

Abstract
Introduction
N-carbamoyl-L-glutamic acid (NCG) is a synthetic analogue of N-acetyl glutamate (NAG) that works effectively as a cofactor for carbamoyl phosphate synthase 1 and enhances ureagenesis by activating the urea cycle. NCG (brand name, Carbaglu) was recently approved by the United States Food and Drug Administration (US FDA) for the management of NAGS deficiency and by the European Medicines Agency (EMA) for the treatment of NAGS deficiency as well as for the treatment of hyperammonenia in propionic, methylmalonic and isovaleric acidemias in Europe.
Areas covered
The history, mechanism of action, and efficacy of this new drug are described. Moreover, clinical utility of NCG in a variety of inborn errors of metabolism with secondary NAGS deficiency is discussed.
Expert commentary
NCG has favorable pharmacological features including better bioavailability compared to NAG. The clinical use of NCG has proven to be so effective as to make dietary protein restriction unnecessary for patients with NAGS deficiency. It has been also demonstrated to be effective for hyperammonemia secondary to other types of inborn errors of metabolism. NCG may have additional therapeutic potential in conditions such as hepatic hyperammonemic encephalopathy secondary to chemotherapies or other liver pathology.
Keywords: N-carbamoyl-L-glutamic acid, Carbaglu, hyperammonemia, N-acetyl glutamate deficiency, NAGS, inborn errors of metabolism, urea cycle
1. Introduction
Inborn errors of ureagenesis are a group of inherited disorders in which there is an enzyme or transporter defect affecting the production or shuttling of biochemical intermediates in the metabolic pathway responsible for the disposal of excess nitrogen. Significant medical problems can arise from excessive accumulation of toxic metabolites such as ammonia or other compounds. Ammonia detoxification through the urea cycle in the periportal hepatocytes is the major route for nitrogen disposal as urea which is excreted from the urine (Figure 1) [1]. There are seven enzymes involved in the disposal of nitrogen via the urea cycle including carbonic anhydrase VA (CAVA), N-acetyl glutamate synthase (NAGS), carbamoyl phosphate synthase 1 (CPS1) and ornithine transcarbamylase (OTC), argininosuccinate synthase (ASS), argininosuccinate lyase, and arginase. A deficiency in any of these enzymes or in the transporters encoded by SLC25A13 or SLC25A15 results in a urea cycle disorder (UCD). Dysfunction of the urea cycle can result in hyperammonemia, leading to encephalopathy, seizures, coma, and death. Hyperammonemia is one of the most significant life-threatening consequences of metabolic decompensation for patients with primary UCDs, but it can also occur in patients with other inborn errors of metabolism (IEM) via secondary inhibition of the urea cycle and substrate deficiency. As a result of secondary urea cycle dysfunction, significant hyperammonemia can be observed in organic acidemias, fatty acid oxidation defects, disorders of amino acid metabolism, and valproic acid toxicity. Appropriate ammonia control is an important part of the medical management of IEM and extensive efforts have been invested to develop new therapies for hyperammonemia over the past decades.
Figure 1.
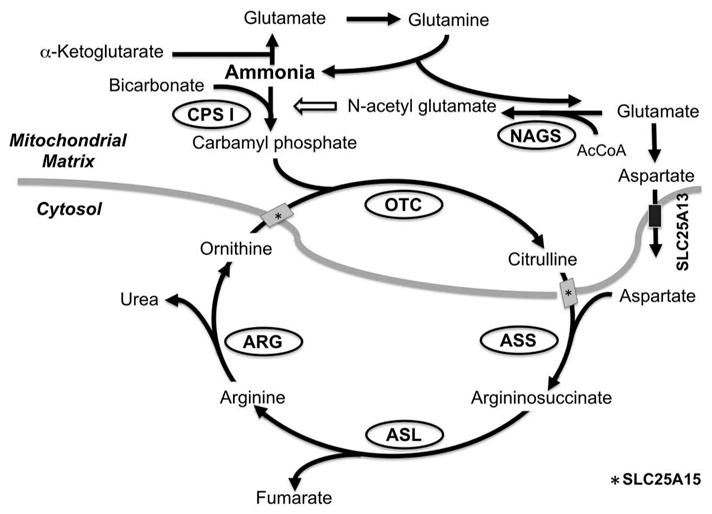
The urea cycle and the mechanism of action of N-carbamoyl-L-glutamic acid (NCG). The enzymes in ovals and the solute transporters depicted by rectangles comprise the urea cycle. Thick gray line depicts mitochondrial membranes. NCG is a synthetic analogue of N-acetylglutamate and activates Carbamoyl phosphate synthase I (CPS1) and enhance ureagenesis through the urea cycle. AcCoA: Acetyl CoA; ASL: Argininosuccinate lyase; ARG: Arginase; ASS: Argininosuccinic acid synthetase; CPS I: Carbamoyl phosphate synthetase I; GPB: Glycerol phenylbutyrate; NAGS: N-acetylglutamate synthase; OTC: Ornithine transcarbamoylase; PAGN: Phenylacetylglutamine.
Traditionally, treatment of hyperammonemia in patients with UCDs includes dietary protein restriction to reduce nitrogen flux and administration of ammonia scavenger medications such as benzoate, phenylacetate, and phenylbutyrate to divert excess nitrogen into a nontoxic, excretable metabolite pool [2,3]. Despite the development of these therapeutic approaches, there has been no complete curative therapy for UCDs except for liver transplantation, which is indicated for the more severe proximal deficiencies in CPS1, OTC, and ASS1. N-carbamoyl-L-glutamic acid (NCG or Carbaglu), a structural analogue of N-acetyl glutamate (NAG), has been used for the treatment of NAGS deficiency from the very description of this UCD [4,5]. By directly replacing NAG, which is an essential activator of the CPS1 enzyme without which this enzyme is inactive [6], NGC functions as an almost curative medication for NAGS deficiency. Additionally, given its capacity to function as a urea cycle activator, NGC has been proposed as a potential therapy for secondary hyperammonemia due to organic acidemias, hepatic encephalopathy, and valproate-induced hyperammonemia [7–9].
