

Abstract
Programmable hydrogels are defined as hydrogels that are able to change their properties and functions periodically, reversibly and/or sequentially on demand. They are different from those responsive hydrogels whose changes are passive or cannot be stopped or reversed once started and vice versa. The purpose of this review is to summarize major progress in developing programmable hydrogels from the viewpoints of principles, functions and biomedical applications. The principles are first introduced in three categories including biological, chemical and physical stimulation. With the stimulation, programmable hydrogels can undergo functional changes in dimension, mechanical support, cell attachment and molecular sequestration, which are introduced in the middle of this review. The last section is focused on the introduction and discussion of four biomedical applications including mechanistic studies in mechanobiology, tissue engineering, cell separation and protein delivery.
1. Introduction
An important law of natural selection is that survival is dependent on the ability to change and adapt to environmental conditions. Otherwise, living beings are eliminated through selection. For example, dinosaurs had been powerful vertebrates for millions of years on the earth; however, they disappeared from this planet as they failed to adapt to the changes of their ecosystem.[1] In contrast, a variety of small animals have the capabilities to survive because they can make diverse changes when necessary. For instance, octopuses and squids living in the undersea realm are skilled in color or pattern change for their protection from predation.[2] These outcomes of natural selection demonstrate the significance of change and adaptation that have become a fundamental guideline for researchers to explore novel materials.
Hydrogels are hydrophilic polymer networks that are formed through crosslinking of monomers or polymer chains via covalent bonds and/or noncovalent interactions such as hydrogen bonding, electrostatic interactions, host-guest complexation and their combinations. These covalent and noncovalent interactions are usually stable with the ability against environmental assaults to maintain the structural or functional integrity of hydrogels. While this stability is critical for various biomaterials (e.g., contact lenses), hydrogels are often required to change their functions to meet special needs as exemplified and discussed herein.
Passive hydrolysis is the simplest and most straightforward strategy used to change the properties of hydrogels. A typical example is degradable poly(ethylene glycol) (PEG) hydrogel.[3–5] Younes et al. synthesized poly (ether ester) block copolymers using PEG and poly(lactic acid) (PLA).[6] The copolymeric PEG/PLA matrices can be hydrolyzed under physiological conditions for the degradation of PLA. When this polymer is functionalized with dimethacrylate, it can form degradable PEG hydrogels that can undergo changes in swelling ratio, mesh size and storage modulus.[3–5] In addition to synthetic polymers, natural biopolymers including proteins, polysaccharides and DNA have been widely used to synthesize degradable hydrogels.[7–9] However, passive hydrolysis is difficult to control once it is started.
Great attention has been paid to explore hydrogels whose functional changes can be controlled. This research area has been inspired not only by the natural selection of living beings, but also the biological mechanisms observed at the molecular and cellular levels. For example, the growth of new blood vessels (i.e., angiogenesis) is a multi-stage procedure, each step of which requires different signaling molecules whose spatial-temporal copresence can lead to the cancellation of their functions in cell regulation.[10] Thus, in such a case, it is desirable to develop hydrogels that can be used to control the release time, dose and order of different angiogenic signaling molecules, which is difficult to achieve with passive hydrolysis, dissolution or swelling. There are two ways of controlling the functional change of hydrogels. One is a one-shot switch, which means that functional properties unidirectionally change for one single time but the functional change is hard to repeat and/or reverse. A typical example is the gel-sol transition for hydrogel dissolution.[11] The other is a periodic, reversible or sequential change of one single or multiple functions. Hydrogels that change in the second way are defined as programmable hydrogels. This review is focused on the introduction of programmable hydrogels in principles, functions and biomedical applications.
2. Principles
Both internal and external factors need to be considered for the development of programmable hydrogels. Internal factors are covalent bonds and/or noncovalent interactions and external factors are environmental stimuli that are applied to trigger internal factors. The change of programmable hydrogels depends on how polymer chains, structures or their functional side groups are responsive to environmental stimulation. Internal and external factors can be rationally combined for the development of a diverse array of hydrogels to change periodically, independently and/or sequentially in functional structures and properties. In this review, the principles will be introduced from the viewpoint of environmental stimulation. Environmental stimuli are classified into three categories including biological, chemical and physical stimuli (Table 1). Instead of introducing all stimuli, this review will focus on those commonly used in the development of programmable hydrogels or those with great potential for this development. Moreover, some of these stimuli are often converted from one to another to achieve one change. In such a situation, the introduction will be focused on the first stimulation that is the driving force for the initiation of the change.
Table 1.
Representative stimuli for potential development of programmable hydrogels.
2.1. Biological Stimuli
Hydrogels have been widely studied for biological and biomedical applications. Thus, hydrogels in response to biological stimuli have been extensively explored in the biomaterials community. Both biomacromolecules and small metabolites can in principle function as biological stimuli to induce the programmable change of hydrogels.
