

Abstract
Background
Leigh Syndrome (LS, OMIM 256000) is an early-onset, progressive neurodegenerative disorder characterized by broad clinical and genetic heterogeneity; it is the most frequent disorder of mitochondrial energy production in children. LS inheritance is complex because patients may present mutations in mitochondrial DNA (mtDNA) or in nuclear genes, which predominantly encode proteins involved in respiratory chain structure and assembly or in coenzyme Q10 biogenesis. However, during the last 15 years, the discovery of several genetic mutations and improved knowledge of the natural history of LS has significantly increased our understanding of this mitochondrial disorder.
Case presentation
Here we describe a 19-year-old male with clinical and neuroimaging LS diagnosed at 3 years of age. Genetic analyses of the whole mtDNA for maternally inherited LS (MILS) and neuropathy ataxia retinitis pigmentosa (NARP) syndrome failed to reveal any pathogenic mutations.
Conclusions
Recently, a missense mutation in ECHS1 and a ~ 35 kb deletion in 10q26.3 involving the region including the gene were identified by WES (whole exome sequencing), uncovering the genetic diagnosis clinically hypothesized for 15 years. We also report the long-term follow-up of this patient, showing a comparison with classical LS or other Leigh-like pictures.
Electronic supplementary material
The online version of this article (10.1186/s12883-018-1103-7) contains supplementary material, which is available to authorized users.
Keywords: Leigh disease, ECHS1 gene, Exome analysis
Background
Leigh Syndrome (LS, OMIM 256000) is a rare, heterogeneous, progressive neurodegenerative disorder caused by mutations in mitochondrial DNA (mtDNA) or in nuclear genes, usually presenting in infancy or early childhood [1, 2]. Atypical or later onset cases have been reported in the literature and are referred to as Leigh-like diseases [3]. The clinical presentation is variable and includes psychomotor delay or regression, acute neurological or acidotic episodes, hypotonia, ataxia, spasticity, movement disorders, and corresponding anomalies of the basal ganglia and brain stem on magnetic resonance imaging (MRI) [3–5]. The prognosis is generally poor with rapid deterioration of cognitive and motor function resulting in death within months or years [6, 7]. Since the identification of the first pathogenic mutation in a LS patient in 1991, more than 75 disease genes have been identified, most of them thanks to the introduction of next-generation sequencing (NGS) technology. Recently, enzymes of the valine degradation pathway have also been shown to cause LS [8, 9]; in particular, mutations in enoyl-CoA hydratase (ECHS1), a nuclear gene encoding a mitochondrial matrix enzyme catalyzing the second step of the ß-oxidation spiral of fatty acids, has been associated with LS in several patients [9–11].
Here we describe a 19-year-old man who was diagnosed at 3 years of age with LS using clinical and neuroimaging data. MILS and NARP syndromes due to mtDNA mutations were excluded. We confirmed the clinical diagnosis hypothesized for 15 years by using whole exome sequencing (WES) analysis, which identified a missense mutation in ECHS1, and by array CGH analysis, which evidenced the deletion of the entire gene. In addition, we report the long-term follow-up of this patient compared with other classical LS or Leigh-like pictures.
Case presentation
The proband is a 19-year-old male born from non-consanguineous parents of Caucasian origin, after a normal pregnancy at 40 weeks of gestation with normal birth measurements (weight 4150 kg, length 52 cm, and cranial circumference 36 cm). Both parents and the 18- year-old brother are healthy (Fig. 1).
Fig. 1.
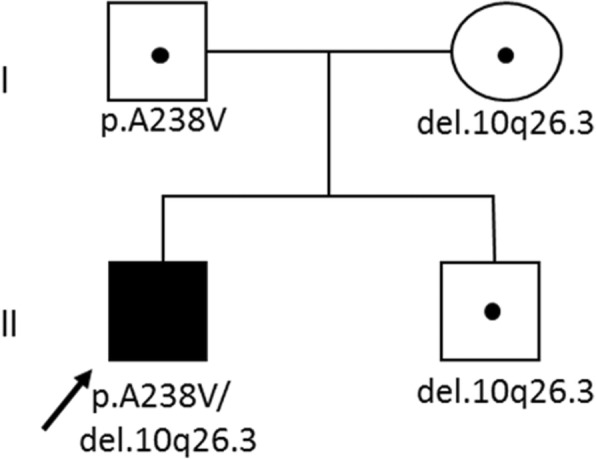
Pedigree of the family. Black symbol represent the proband; healthy family members are shown with open symbols. Small dot: ECHS1 mutation carriers
Nystagmus, convergent strabismus, and mild lower spasticity appeared at 7 months of age as the first symptoms of the disease, and subsequently, developmental psychomotor regression was observed. In particular, the child at 14 months lost the ability to walk alone, presenting ataxic signs, and at 16 months chorea of the arms and dystonia of the trunk appeared.