2. NAG and NAGS deficiencies
The first enzymatic step of the urea cycle is normally catalyzed by CPS1 to form carbamoyl phosphate from bicarbonate, ammonia, and two molecules of ATP (Figure 1). Human CPS1 protein was first purified from the liver by Rubio and colleagues [10]. CPS1 is the rate-limiting enzyme of the overall activity of the urea cycle, and NAG is essential as an activating cofactor in order to achieve full activation of the urea cycle [11], and recent identification of crystal structure of human CPS1 protein enhanced the understanding of its mechanism [6]. NAG is biosynthesized in the mitochondrial matrix by a reaction which produces NAG from glutamate and acetyl-CoA [12]. Deficiency of NAGS leads to hyperammonemia due to inadequate activation of urea cycle function despite normal activities of all of the component enzymatic steps. The onset and severity of clinical symptoms vary depending on the residual activity of the mutant enzyme. NAGS deficiency is inherited in an autosomal recessive manner and the human NAGS gene was identified on chromosome 17q21.31 in 2002 [12].
The first NAGS deficiency case was described in 1981 in a male infant from Switzerland with a family history of infantile deaths of his siblings with hyperammonemia [13]. This patient’s biochemical profile suggested a blockage of the urea cycle proximal to OTC and the diagnosis was confirmed by non-detectable activity of NAGS enzyme in the liver tissue from biopsy, while CPS1 activity was intact [13]. Subsequently, another NAGS deficiency case was identified in a neonate with a similar lethal presentation [14]. Since then, more than 50 patients have been reported worldwide [15,16]. Of interest, clinical symptoms of NAGS deficiency are diverse ranging from irritability, vomiting, and lethargy to behavioral abnormalities. In general, patients with the severe form of the disorder become symptomatic during the neonatal period with hyperammonemic encephalopathy, while milder cases present in adulthood with neuropsychiatric or behavioral symptoms [15]. A recent cohort and biochemical study identified that mutations in acetyltransferase (GNAT) domain are more frequent than in the other kinetic amino acid kinase domain [16]. Additionally, mutations affecting GNAT domain are more deleterious and cause severe neonatal onset form [16]. Because of its rarity, the prevalence of this disorder is unclear but the distribution of cases is panethnic [15,17]. The true incidence of the disorder is likely underestimated for various reasons: newborn screening by tandem-mass spectrometry does not detect NAGS deficiency, mildly affected individuals may remain symptom-free for years, and severely affected individuals may die without being diagnosed. The clinical and biochemical presentation of NAGS deficiency may be indistinguishable from that of CPS1 deficiency and molecular genetic analysis is particularly helpful to confirm the diagnosis of NAGS deficiency, supplanting enzymatic assay as the preferred confirmatory method [18].
In addition to primary NAG deficiency due to alterations in the NAGS gene, hyperammonemia as a result of secondary NAG deficiency is observed. This can be caused by inhibition of NAGS activity through excessive accumulation of toxic metabolites due to fatty acid oxidation defects, organic acid-emias, valproic acid, and mitochondrial dysfunction [19–21].
2.1. NCG as a therapeutic drug for NAGS deficiency
Studies investigating the mechanism of citrulline biosynthesis led to the discovery that NCG activates ureagenesis [22], later finding that this compound enhances the activity of CPS1 with a higher Km than the natural activator, NAG [10,11,23,24]. In 1972, Kim and colleagues reported that rats intoxicated with a lethal dose of ammonium acetate were protected from lethality by a preloading injection of NCG along with arginine [25]. Of interest, this protective effect was much higher with NCG compared to NAG [25]. The effect was explained in part by the higher resistance to degradation by cytosolic amino acylase of NCG relative to NAG, a pharmacokinetic property that allows NGC to reach the mitochondria more effectively than NAG [26].
Based on these preclinical studies, Bachmann and colleagues used NCG to treat a patient with NAGS deficiency and reported a significant reduction in blood ammonia levels [5,13]. In light of these observations of remarkable improvement of hyperammonemia in neonatal cases, NCG was tried as an experimental therapy for neonates and infants who were suspected to have UCDs [27]. Interestingly, rapid normalization of ammonia occurred in patients who were subsequently confirmed by molecular or enzymatic assays to be affected with NAGS deficiency. Long-term follow-up of these patients demonstrated stabilization of ammonia levels and improved nitrogen balance using experimental doses of NCG [27–29]. A more recent clinical study using stable isotope nitrogen labeling in an adolescent patient with NAGS deficiency proved that NCG was able to restore ureagenesis [30]. The effect to increase ureagenesis was further confirmed in a study using healthy subjects who received a single dose of NCG without significant complications, suggesting a rationale for the potential use of this compound for inborn errors other than NAGS deficiency [8]. It has been also speculated that NCG can be useful for the prevention and treatment of hyperammonemia due to non-metabolic clinical conditions such as liver disease [25].
NAGS deficiency is an extremely rare disorder. With fewer than 20 patients in the US, it was difficult to conduct clinical trials to prove the efficacy of NCG for the treatment of hyperammonemia secondary to NAGS deficiency. Therefore, using the data from historical cases of successful management with NCG [27,28,31–34], the FDA approved Carbaglu (NCG) as an orphan drug for NAGS deficiency in the US through its flexible approval process in March 2010 [35]. In Europe, a marketing authorization for Carbaglu was validated by the European Commission in January 2003.
