P12 wild-type C57BL/6 male mice was injected with 50 μl of lentiviral preparation or saline solution. The injection was repeated on P15 and P18. Tamoxifen (Tam) was dissolved in sterile corn oil and administered subcutaneously at 100mg/kg per injection for five consecutive days. JQ1 was dissolved in DMSO and administered by intraperitoneal injection at 25mg/kg body weight every other day. This concentration of JQ1 was chosen because it prevented muscle cachexia in KP53 mice during the first three weeks of treatment without affecting tumor growth. D-luciferin was dissolved in PBS and administered by intraperitoneal injection at 75mg/kg before imaging. Control mice received solvent only. To evaluate the changes in muscle mass, the weights of the GC, EDL, Sol, and TA muscles were normalized by the body weight (BW). To measure the levels of circulating Act-A, serum was analyzed using an ELISA kit (R&D systems).
To assess muscle wasting in the xenotransplantation studies, 3-month-old NOD-SCID, C57BL/6, or Balb/C were used. A total of 5 × 106 or 2 × 106 cells were injected subcutaneously. For pharmacological studies, mice with established disease were divided into treatment groups and treated every other day with JQ1. For cachexia experiments, PDAC mice were enrolled into experiments as soon as they exhibit weight stagnation or beginning of weight loss. For in vivo imaging, mice were anaesthetized by isoflurane inhalation and imaged after injection of D-luciferin using a Xenogen IVIS Spectrum (Caliper Life Sciences, Hopkinton, MA). Tumor burden was assessed by serial bioluminescence imaging and confirmed by measuring the tumor weight. Carcass weight was calculated by subtracting tumor weight from the total weight.
Grip Strength Measurement
Forelimb grip strength was assessed at the end of the study following published protocols (White et al., 2013). Each mouse was allowed to grab a bar attached to a force transducer as it was pulled by the tail horizontally away from the bar (DFX II Series force gauge; Chatillon). The results from 5 repetitions with a 30-s pause between each were averaged to determine the grip strength of each mouse.
Metabolic Phenotyping
Whole body energy metabolism was evaluated using a Comprehensive Lab Animal Monitoring System (CLAMS, Accuscan Instruments). Mice were acclimated in the metabolic chambers for 2 days before starting the experiment. CO2 and O2 levels were collected for each mouse over a period of 4 days and normalized to total body weight. Movement and food intake were measured at regular intervals.
Real-Time PCR (RT-PCR)
Total RNA was extracted from frozen mice tissue samples or cultured cells using TRIzol, purified with QIAGEN RNeasy minicolumns, and reverse transcribed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). The resulting cDNA was analyzed by RT-PCR. Briefly, 25 ng cDNA and 150 nmol of each primer were mixed with SYBR GreenER qPCR SuperMix (Invitrogen). Reactions were performed in the 96-well format using an ABI PRISM 7900HT instrument (Applied Biosystems). Relative mRNA levels were calculated using the comparative CT method and normalized to Gapdh mRNA.
For clinical samples, total RNA was extracted from frozen muscle samples using TRIzol and reverse transcribed using a Revert Aid H Minus First Strand cDNA synthesis kit (Thermo Scientific). Then, the cDNA was analyzed by RT-PCR. Briefly, 6.6 ng cDNA and 10nM of each primer were mixed with the mix Fast Start Universal SYBR Green Master with Rox (Roche). Reactions were performed in 48-well format using a Step One instrument (Applied Biosystems). Relative TWIST1 mRNA levels were calculated using the comparative CT method and normalized to GAPDH mRNA.