We performed the first MRI showing the typical pattern of Leigh Syndrome with the presence of bilaterally hyperintense signals in the basal ganglia and thalami and of periventricular white matter (Fig. 2a-b); proton magnetic resonance spectroscopy (1H MRS) showed a lactate peak at 1.33 ppm.
Fig. 2.
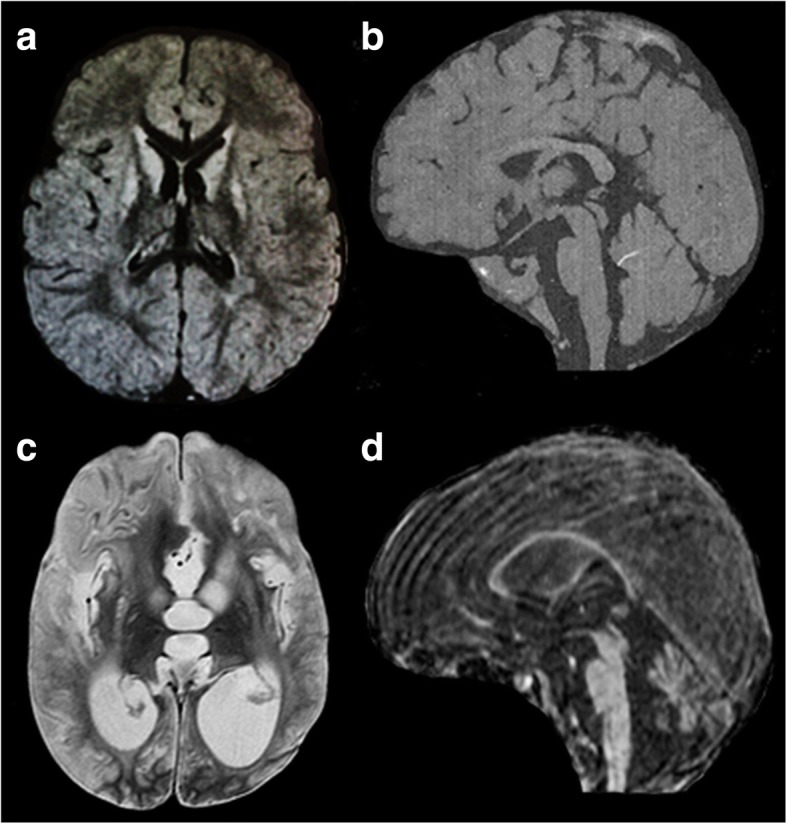
1.5 T low-quality images of the first MRI examination of the boy (16 months); the best available images were chosen. Axial FLAIR (a) slice shows the typical pattern of LS with basal ganglia involvement (caudate and lenticular nuclei) and mild involvement of thalami bilaterally. Sagittal T1 slice (b) shows normal tropism of the corpus callosum and cerebellum. Last MRI examination at 12 yrs. (c-d) shows marked progression of basal ganglia involvement with enlargement of lateral and third ventricles, indicating global atrophy. Sagittal FLAIR slice (d) highlights marked thinning of the corpus callosum, a consequence of brain atrophy, and severe cerebellum atrophy. The presence of significant movement artifacts is related to MRI without anesthesia, because of critical conditions of the patient
The metabolic pattern was characterized by increased levels of plasma lactate, alanine, and valine and reduced levels of citrulline; an increase in 3 methylglutaconic acid was observed in urinary organic acids. Electroencephalogram (EEG) did not show epileptic discharges. A muscle biopsy performed at 18 months showed signs of a mild myopathic process with non-specific abnormalities of oxidative reactions. Respiratory chain activities revealed a slight reduction of complexes II and III.
At 26 months the child presented a dyskinetic tetraparesis associated with hyposthenia of the trunk and limbs. No cardiac, endocrine, gastrointestinal nor renal involvement was observed. The Griffiths scale, which examines the cognitive profile, showed a moderate intellectual disability. The clinical condition remained stable up to 9 years of life, when he presented drug resistance and generalized tonic clonic and myoclonic seizures. Spastic tetraparesis worsened and neurological changes, such as dysarthria/dysphagia, loss of eye contact, and axial and limb dystonia were occurring during acute viral infections. Cognitive deterioration was progressively evident in association with a regression of language skills.