3. Further characterization of efficacy of NCG in various conditions
3.1. NAGS deficiency
Following the early cases described in Section 4, additional cases of successful management of hyperammonemia using NCG have been reported, supporting its effectiveness for both acute and long-term management of hyperammonemia, particularly in neonates with NAGS deficiency, and also demonstrating dosing requirements for optimal control of hyperammonemia. Severe neonatal cases of hyperammonemia that were refractory to traditional management were successfully treated with 100 mg/kg/day dose of NCG in two independent reports [36,37]. In each case, 100 mg/kg/day of NCG was sufficient to control ammonia levels without protein restriction and appeared to improve growth velocity. Dose reduction of NCG with a lower than standard 10 mg/kg/day was attempted in the patient with a homozygous missense mutation in exon 6 of the NAGS gene, c. 1450 T > C, p. Trp484Arg [37]. This dose was adequate to control ammonia levels in a non-decompensated state but was inadequate to prevent hyperammonemia during episodes of illness. After adjusting the dose of NCG to 30 mg/kg/day, ammonia levels were well controlled without protein restriction and no developmental abnormalities were detected at three years of age. For a Korean boy who was diagnosed with NAGS deficiency at two years of age with confirmed compound heterozygous mutations, c.929T>C and c.1464_1465del (p.V310A and p. H488Qfs*2), a dose reduction down to 75 mg/kg/day was successfully achieved without recurrent hyperammonemic episodes [38]. Currently, the consensus guidelines recommend 100 mg/kg of NCG as initial standard dosage for management of acute hyperammonemia for patients with NAGS deficiency and the dose can be adjusted for chronic management [39].
Recently, additional adult cases of NAGS have been reported [15,32,40–42]. These were either late-onset forms with hypomorphic alleles or late-identified cases that were not diagnosed during childhood despite multiple episodes of hyperammonemia with fluctuating behavioral changes. Even in these adult cases, the efficacy of NCG was promising, with stabilization of ammonia control and complete protein liberalization observed in treated patients [15]. In our personal experience, an adult who presented with hyperammonemic encephalopathy triggered by surgical stress was found to have molecularly confirmed NAGS deficiency. His baseline ammonia levels were persistently elevated with high plasma glutamine but these values normalized after starting NCG.
In aggregate, these clinical reports demonstrated that treatment of NAGS deficiency with NCG in the acute and long-term settings was effective in treating and avoiding hyperammonenmic episodes, preventing neurologic deterioration when given during early stages of decompensation and at an early age and helped to improve growth parameters. Most patients with primary NAGS deficiency who are treated with NCG do not need additional drugs or protein restriction at baseline, but may require additional interventions during an acute illness or metabolic decompensation [4].
3.2. Organic acidemias
Hyperammonemia in patients with organic acidemias, including methylmalonic acidemia (MMA), propionic acidemia (PA), and isovaleric acidemia (IVA), appears to be caused by reduced NAG synthesis secondary to lowered acetyl-CoA and/or glutamate levels and to the accumulation of inhibitory metabolites [20]. As a result, patients with organic acidemias frequently experience hyperammonemia along with severe metabolic acidosis during decompensation episodes which can lead to acute and/or chronic neurologic damage. Conventional treatments including dietary protein restriction, caloric supports, ammonia scavenger medications, and dialysis have been employed for the management or hyperammonemia in organic acidemias. The success of scavengers such as benzoate or phenylacetate to increase nitrogen disposal in the management of hyperammonemia caused by UCDs has led to their application for the treatment of refractory hyperammonemia in cases of organic acidemias [3]. However, the use of these medications in organic acidemias is controversial. While no controlled studies have been performed, some reported cases suggest that scavenger medications may have limited effectiveness to lower ammonia levels during acute hyperammonemic crisis in children in PA [43–45]. Therefore, currently proposed guidelines for the management of MMA and PA caution that phenylbutyrate/phenylacetate should be used with extreme caution and should be discontinued once the diagnosis of MMA and PA is established [46].
Based on the currently recognized pathophysiology, the application of NCG as a therapeutic option for the secondary hyperammonemia caused by organic acidemias has emerged [7,20]. Successful control of hyperammonemia in patients with decompensated PA, MMA, and IVA with use of NGC trials has been reported without significant adverse events [44,47–52]. The effect of NCG in these cases was quite remarkable and significant reduction of ammonia levels was achieved within 12 h in most cases with a dose of 100–250 mg/kg/day. The observed responses obviated the use of scavenger medications or hemodialysis after the stabilization of ammonia levels [43,47,49,53]. Recent biochemical studies using heavy-isotope labeled urea precursors revealed that the administration of NCG accelerated the production of urea, validating the effectiveness of NGC in PA [8,9,54].
In 2008, NCG was approved for the management of hyperammonemia due to IVA, PA, and MMA in addition to its indication for primary NAGS deficiency by the European Medicines Agency. Since then, NCG has been used for the management of hyperammonemia from organic acidemias in Europe; a retrospective outcome review was recently published [55]. This open-labeled retrospective, multi-center clinical study revealed that NCG had a significant impact on reducing ammonia levels with or without scavenger medications in both neonatal and older patient populations with IVA, PA, and MMA. The findings obtained in the study were consistent with the previously reported clinical efficacy of NCG in organic acidemias, indicating that NCG is an effective therapy for acute secondary hyperammonemia and that it reduced the need for more aggressive medical treatments such as hemodialysis. In the US, a clinical trial of Carbaglu for the treatment of hyperammonemia in organic acidemias is currently ongoing (NCT00843921).