Primers used for mice tissue or cell samples:
Twist1-For 5′-GCAAGATCATCCCCACGCTG-3′
Twist1-Rev 5′-GCAGGACCTGGTAGAGGAAG-3′
MuRF1-For 5′-ACCTGCTGGTGGAAAACATC-3′
MuRF1-Rev 5′-CTTCGTGTTCCTTGCACATC-3′
Atrogin1-For 5′-GCAAACACTGCCACATTCTCTC-3′
Atrogin1-Rev 5′-CTTGAGGGGAAAGTGAGACG-3′
MyoD- For 5′-GATGGCATGATGGATTACAG-3′
MyoD-Rev 5′-GCAGTCGATCTCTCAAAGCAC-3′
Myf5-For 5′-GCATTGTGGATCGGATCACG-3′
Myf5-Rev 5′-GAGTGTCCTTGAGGATGCCTG-3′
myogenin-For 5′-GCCAAGCATTTGCAGTGGATC-3′
myogenin-Rev 5′-TCAGGCGGCAGCTTTACAAAC-3′
Mstn-For 5′-AGTGGATCTAAATGAGGGCAGT-3′
Mstn-Rev 5′-GTTTCCAGGCGCAGCTTAC-3′
Mck-For 5′-ACAGCACAGACAGACACTCA-3′
Mck-Rev 5′-GTGTCTCCTTATCGCGAAGC-3′
MyHC-For 5′-ACTGAGGAAGACCGCAAGAA-3′
MyHC-Rev 5′-TTTGGCCAGGTTGACATTGG-3′
Pax7-For 5′-CCACAGCTTCTCCAGCTACTCTG-3′
Pax7-Rev 5′-CACTCGGGTTGCTAAGGATGCTC-3′
Gapdh-For 5′-CACCATCTTCCAGGAGCGAG-3′
Gapdh-Rev 5′-CACCATCTTCCAGGAGCGAG-3′
Primers used for human samples:
TWIST1-For 5′-AGCTGAGCAAGATTCAGACC-3′
TWIST1-Rev 5′-AGCTTGCCATCTTGGAGTC-3′
GAPDH-For 5′-CGCTGAGTACGTGGAGTC-3′
GAPDH-Rev 5′-GCAGGAGGCATTGCTGATGA-3′
Immunoblotting
Cell extracts were prepared in lysis buffer containing 50mM Tris HCl (pH = 8.0), 120mM NaCl, 5mM EDTA, 1% Igepal, protease inhibitors (Roche Diagnostics), and phosphatase inhibitors (Calbiochem). Protein concentrations were determined by using the Bradford reagent (Sigma-Aldrich) and samples were denatured using SDS sample buffer (Invitrogen). Samples were loaded into a NuPAGE Bis-Tris Gel (Invitrogen) and separated by electrophoreses at 200 V. The gels were then transferred onto a nitrocellulose membrane (BioRad) by a wet transfer system (BioRad) and blocked by incubation with 5% dry milk in TBST (TBS with 0.2% Tween20). Membranes were probed with the primary antibody for 2 hr at room temperature in the blocking buffer, washed with TBST, and incubated with the peroxidase-conjugated secondary antibody. Enhanced chemiluminescence (ECL) western blotting substrates (Pierce) were used for visualization of the results. Primary antibodies against the following antigens were used at specified dilutions: Twist1, 1:500; Smad4, 1:1,000; pSmad2, 1:1,000; Smad2, 1:1,000; Ubiquitin, 1:500; Mstn, 1:200; MuRF1, 1:200; Atrogin1, 1:500; MHC, 1:500; elF3-f, 1:200, Pax7, 1:500; β-Actin, 1:5,000
Immunofluorescence and Histology
Tissue samples were fixed in 10% formalin and embedded in paraffin. Tissue sections were deparaffinised with xylene and rehydrated in a graded series of ethanol. Antigen-retrieval was performed for 30 min at high temperature in citrate buffer. Then, slides were blocked and incubated overnight with anti-dystrophin (1:50), anti-Myc (1:100), anti-Twist1 (1:50), or IgG-matched isotype control antibody (negative control) at 4°C. Finally, slides were incubated with the secondary antibodies conjugated to Alexa-Fluor®568 or Alex-Fluor®448 and costained with DAPI. Slides were viewed on a fluorescence microscope.
For muscle or pancreatic tissue histology, formalin-paraffin embedded sections were stained with H&E. For pancreatic tissue immunohistochemistry, formalin-paraffin embedded sections were immunostained with anti-Cytokeratin 19 antibody (CK19, 1:100) followed by incubation with secondary antibody conjugated with peroxidase and revealed by DAB using standard techniques. Myofiber cross-sectional areas were examined in at least six animals for each determination and the myofibers diameters were quantified using at least 200 myofibers.