In order to treat the severe spasticity and associated dystonia, an intratecal baclofen pump was implanted, and at 10 years the child was continuously fed by parenteral gastrostomy (PEG). During this period, he also presented constant rhythmic jerky movements of the right arm and of the soft palate, which were indicative of palatal myoclonus status. EEG recording over time showed recurrent focal seizures clinically associated with motor manifestations and during palatal myoclonus, suggesting an ongoing non-convulsive status epilepticus, treated with carbamazepine and benzodiazepine in combination with phenytoin, already administered. No adverse effects were observed, and remission of the seizures was completely obtained; subsequently, phenytoin was gradually reduced and stopped.
At 11 years and 8 months of age, the spastic tetraparesis associated with dystonic movement disorders was complicated by severe scoliosis. The boy also presented a severe intellectual disability, an epileptic encephalopathy, and cerebral visual impairment. Bilateral optic atrophy and sensorineuronal deafness were discovered at 13 years: flash visual evoked potential was not identifiable, whereas brainstem acoustic evoked potential was altered with pons and mesencephalon wave involvement. At around 12 years of age, a second MRI was performed, which showed marked progression of basal ganglia involvement with enlargement of lateral and third ventricles, evident signs of global cerebral atrophy (Fig. 2c-d). A mild double peak of lactate was evident on 1H MRS. A new analysis of muscle tissue and oxidative-phosphorylation enzymes was performed at the age of 13 years, and despite the severe clinical picture, only mild changes in the reduction of complexes II and III were confirmed. Over the course of the disease, limb spasticity with stable elbow and knee contractures were evident, but dystonic movements diminished, and no spontaneous movements were visible. Cardiac and renal function remained unchanged. Constipation was observed.
A tracheostomy was performed at 17 years when he had a bronchopneumonic infection, and his respiratory condition worsened. At 19 years, the patient showed a persistent vegetative state with partial symptomatic drug-resistant epilepsy, pendular nystagmus as seen in blind patients, spastic tetraparesis with hip dislocation, valgus pronatus deformation of the feet, severe scoliosis, and stable flexion of the right arms.
WES analysis was performed on the proband and the asymptomatic father’s and mother’s DNA. After filtering criteria (described in detail in Additional file 1) were applied, only four missense mutations were left. None of the missense variants were exclusive of the affected individual (Table 1) except one in the ECHS1 gene: the c.713C > T/p.Ala238Val mutation was present in the proband in an apparent homozygous state, whereas the father was found to be heterozygous. This mutation, present in the ExAC Browser (http://exac.broadinstitute.org/; MAF = 0.000016), is predictably damaging when examined in silico using Polyphen2 and has already been reported in the compound heterozygous form in a family presenting with LS [3].
Table 1.
Presence of Leigh-related missense mutations in samples
| Genes | Designation | Proband | Mother | Father |
|---|---|---|---|---|
| PNPT1 | c.1519G > T; p.Ala507Ser | Het | – | Het |
| SYNE1 | c.23315G > A; p.Arg7772Gln | Het | Het | – |
| SYNE1 | c.13909G > A; p.Asp4637Asn | Het | Het | – |
| ECHS1 | c.713C > T; p.Ala238Val | Homo | – | Het |
Manual inspection of bam files by Integrative Genome Viewer (IGV) as well as Sanger sequencing of the flanking region revealed the lack of this mutation in the mother (Fig. 3a and b). To further investigate the chromosomal region containing ECHS1, we used CeQer, a software program able to detect copy number variation from exome data. We detected a deletion in an extended region of chromosome 10 (from 135,120,573 to 135,187,238) involving five genes: ZNF511, CALY, PRAP1, FUOM, and ECHS1. This deletion was present in the proband and in his mother but not in the father (Additional file 1: Figure S1).
Fig. 3.

IGV visualization (a) and sequence chromatographs (b) of the ECHS1 region flanking the missense mutation c.713C > T/p.Ala238Val from the affected patient (II-1, indicated by an arrow), his father (I-1) and mother (I-2). In IGV the green color corresponds to the nucleotide base A; the brown to the nucleotide base G
The array CGH identified in the proband a microdeletion of at least 35 kb in 10q26.3 (Additional file 1: Figure S2). This deletion was also detected in the mother. The maternal grandmother was negative; therefore, it is likely that this deletion was inherited from the grandfather (sample not available). In order to confirm whether the deletion led to a loss of copy number of the five included genes, a real-time PCR assay was performed. The results showed that the patient and his mother had a decreased copy number in all genes compared with that of the father (Fig. 4). The Additional file 1 describes all the methods used in this paper and the additional file figures not shown in the text.