3.3. Other urea cycle defects—OTC and CPS1 deficiencies
At present, NCG is only approved for use in NAGS deficiency but it has been proposed that the medication may have clinical efficacy for other UCDs such as OTC and CPS1 deficiencies [8]. The mechanism that has been hypothesized to support this use is that carbamoyl phosphate may act as a chaperone to stabilize the OTC enzyme, therefore increasing the pool of available carbamoyl phosphate by using NCG may increase the residual function of the urea cycle and thus reduce ammonia levels [8]. Additionally, a single dose of NCG has been proven to augment ureagenesis in control subjects, supporting the potential utility of NCG to increase flux through the urea cycle [8]. Tummolo’s group described a 13-year-old female with OTC deficiency in whom hyperammonemia was successfully treated with NCG, suggesting the potential usefulness of NCG in symptomatic heterozygous OTC females or in OTC patients with hypomorphic alleles [56].
For CPS1 deficiency, a recent in vitro study revealed that NCG stabilizes CPS1 protein [57]. It would be based on a pharmacochaperone effect of NCG on CPS1 deficiency, which is stabilized by this compound in the presence of ATP [57]. Furthermore, a more recent in vitro study provided an interesting insight that even in patients carrying CPS1 mutations that decrease kinetic activity may benefit from NCG by saturating the NAG binding site; however, this effect seems mutation-specific [58]. Ah Mew and colleagues tested the effects of NCG for enhancing ureagenesis in five patients with late-onset CPS1 deficiency and they found that four of them showed increased urea production and reduction of ammonia levels [59]. Because of the variable degrees of improvement in urea-genesis and ammonia levels among patients, they concluded that the response to NCG in patients with CPS1 is mutation-specific as suggested in the in vitro studies [57–59]. In the US, a clinical trial of Carbablu for OTC and CPS1 deficiencies is currently ongoing in the same protocol as described for organic acidemias (NCT00843921).
4. Other conditions and future potential
There might be potential efficacy of NCG in the treatment of hyperammonemia secondary to other disorders [8]. Maple syrup urine disease (MSUD) is one of the aminoacidopathies with hyperleucinemia resulting in metabolic encephalopathy [60]. It can also rarely cause hyperammonemia secondary to metabolic decompensation. Intriguingly, there is a case report of successful management of hyperammonemia by using NCG during an acute decompensation episode triggered by a viral illness in a 4-year-old female with MSUD [61]. Since biochemical basis for its effectiveness has not been explained and it was only reported in a single case, the rationale of the usage of NCG for MSUD has not been justified yet but it raises the possibility that NCG may be an effective adjunctive therapy along with standard treatments for MSUD. Similarly, there is a report of three children with a successful resolution of hyperammonemia caused by CAVA deficiency by using NCG [62]; however, the pathophysiological rationale for the clinical usage of NCG is again still unknown in this case.
Apart from IEM, another potential use for NCG is to treat valproate-induced hyperammonemia. This well-known adverse effect of valproic acid is caused by inhibition of NAGS by the drug metabolite, valproyl-CoA [63]. Hyperammonemia caused by short- or long-term use of valproic acid in children has been successfully managed with NCG [64].
Moreover, there have been reports of hyperammonemic encephalopathy after the use of various chemotherapeutic agents such as cisplatin, fluorouracil, cytarabine, vincristine, amsacrine, etoposide, L-asparaginase, cyclophosphamide, and their various combinations for patients with malignancies [65]. The underlying mechanism of hyperammonemia secondary to chemotherapy seems multifactorial and distinctive to primary defects in the urea cycle in the majority of cases [65]. However, a subset of patients who underwent chemotherapies for hepatocellular carcinoma and gastric adenocarcinoma revealed low plasma concentration of arginine, suggesting involvement of altered ureagenesis through the urea cycle [66–69]. One of those cases with hepatocellular carcinoma treated with doxorubicine, cisplatin, and fluorouracil showed biochemical profile emulating OTC [66], implying a potential utility of NCG. Of interest, hyperammonemia for the patient with gastric adenocarcinoma treated with cisplatine, epirubicine, and capecitabine was successfully managed with NCG [68]. However, it is possible that NCG may be effective only to a particular subset of patients with specific biochemical profiles reflecting depression of the urea cycle. Further clinical evidence and studies are necessary in order to better describe the underlying mechanism of chemotherapy-associated hyperammonemic encephalopathy.
Of particular interest, recent in vitro and in vivo mouse studies suggested that NCG may function as an anticancer drug by promoting apoptosis and suppressing proliferation for pancreatic ductal carcinoma, triple-negative breast cancer, hepatoma, and lung cancer [70]. Given its safety, toxicity, and mutagenicity profiles [71], NCG may have a potential role as a cancer treatment. However, additional preclinical studies confirming this effect will need to be done.
5. Conclusions
NCG (Carbaglu) is an orphan drug for the acute- and long-term management of hyperammonemia caused by NAGS deficiency that was recently approved for this use in the US, while in Europe it was approved for such use in 2010 and for the hyperammonemia of PA, MMA, and IVA at later dates. NAGS patients receiving this treatment are able to maintain normal ammonia levels without dietary protein restriction. Given its oral route of administration, NCG can be also effective for management of hyperammonemia in an outpatient setting along with conventional treatment including dietary protein restriction [72]. Due to the rarity of the disorders and its relatively recent introduction to the market, long-term data on safety and efficacy are still limited. Nonetheless, the available literature suggests that NCG has proven to be quite effective for the management of hyperammonemia in NAGS deficiency and in non-UCD IEM. It can be speculated that prevention of secondary hyperammonemia in IEM may have beneficial effects including slowing of chronic neurological deterioration and improvement of growth parameters for children. NCG has been proven to be safe in all age groups and represents a life-changing treatment in the management of NAGS deficiency. Future studies are necessary to validate its use for other IEM or conditions such as hepatic encephalopathy as proposed by the studies in healthy human subjects and a rat cirrhotic model [8,73]. In general, hyperammonemia caused by conditions other than defects in the urea cycle are complex with involvement of multiple factors and the contribution of secondary NAGS deficiency may be fractional. The mechanism of action of NCG is simply activating the function of CPS1; therefore, there might be a limitation of its use, while NCG has been shown to increase ureagenesis [8].