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed using the chromatin immunoprecipitation assay kit following the manufacturer’s instructions (Millipore). Briefly, chromatin was extracted from tissue or cell samples, sonicated, and immunoprecipitated with antibodies against Twist1 or Smad4 or isotype matched control IgG. PCR was run on the chromatin and the products were analyzed on a 2% agarose gel. Relative DNA binding was determined by quantitative PCR.
The following primers were used:
MuRF1, E1-For 5′-CGGAGACCAACTTCTGCAGG-3′
MuRF1, E1-Rev 5′-CAGGCTGCTGGCAGGGCTTG-3′
MuRF1, E2-For 5′-CGGCAGGGCAACAGCGATTG-3′
MuRF1, E2-Rev 5′-GAGCCCCAGGAGGGGACCAG-3′
MuRF1, E3-For 5′-CTGGTCCCCTCCTGGGGCTC-3′
MuRF1, E3-Rev 5′-CGACTGTCTCGCCCACCTGC-3′
Atrogin1, EA-For 5′-GCCTGGCCATGCGGCTTCAAG-3′
Atrogin1, EA-Rev 5′-GCCGCAAGAGCCGCCTGCTC-3′
Mstn, EMS1-For 5′-CACAGGATCAATTTCCTCTGAG-3′
Mstn, EMS1-Rev 5′-CCATAATGTACTAGATTTGC-3′
Mstn, EMS2-For 5′-CCCATTCGATCTTGTGGCTCC-3′
Mstn, EMS2-Rev 5′-GAGCAAGTCGCATGTAGAGG-3′
Twist1, SBE-For 5′-CGTCAGGCCAATGACACCGC-3′
Twist1, SBE-Rev 5′-CCGCAAGCGCGCCCCGTTCC-3′
Gapdh-For 5′-ATCCACGACGGACACATTGG-3′
Gapdh-Rev 5′-TGGTGCTGCCAAGGCTGTGG-3′
Luciferase Reporter Assay
C2C12 cells were plated in 6-well plates and transfected using Lipofectamine 2000. The pRL-SV40 plasmid (Promega) was cotransfected as a normalization control. Cells were then incubated for 24 hr with the transfection mixtures and allowed to recover for another 24 hr before measuring luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). Firefly Luciferase activity was normalized on the basis of Renilla luciferase expressed from the pRL-SV40 plasmid.
Statistical Analysis
Sample size for in vivo experiments was determined empirically using experimental data from other studies to ensure that the desired statistical power could be achieved (>85% confidence for a two-fold change in any given parameter at the P < 0.05 significance level). Significant differences between two groups were evaluated using a two tailed, unpaired t-test T. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant. All data are presented as mean ± SEM or as boxplots. The error bars (SEM) were derived from at least three independent biological replicates. Although the majority of the data presented have normal distribution and similar variance, we systematically examined variance using an F-test, and Mann–Whitney U-test or Student’s t-test employed accordingly. Kaplan–Meier survival curves were calculated using the survival time for each mouse from all treatment groups, and significance was determined by log-rank test.
Supplementary Material
Highlights.
Enforced expression of Twist1 triggers muscle atrophy
Twist1 expression is de-repressed in skeletal muscle undergoing cancer cachexia
Twist1 functions in Activin/Myostatin signaling to mediate muscle cachexia
Targeting of Twist1 prevents cancer-induced muscle cachexia and prolongs survival
Acknowledgments
We thank Drs. Xu K., Sharpless N., and Xu X. for providing Twist1fl/fl, p16Inkk4a-Luc, LSL.KrasG12D and Pdx1.Cre mice, respectively. We thank Dr. Hall’s lab members for help with phenotyping analyses. This work was supported by grants from the NIH (R01-AR059070 and R01-CA210911), Department of Defense (PR162051 and PR162051), and Association pour la Recherche sur le Cancer (ARC) to A.A., and the Cancer Plan, Belgian Ministry of Public Health, Belgian Foundation against Cancer, Saint-Luc Foundation, and Fonds de la Recherche Scientifique Médicale (FNRS-FRSM) to J-P.T.