Fig. 4.

RT-PCR shows gene profiles on genomic DNA. To determine relative gene load, a ∆∆Cq method was performed from qPCR data with GAPDH as an endogenous reference gene. These data show a depletion of the ZNF511, CALY, PRAP1, FUOM, and ECHS1 genes in two cases (red and purple histograms) with respect to the father (green histogram), who does not present a deletion of the region of interest
Discussion
In this study, we report the case of a 19-year-old man with a diagnosis of Leigh Syndrome. By WES analysis, a missense mutation in the nuclear gene ECHS1 was identified in the proband and his father in the heterozygous state and a deletion in 10q26.3 involving the region including ECHS1 was found in the proband and his mother. CGH analysis and RT-PCR confirmed this deletion.
In the recent years, WES technology has been extensively used for clinical studies; since a significant part of mutations exists in the exons, this approach has achieved great results for identifying Mendelian disease genes. Success of exome sequencing in revealing pathogenic mutations and identifying genes have been demonstrated by several studies [12, 13].
Using WES, Haack et al. [10] identified 10 unrelated individuals carrying compound heterozygous or homozygous mutations in ECHS1 and exhibiting very heterogeneous phenotypes. Recently, two siblings were described with fatal Leigh disease who had increased excretion of several metabolites that are features of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency [9]. Further papers described ECHS1 mutations variously associated with vacuolating leukoencephalopathy, basal ganglia abnormalities or a slow neurodegenerative condition with global brain atrophy or a single episode of metabolic acidosis. All patients presented with lactic acidosis [3, 13, 14].
Our patient presented a clear clinical overlap with the reported cases with ECHS1 or HIBCH deficiency, and the same mutation (p.Ala238Val) was found in two patients [3]. This variant, together with other four missense mutations, is present in the large enoyl-CoA hydratase/isomerase domain, responsible for substrate binding and the catalytic activity of the enzyme and is predicted by the MuPro and Auto-Mute software programs to decrease protein stability [15, 16]. ECHS1 is a nuclear gene involved in several metabolic pathways involving fatty acids and amino acids. The substantial discrepancy between an expected moderate impairment of mitochondrial fatty acid oxidation and the severe clinical presentation of ECHS1-deficient individuals may account for additional pathogenic mechanisms. In addition, ECHS1 has been suggested to be involved in L-isoleucine, L-valine, and L-lysine oxidation using tiglyl-CoA, 2-methacrylyl-CoA, or crotonyl-CoA, respectively, as a substrate. The mitochondrial respiratory chain does not appear to be severely impaired in ECHS1 patients, as demonstrated also in our case. Possible biomarkers of the disease are not currently available; the methacrylate metabolites and/or 2-methyl-2,3-dihydroxybutyric acid could represent an important hint. In our patient, an increase of plasma levels of alanine and valine and the presence of 3-methylglutaconic acid in urine suggested an inherited disorder of mitochondrial energy metabolism [10]. Our patient presented with many clinical characteristics associated with LS, including general (failure to thrive), neurologic (hypotonia, dystonia, developmental delay, MRI findings), audiologic (sensorineural hearing loss) and ophthalmologic (nystagmus, optic atrophy) features but, given the age of onset of the disease and the severity of the clinical picture his outcome was unexpected. The young man is still alive and the interruption of epilepsy in recent years may be related to the severe brain atrophy developed over time.
Conclusions
Despite the advent of NGS technologies, proved to be successful in the identification of disease-causing genes, the exact diagnosis of genetically heterogeneous diseases, such as LS, remains difficult. In our case, the application of complementary genetic methodologies has allowed the molecular diagnosis in a case that would have not be resolved with standard analyses.
The description of additional ECHS1 patients with their longitudinal follow up, could contribute to a better definition of the clinical spectrum of ECHS1 deficiency and to reach an early diagnosis, helpful for optimal genetic counselling.
Additional file
Methods used to perform genetic analyses. (DOCX 350 kb)
Availability of data and materials
All data are contained within the article.