6. Expert commentary
NCG, originally used as an experimental drug to treat hyperammonemia by enhancing ureagenesis, is now approved as a therapeutic medication as Carbaglu by the authorizing agencies. Emerging evidence supported by biochemical studies along with the past clinical reports proved the efficacy of NCG in the acute- and long-term management of hyperammonemia caused by not only primary NAGS deficiency but also other types of IEM. The use of NCG in appropriate settings is life-saving for patients with severe hyperammonemia, particularly for neonates with acute hyperammonemia crisis. Making an immediate diagnosis in this setting is often very difficult and using this drug to rapidly reduce ammonia levels may help to reduce the use of more invasive medical procedures associated with initiating scavenger medications or hemodialysis.
The route of administration of this medication is limited to oral or enteral routes. NCG is a highly effective outpatient-based drug for ammonia control caused by NAGS deficiency. As administration of the synthetic cofactor induces the latent urea cycle activity in the hepatocytes of NAGS patients, no other treatments are generally required. By replacing strict restriction of dietary protein and scavenger medication with a single agent that achieves better ammonia control, it is expected that the quality of life of these patients will be substantially improved. In addition to allowing a more flexible dietary regimen, early diagnosis and treatment of these patients is likely to lead to improved overall neurocognitive outcome. Because the drug is only approved as an oral formulation, it is challenging to use NCG for those who are in a severe hyperammonemic crisis with neurological depression and with a risk of aspiration. Early initiation of the drug before the development of severe neurological symptoms such as seizures is preferable but administration via nasogastric tube can be considered unless clinically contraindicated.
7. Five-year view
Regional differences in terms of the currently approved indications of Carbaglu still exist. As of April 2016, while Carbaglu is currently approved for the acute and long-term management of patients with hyperammonemia due to primary NAGS deficiency and the treatment of hyperammonemia from organic acidemias in Europe, it is only approved for the management of NAGS deficiency in the US. Its use is also not yet approved in other countries such as in Japan. A clinical trial of Carbaglu for conditions including acute hyperammonemia in PA, MMA, and other UCDs is currently in process in the US. Given the favorable outcome supported by previously reported cases and biochemical studies, it is reasonably likely that the usage of NCG will be expanded to those disorders.
NAGS enzyme activity can be affected by multiple factors which can, under a variety of clinical conditions, manifest as secondary hyperammonemia [74]. In addition to its use for IEM, NCG may be useful to treat secondary NAGS deficiency caused by medications such as valproate or chemotherapeutics [64,68]. Additional indications for the use of this drug may arise as research into causes of drug-induced hyperammonemia advances.
Key issues.
NCG is a structural analogue of NAG that acts as an activator of CPS1, enhancing the activity of the urea cycle and promoting ureagenesis.
NCG has been recently approved for the acute and chronic management of patients with NAGS deficiency.
NCG is highly effective for the treatment of NAGS deficiency and can eliminate the requirement for dietary protein restriction and ammonia scavenger medication.
NCG has been approved in Europe for the acute management of hyperammonemia due to secondary NAGS deficiency induced by organic acidemias.
NCG may have therapeutic potential to treat hyperammonemia caused by conditions other than IEM in which activation of the urea cycle is compromised by inhibition of NAGS enzymatic activity.
Acknowledgments
Funding
This paper was not funded.
Footnotes
Declaration of interest
C. Chapel-Crespo and K. Oishi are employees of the Icahn School of Medicine at Mount Sinai. G. A. Diaz is an employee of the Icahn School of Medicine at Mount Sinai and serves on the medical advisory board of Genepeeks and has consulted for SynLogic Therapeutics, Aeglea Biotherapeutics and the Gerson Lehrman Group. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Brusilow SW, Horwich AL. Urea cycle enzymes. The online metabolic and molecuar bases of inherited disease. [cited 2014 Jan] Available from: http://www.ommbid.com.