Footnotes
AUTHOR CONTRIBUTIONS
P.P. and S.K. were responsible for the experimental design and data analysis. A.A., P.S., S.E., T.L.N, and S.O. were responsible for mice breeding, genotyping, and muscle preparation. P.S. and C.P. contributed to the in vitro experiments. M.S. contributed to muscle-related experiments. A.L. and J-P.T. collected human muscle samples and performed gene expression analysis. M.R., J-P.T., and A.A. wrote the manuscript with input from other authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–351. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Gene Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Cheung AF, Dupage MJ, Dong HK, Chen J, Jacks T. Regulated expression of a tumor-associated antigen reveals multiple levels of T-cell tolerance in a mouse model of lung cancer. Cancer Res. 2008;68:9459–9468. doi: 10.1158/0008-5472.CAN-08-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Muentener C, Mercado MA, Spicer DB. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dynam. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–495. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 2012;16:153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Gallot YS, Durieux AC, Castells J, Desgeorges MM, Vernus B, Plantureux L, Remond D, Jahnke VE, Lefai E, Dardevet D, et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res. 2014;74:7344–7356. doi: 10.1158/0008-5472.CAN-14-0057. [DOI] [PubMed] [Google Scholar]
- Hamamori Y, Wu HY, Sartorelli V, Kedes L. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol Cell Biol. 1997;17:6563–6573. doi: 10.1128/mcb.17.11.6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J of Biochem Cell Biol. 2013;45:2333–2347. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- Keefe AC, Lawson JA, Flygare SD, Fox ZD, Colasanto MP, Mathew SJ, Yandell M, Kardon G. Muscle stem cells contribute to myofibres in sedentary adult mice. Nat Commun. 2015;6:7087. doi: 10.1038/ncomms8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 2010;18:448–458. doi: 10.1016/j.ccr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, Trefois P, Gruson D, Thissen JP. Role of Activin A and myostatin in human cancer cachexia. J Clin Endocrinol M. 2015;100:2030–2038. doi: 10.1210/jc.2014-4318. [DOI] [PubMed] [Google Scholar]
- Ma K, Mallidis C, Artaza J, Taylor W, Gonzalez-Cadavid N, Bhasin S. Characterization of 5′-regulatory region of human myostatin gene: regulation by dexamethasone in vitro. Am J Physiol Endocrinol Metab. 2001;281:E1128–1136. doi: 10.1152/ajpendo.2001.281.6.E1128. [DOI] [PubMed] [Google Scholar]
- Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ. Twist is a potential oncogene that inhibits apoptosis. Gene Dev. 1999;13:2207–2217. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Pawlikowski B, Pulliam C, Betta ND, Kardon G, Olwin BB. Pervasive satellite cell contribution to uninjured adult muscle fibers. Skelet muscle. 2015;5:42. doi: 10.1186/s13395-015-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, Zanatta L, Pivetta F, Grizzo A, Sonego M, et al. A “twist box” code of p53 inactivation: twist box: p53 interaction promotes p53 degradation. Cancer Cell. 2012;22:404–415. doi: 10.1016/j.ccr.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90–106. doi: 10.1038/cr.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt JD, Lunt AI, Parry DJ, Partridge TA. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Animal. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Gregorevic P, Sandri M. TGFβ and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends Endocrinol Metab. 2014;25:464–471. doi: 10.1016/j.tem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer DB, Rhee J, Cheung WL, Lassar AB. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Reversing cachexia. Cell. 2010;142:511–512. doi: 10.1016/j.cell.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P, Puisieux A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell. 2004;6:625–630. doi: 10.1016/j.ccr.2004.09.033. [DOI] [PubMed] [Google Scholar]
- White JP, Gao S, Puppa MJ, Sato S, Welle SL, Carson JA. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol Cell Endocrinol. 2013;365:174–186. doi: 10.1016/j.mce.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.