Abbreviations
- 1H MRS
Proton magnetic resonance spectroscopy
- EEG
Electroencephalogram
- IGV
Integrative genome viewer
- LS
Leigh syndrome
- MILS
Maternally inherited Leigh syndrome
- MRI
Magnetic resonance imaging
- mtDNA
mitochondrial DNA
- NARP
Neuropathy ataxia retinitis pigmentosa
- NGS
Next generation sequencing
- PEG
Parenteral gastrostomy
- WES
Whole exome sequencing
Authors’ contributions
PA and CMM are co-first authors and drafted the manuscript. MLF, SF, FL, VDG, CN, AV, VB, and BT acquired, analyzed and interpreted the genetic data. PA, CN and RB were the main contributor in writing the paper. RB and MAC are co-last authors, and critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Regional Pediatric Ethics Committee (Meyer Hospital, Florence).
Written informed consent was obtained from the patient’s parent for participation in this study.
Consent for publication
Written informed consent was obtained from the patient’s parent for publication of this case report and any accompanying images. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Paolo Aretini, Chiara Maria Mazzanti, Roberta Battini and Maria Adelaide Caligo contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s12883-018-1103-7) contains supplementary material, which is available to authorized users.
Contributor Information
Paolo Aretini, Email: p.aretini@fpscience.it.
Chiara Maria Mazzanti, Email: c.mazzanti@fpscience.it.
Marco La Ferla, Email: m.laferla@fpscience.it.
Sara Franceschi, Email: s.franceschi@fpscience.it.
Francesca Lessi, Email: f.lessi@fpscience.it.
Veronica De Gregorio, Email: veronicadegregorio@libero.it.
Claudia Nesti, Email: cnesti@fsm.unipi.it.
Angelo Valetto, Email: a.valetto@ao-pisa.toscana.it.
Veronica Bertini, Email: v.bertini@ao-pisa.toscana.it.
Benedetta Toschi, Email: b.toschi@ao-pisa.toscana.it.
Roberta Battini, Phone: + 39 050 886229, Email: rbattini@fsm.unipi.it.
Maria Adelaide Caligo, Email: adelaide.caligo@do.unipi.it.
References
- 1.Ronchi D, Cosi A, Tonduti D, Orcesi S, Bordoni A, Fortunato F, et al. Clinical and molecular features of an infant patient affected by Leigh disease associated to m.14459G>a mitochondrial DNA mutation: a case report. BMC Neurol. 2011;11:85. doi: 10.1186/1471-2377-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2015;79(2):190-203. [DOI] [PubMed]
- 3.Tetreault M, Fahiminiya S, Antonicka H, Mitchell GA, Geraghty MT, Lines M, et al. Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum Genet. 2015;134:981–991. doi: 10.1007/s00439-015-1577-y. [DOI] [PubMed] [Google Scholar]
- 4.Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014;7:221–234. doi: 10.2147/TACG.S46176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 2008;39:223–235. doi: 10.1016/j.pediatrneurol.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Gerards M, Sallevelt SCEH, Smeets HJM. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. 2016;117:300–312. doi: 10.1016/j.ymgme.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir L, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52. doi: 10.1186/1750-1172-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferdinandusse S, Waterham HR, Heales SJR, Brown GK, Hargreaves IP, Taanman J-W, et al. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J Rare Dis. 2013;8:188. doi: 10.1186/1750-1172-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, et al. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–2908. doi: 10.1093/brain/awu216. [DOI] [PubMed] [Google Scholar]
- 10.Haack TB, Jackson CB, Murayama K, Kremer LS, Schaller A, Kotzaeridou U, et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol. 2015;2:492–509. doi: 10.1002/acn3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakai C, Yamaguchi S, Sasaki M, Miyamoto Y, Matsushima Y, Goto Y-I. ECHS1 mutations cause combined respiratory chain deficiency resulting in Leigh syndrome. Hum Mutat. 2015;36:232–239. doi: 10.1002/humu.22730. [DOI] [PubMed] [Google Scholar]
- 12.Hedges D, Burges D, Powell E, Almonte C, Huang J, Young S, et al. Exome sequencing of a multigenerational human pedigree. PLoS One. 2009;4:e8232. doi: 10.1371/journal.pone.0008232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhlenbäumer G, Hullmann J, Appenzeller S. Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat. 2011;32:144–151. doi: 10.1002/humu.21400. [DOI] [PubMed] [Google Scholar]
- 14.Ferdinandusse S, Friederich MW, Burlina A, Ruiter JPN, Coughlin CR, Dishop MK, et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J Rare Dis. 2015;10:79. doi: 10.1186/s13023-015-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 16.Masso M, Vaisman II. AUTO-MUTE 2.0: a portable framework with enhanced capabilities for predicting protein functional consequences upon mutation. Adv Bioinforma. 2014;2014:278385–278387. doi: 10.1155/2014/278385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods used to perform genetic analyses. (DOCX 350 kb)