- 2.Matoori S, Leroux JC. Recent advances in the treatment of hyperammonemia. Adv Drug Deliv Rev. 2015;90:55–68. doi: 10.1016/j.addr.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Oishi K, Diaz GA. Glycerol phenylbutyrate for the chronic management of urea cycle disorders. Expert Rev Endocrinol Metab. 2014;9(5):427–434. doi: 10.1586/17446651.2014.945908. [DOI] [PubMed] [Google Scholar]
- 4.Haberle J. Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther Clin Risk Manag. 2011;7:327–332. doi: 10.2147/TCRM.S12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bachmann C, Colombo JP, Jaggi K. N-acetylglutamate synthetase (NAGS) deficiency: diagnosis, clinical observations and treatment. Adv Exp Med Biol. 1982;153:39–45. doi: 10.1007/978-1-4757-6903-6_6. [DOI] [PubMed] [Google Scholar]
- 6.de Cima S, Polo LM, Diez-Fernandez C, et al. Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis. Sci Rep. 2015;5:16950. doi: 10.1038/srep16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniotti M, La Marca G, Fiorini P, et al. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int J Gen Med. 2011;4:21–28. doi: 10.2147/IJGM.S10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Ah Mew N, Payan I, Daikhin Y, et al. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009;98(4):325–330. doi: 10.1016/j.ymgme.2009.07.010. Describes a clinical study confirming that increase ureagenesis can be obtained by using NCG in healthy subjects as well. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuchman M, Caldovic L, Daikhin Y, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64(2):213–217. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubio V, Ramponi G, Grisolia S. Carbamoyl phosphate synthetase I of human liver. Purification, some properties and immunological cross-reactivity with the rat liver enzyme. Biochim Biophys Acta. 1981;659(1):150–160. doi: 10.1016/0005-2744(81)90279-5. [DOI] [PubMed] [Google Scholar]
- 11.Fahien LA, Schooler JM, Gehred GA, et al. Studies on the mechanism of action of acetylglutamate as an activator of carbamyl phosphate synthetase. J Biol Chem. 1964;239:1935–1941. [PubMed] [Google Scholar]
- 12.Caldovic L, Morizono H, Gracia Panglao M, et al. Cloning and expression of the human N-acetylglutamate synthase gene. Biochem Biophys Res Commun. 2002;299(4):581–586. doi: 10.1016/s0006-291x(02)02696-7. [DOI] [PubMed] [Google Scholar]
- 13•.Bachmann C, Krahenbuhl S, Colombo JP, et al. N-acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304(9):543. doi: 10.1056/NEJM198102263040918. First report of NAGS deficiency patient. [DOI] [PubMed] [Google Scholar]
- 14.Bachmann C, Brandis M, Weissenbarth-Riedel E, et al. N-acetylglutamate synthetase deficiency, a second patient. J Inherit Metab Dis. 1988;11(2):191–193. doi: 10.1007/BF01799871. [DOI] [PubMed] [Google Scholar]
- 15.Cartagena A, Prasad AN, Rupar CA, et al. Recurrent encephalopathy: NAGS (N-acetylglutamate synthase) deficiency in adults. Can J Neurol Sci. 2013;40(1):3–9. doi: 10.1017/s0317167100012877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Sancho-Vaello E, Marco-Marin C, Gougeard N, et al. Understanding N-acetyl-L-glutamate synthase deficiency: mutational spectrum, impact of clinical mutations on enzyme functionality, and structural considerations. Hum Mutat. 2016;37(7):679–694. doi: 10.1002/humu.22995. Most recent comprehensive report of clinical mutation in patients with NAGS deficiency. [DOI] [PubMed] [Google Scholar]
- 17.Ah Mew N, Caldovic L. N-acetylglutamate synthase deficiency: an insight into the genetics, epidemiology, pathophysiology, and treatment. Appl Clin Genet. 2011;4:127–135. doi: 10.2147/TACG.S12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haberle J, Schmidt E, Pauli S, et al. Mutation analysis in patients with N-acetylglutamate synthase deficiency. Hum Mutat. 2003;21(6):593–597. doi: 10.1002/humu.10216. [DOI] [PubMed] [Google Scholar]
- 19.Coude FX, Grimber G, Parvy P, et al. Inhibition of ureagenesis by valproate in rat hepatocytes. Role of N-acetylglutamate and acetyl-CoA. Biochem J. 1983;216(1):233–236. doi: 10.1042/bj2160233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest. 1979;64(6):1544–1551. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams CA, Tiefenbach S, McReynolds JW. Valproic acid-induced hyperammonemia in mentally retarded adults. Neurology. 1984;34(4):550–553. doi: 10.1212/wnl.34.4.550. [DOI] [PubMed] [Google Scholar]
- 22.Grisolia S, Cohen PP. The catalytic role of carbamyl glutamate in citrulline biosynthesis. J Biol Chem. 1952;198(2):561–571. [PubMed] [Google Scholar]
- 23.Schooler JM, Fahien LA, Cohen PP. 2-Acetoxyglutarate as an activator of carbamyl phosphate synthetase. J Biol Chem. 1963;238:1909–1910. [PubMed] [Google Scholar]
- 24.Grisolia S, Cohen PP. Catalytic role of of glutamate derivatives in citrulline biosynthesis. J Biol Chem. 1953;204(2):753–757. [PubMed] [Google Scholar]
- 25•.Kim S, Paik WK, Cohen PP. Ammonia intoxication in rats: protection by N-carbamoyl-L-glutamate plus L-arginine. Proc Natl Acad Sci U S A. 1972;69(12):3530–3533. doi: 10.1073/pnas.69.12.3530. First repot to indicate a protective effect of NCG for hyperammonemia using an animal model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reglero A, Rivas J, Mendelson J, et al. Deacylation and transacetylation of acetyl glutamate and acetyl ornithine in rat liver. FEBS Lett. 1977;81(1):13–17. doi: 10.1016/0014-5793(77)80917-4. [DOI] [PubMed] [Google Scholar]
- 27.Guffon N, Schiff M, Cheillan D, et al. Neonatal hyperammonemia: the N-carbamoyl-L-glutamic acid test. J Pediatr. 2005;147(2):260–262. doi: 10.1016/j.jpeds.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 28.Schubiger G, Bachmann C, Barben P, et al. N-acetylglutamate synthetase deficiency: diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur J Pediatr. 1991;150(5):353–356. doi: 10.1007/BF01955939. [DOI] [PubMed] [Google Scholar]
- 29.Gessler P, Buchal P, Schwenk HU, et al. Favourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiency. Eur J Pediatr. 2010;169(2):197–199. doi: 10.1007/s00431-009-1006-0. [DOI] [PubMed] [Google Scholar]
- 30••.Caldovic L, Morizono H, Daikhin Y, et al. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145(4):552–554. doi: 10.1016/j.jpeds.2004.06.047. An important clinical study to prove that NCG was able to restore ureagenesis in NAGS deficiency. [DOI] [PubMed] [Google Scholar]
- 31.Guffon N, Vianey-Saban C, Bourgeois J, et al. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18(1):61–65. doi: 10.1007/BF00711374. [DOI] [PubMed] [Google Scholar]
- 32.Hinnie J, Colombo JP, Wermuth B, et al. N-Acetylglutamate synthetase deficiency responding to carbamylglutamate. J Inherit Metab Dis. 1997;20(6):839–840. doi: 10.1023/a:1005344507536. [DOI] [PubMed] [Google Scholar]
- 33.Morris AA, Richmond SW, Oddie SJ, et al. N-acetylglutamate synthetase deficiency: favourable experience with carbamylglutamate. J Inherit Metab Dis. 1998;21(8):867–868. doi: 10.1023/a:1005478904186. [DOI] [PubMed] [Google Scholar]
- 34.Elpeleg O, Shaag A, Ben-Shalom E, et al. N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy. Ann Neurol. 2002;52(6):845–849. doi: 10.1002/ana.10406. [DOI] [PubMed] [Google Scholar]
- 35.Sasinowski FJ. Quantum of effectiveness evidence in FDA’s approval of orphan drugs. Drug Information Journal. 2012;46(2):238–263. [Google Scholar]
- 36.Van Leynseele A, Jansen A, Goyens P, et al. Early treatment of a child with NAGS deficiency using N-carbamyl glutamate results in a normal neurological outcome. Eur J Pediatr. 2014;173(12):1635–1638. doi: 10.1007/s00431-013-2205-2. [DOI] [PubMed] [Google Scholar]
- 37.Kiykim E, Zubarioglu T. Low dose of carglumic acid for treatment of hyperammonemia due to N-acetylglutamate synthase deficiency. Indian Pediatr. 2014;51(9):755–756. [PubMed] [Google Scholar]
- 38.Kim JH, Kim YM, Lee BH, et al. Short-term efficacy of N-carbamylglutamate in a patient with N-acetylglutamate synthase deficiency. J Hum Genet. 2015;60(7):395–397. doi: 10.1038/jhg.2015.30. [DOI] [PubMed] [Google Scholar]
- 39.Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caldovic L, Morizono H, Panglao MG, et al. Late onset N-acetylglutamate synthase deficiency caused by hypomorphic alleles. Hum Mutat. 2005;25(3):293–298. doi: 10.1002/humu.20146. [DOI] [PubMed] [Google Scholar]
- 41.Caldovic L, Morizono H, Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28(8):754–759. doi: 10.1002/humu.20518. [DOI] [PubMed] [Google Scholar]
- 42.Tuchman M, Lee B, Lichter-Konecki U, et al. Cross-sectional multi-center study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abacan M, Boneh A. Use of carglumic acid in the treatment of hyperammonaemia during metabolic decompensation of patients with propionic acidaemia. Mol Genet Metab. 2013;109(4):397–401. doi: 10.1016/j.ymgme.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 44.Jones S, Reed CA, Vijay S, et al. N-carbamylglutamate for neonatal hyperammonaemia in propionic acidaemia. J Inherit Metab Dis. 2008;31(Suppl 2):S219–S222. doi: 10.1007/s10545-008-0777-1. [DOI] [PubMed] [Google Scholar]
- 45.Levesque S, Lambert M, Karalis A, et al. Short-term outcome of propionic aciduria treated at presentation with N-carbamylglutamate: a retrospective review of four patients. JIMD Rep. 2012;2:97–102. doi: 10.1007/8904_2011_54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumgartner MR, Horster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Filippi L, Gozzini E, Fiorini P, et al. N-carbamylglutamate in emergency management of hyperammonemia in neonatal acute onset propionic and methylmalonic aciduria. Neonatology. 2010;97(3):286–290. doi: 10.1159/000255168. [DOI] [PubMed] [Google Scholar]
- 48.Gebhardt B, Dittrich S, Parbel S, et al. N-carbamylglutamate protects patients with decompensated propionic aciduria from hyperammonaemia. J Inherit Metab Dis. 2005;28(2):241–244. doi: 10.1007/s10545-005-5260-7. [DOI] [PubMed] [Google Scholar]
- 49.Gebhardt B, Vlaho S, Fischer D, et al. N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria. Mol Genet Metab. 2003;79(4):303–304. doi: 10.1016/s1096-7192(03)00095-7. [DOI] [PubMed] [Google Scholar]
- 50.Levrat V, Forest I, Fouilhoux A, et al. Carglumic acid: an additional therapy in the treatment of organic acidurias with hyperammonemia? Orphanet J Rare Dis. 2008;3:2. doi: 10.1186/1750-1172-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwahn BC, Pieterse L, Bisset WM, et al. Biochemical efficacy of N-carbamylglutamate in neonatal severe hyperammonaemia due to propionic acidaemia. Eur J Pediatr. 2010;169(1):133–134. doi: 10.1007/s00431-009-1036-7. [DOI] [PubMed] [Google Scholar]
- 52.Kasapkara CS, Ezgu FS, Okur I, et al. N-carbamylglutamate treatment for acute neonatal hyperammonemia in isovaleric acidemia. Eur J Pediatr. 2011;170(6):799–801. doi: 10.1007/s00431-010-1362-9. [DOI] [PubMed] [Google Scholar]
- 53.Yap S, Leong HY, Abdul Aziz F, et al. N-Carbamylglutamate is an effective treatment for acute neonatal hyperammonaemia in a patient with methylmalonic aciduria. Neonatology. 2016;109(4):303–307. doi: 10.1159/000443630. [DOI] [PubMed] [Google Scholar]
- 54.Ah Mew N, McCarter R, Daikhin Y, et al. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126(1):e208–e214. doi: 10.1542/peds.2010-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55••.Valayannopoulos V, Baruteau J, Delgado MB, et al. Carglumic acid enhances rapid ammonia detoxification in classical organic acidurias with a favourable risk-benefit profile: a retrospective observational study. Orphanet J Rare Dis. 2016;11(1):32. doi: 10.1186/s13023-016-0406-2. The most recent retrospective outcome review of NCG in organic acidemias. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tummolo A, Favia V, Bellantuono R, et al. Successful early management of a female patient with a metabolic stroke due to ornithine transcarbamylase deficiency. Pediatr Emerg Care. 2013;29(5):656–658. doi: 10.1097/PEC.0b013e31828ec2b9. [DOI] [PubMed] [Google Scholar]
- 57.Diez-Fernandez C, Martinez AI, Pekkala S, et al. Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Hum Mutat. 2013;34(8):1149–1159. doi: 10.1002/humu.22349. [DOI] [PubMed] [Google Scholar]
- 58.Diez-Fernandez C, Gallego J, Haberle J, et al. The study of carbamoyl phosphate synthetase 1 deficiency sheds light on the mechanism for switching on/off the urea cycle. J Genet Genomics. 2015;42(5):249–260. doi: 10.1016/j.jgg.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 59.Ah Mew N, McCarter R, Daikhin Y, et al. Augmenting ureagenesis in patients with partial carbamyl phosphate synthetase 1 deficiency with N-carbamyl-L-glutamate. J Pediatr. 2014;165(2):e401–e403. doi: 10.1016/j.jpeds.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strauss KA, Puffenberger EG, Morton DH. GeneReviews at GeneTests Medical Genetics Information Resource (database online) Copyright, University of Washington; Seattle: 1997–2013. Maple Syrup Urine Disease. 2006 [updated 2013 May 9; cited 2016 May 29]. Available from: http://www.genetests.org. according to NCBI citation guideline. [PubMed] [Google Scholar]
- 61.Kalkan Ucar S, Coker M, Habif S, et al. The first use of N-carbamylglutamate in a patient with decompensated maple syrup urine disease. Metab Brain Dis. 2009;24(3):409–414. doi: 10.1007/s11011-009-9155-4. [DOI] [PubMed] [Google Scholar]
- 62.van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014;94(3):453–461. doi: 10.1016/j.ajhg.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aires CC, van Cruchten A, Ijlst L, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426–434. doi: 10.1016/j.jhep.2010.11.031. [DOI] [PubMed] [Google Scholar]
- 64.Kasapkara CS, Kangin M, Tas FF, et al. Unusual cause of hyperammonemia in two cases with short-term and long-term valproate therapy successfully treated by single dose carglumic acid. J Pediatr Neurosci. 2013;8(3):250–252. doi: 10.4103/1817-1745.123697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nott L, Price TJ, Pittman K, et al. Hyperammonemia encephalopathy: an important cause of neurological deterioration following chemotherapy. Leuk Lymphoma. 2007;48(9):1702–1711. doi: 10.1080/10428190701509822. [DOI] [PubMed] [Google Scholar]
- 66.Chan JS, Harding CO, Blanke CD. Postchemotherapy hyperammonemic encephalopathy emulating ornithine transcarbamoylase (OTC) deficiency. South Med J. 2008;101(5):543–545. doi: 10.1097/SMJ.0b013e31816bf5cc. [DOI] [PubMed] [Google Scholar]
- 67.Jeffers LJ, Dubow RA, Zieve L, et al. Hepatic encephalopathy and orotic aciduria associated with hepatocellular carcinoma in a non-cirrhotic liver. Hepatology. 1988;8(1):78–81. doi: 10.1002/hep.1840080116. [DOI] [PubMed] [Google Scholar]
- 68.Lazier J, Lupichuk SM, Sosova I, et al. Hyperammonemic encephalopathy in an adenocarcinoma patient managed with carglumic acid. Curr Oncol. 2014;21(5):e736–e739. doi: 10.3747/co.21.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winter SS, Rose E, Katz R. Hyperammonemia after chemotherapy in an adolescent with hepatocellular carcinoma. J Pediatr Gastroenterol Nutr. 1997;25(5):537–540. doi: 10.1097/00005176-199711000-00010. [DOI] [PubMed] [Google Scholar]
- 70.Chen CT, Chen YC, Yamaguchi H, et al. Carglumic acid promotes apoptosis and suppresses cancer cell proliferation in vitro and in vivo. Am J Cancer Res. 2015;5(12):3560–3569. [PMC free article] [PubMed] [Google Scholar]
- 71.Wu X, Wan D, Xie C, et al. Acute and sub-acute oral toxicological evaluations and mutagenicity of N-carbamylglutamate (NCG) Regul Toxicol Pharmacol. 2015;73(1):296–302. doi: 10.1016/j.yrtph.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 72.Soyucen E, Demirci E, Aydin A. Outpatient treatment of propionic acidemia-associated hyperammonemia with N-carbamoyl-L-glutamate in an infant. Clin Ther. 2010;32(4):710–713. doi: 10.1016/j.clinthera.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 73.Kim WH, Park H, Yun C, et al. Mixture of N-carbamoyl-L-glutamate plus L-arginine can protect rats with liver cirrhosis from acute ammonia intoxication. J Hepatol. 2001;35(6):719–725. doi: 10.1016/s0168-8278(01)00199-4. [DOI] [PubMed] [Google Scholar]
- 74.Caldovic L, Ah Mew N, Shi D, et al. N-acetylglutamate synthase: structure, function and defects. Mol Genet Metab. 2010;100(Suppl 1):S13–S19. doi: 10.1016/j.ymgme.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]