DNA and aptamer
While nucleic acids are genetic materials in the living systems, they have been recently used as building elements to develop a variety of synthetic materials.[9] Nagahara and Matsuda reported the first study to replace chemical crosslinkers with double-stranded DNA helices to synthesize a polyacrylamide hydrogel.[12] The ability to use double-stranded DNA as a crosslinker to develop hydrogels has been confirmed by many other research groups.[13–19] Notably, a double-stranded DNA helix can interact with a third strand that has the ability to displace one original strand of the helix. This procedure is called strand displacement and migration.[20,21] Thus, DNA can be used as not only a crosslinker but also a triggering molecule to reverse the DNA crosslinker. In the presence of a third DNA strand, the DNA crosslinker can be dissociated through competitive DNA hybridization via strand displacement.[19] As a result, the hydrogel can undergo structural and functional change owing this variation of crosslinking density in the presence of a triggering DNA from a gel to a solution.[17,19]
DNA not only recognizes its complementary sequences, but also non-nucleic acid molecules with rational discovery and design. Tuerk, Gold, Ellington and Szostak have developed a technology called systematic evolution of ligands by exponential enrichment (SELEX),[22,23] through which nucleic acid aptamers can be identified from DNA/RNA libraries to recognize a variety of molecules ranging from proteins to small chemicals with high affinities and specificities.[24] Nucleic acid aptamers are small and structurally stable with little immunogenicity. In addition, different from other affinity ligands such as antibodies, aptamers are chemically synthesized using standard solid state phosphoramidite reactions. It minimizes the batch-to-batch variation and improves the reproducibility of hydrogel systems. Aptamers have been used to functionalize hydrogels using different methods. For instance, aptamers with Acrydite can be chemically conjugated to the hydrogel matrix during free radical polymerization.[25,26] Aptamers can also be chemically or physically integrated onto the surface of particles that are physically mixed with a pre-gel solution to form particle/hydrogel composites.[27–29] It is important to note that nucleic acid aptamers have features of both ligands and single-stranded oligonucleotides and can recognize both target molecules and triggering complementary sequences (CSs).[30,31] Thus, rationally designed aptamers that can be used as either a crosslinker[16,17] or a functional side group of a hydrogel [25,26] can undergo a structural change in the presence of target molecules or CSs.[27,32,33] Worthy of noting is that DNA hybridization and aptamer-target recognition both have very high specificity. Thus, it is in principle feasible to incorporate an unlimited number of aptamers as functional side groups into one hydrogel to recognize different targets and/or their triggering DNA sequences for a diverse array of programmable changes, which cannot be easily achieved using any other ligands.[28]
Protein and peptide
Proteins and peptides have also been studied either as functional crosslinkers or functional side groups. Miyata et al. pioneered the development of antigen-responsive hydrogels with grafted antigens and corresponding antibodies.[34] The interactions between grafted antigens and antibodies allow for the formation of hydrogel through polyvalent antigen-antibody binding. As molecular recognition is controlled by physical forces, the bound two proteins can be exchanged with any of their free forms. Indeed the presence of free antigens in the surrounding of the antigen-antibody hydrogel can replace the antigens grafted to hydrogel network through binding competition.[34] Moreover, stepwise exposure of the hydrogel to free antigens can induce pulsatile change of the hydrogel in volume. Similar to the antibody systems, grafted peptides can be replaced by free peptides.[35]
While both DNA and proteins/peptides can be used as stimuli to program the change of hydrogels, it is also important to emphasize that these two families of biomolecules are different in system design and working efficiency. When a triggering DNA is applied to stimulate a DNA duplex, the length of this triggering DNA can be extended to form more base pairs with its target DNA than the displaced DNA duplex.[36] Thus, molecular competition is thermodynamically favorable for the triggered dissociation of the duplex. In contrast, free proteins and peptides are the same as grafted counterparts without favorable priority in molecular competition. Thus, a very large amount of free proteins or peptides have to be used to induce a change.[34,35]
Enzyme (e.g., MMP and DNase)
Enzymes play essential roles in many biochemical reactions. The human body has various enzymes and these enzymes can be used as triggering elements for the development of enzyme-responsive hydrogels. To develop these hydrogels, the substrates of enzymes need to be incorporated into hydrogels either as pendent groups or crosslinkers.[37] Notably, the hydrogels should possess the properties to allow enzymes to get access to the incorporated substrates. Depending on whether the substrates are incorporated into the hydrogels as crosslinkers or functional side groups, enzymatic reactions will lead to either changes in bulk properties or side groups.[38]
Matrix metalloproteinases (MMPs) are an important family of proteases that are responsible for the degradation of most extracellular matrix (ECM) proteins through MMP-catalyzed hydrolysis of peptide bonds during organogenesis or the development of human diseases.[39] Specific peptide domains have been incorporated into PEG hydrogels so that inert synthetic PEG hydrogels can be converted into MMP-responsive PEG hydrogels for dynamic change.[40] In addition to synthetic hydrogels such as PEG, peptide domains can be applied to functionalize natural polymers (e.g., hyaluronic acid) for the development of MMP-responsive hydrogels.[41] MMPs come from either the cells incorporated into the hydrogels or the microenvironment of the hydrogels, which depends on the requirement of an application.
DNases are enzymes with the ability to catalyze the hydrolysis of phosphodiester bonds of DNA.[42,43] Exonucleases cleave residues at the end of DNA molecules whereas endonucleases cleave phosphodiester bonds in the middle. Some of them cut DNA molecules without discrimination whereas others are very sequence-specific. DNases have been used to induce dynamic changes of DNA-functionalized hydrogels.[44] It is important to note that similar to MMP-responsive hydrogels, the accessibility of DNA incorporated into the hydrogels to DNases is critical to determine the sensitivity of the change. When DNA is tethered on the hydrogel surface, it would be easier for DNases to approach DNA.[44] In contrast, for the DNA molecules inside a hydrogel, the hydrogel chains may protect them from DNase degradation.[45]
Small metabolite (e.g., glucose and ATP)
Glucose is an important metabolite as it is a source of energy of the body. The concentration of glucose in the blood stream periodically goes up and down following the pattern of meals every day. Thus, glucose can be a good candidate as a triggering molecule for the development of programmable hydrogels. Three types of glucose-responsive hydrogels have been developed by using phenylboronic acid (PBA),[46,47] concanavalin A (Con A)[48] and glucose oxidase (GOD).[49–51] PBA and its functionalized polymers can form reversible covalent complexes with two hydroxyl groups of glucose.[46,47] PBA exhibits two states including the uncharged and charged states in equilibrium. In the presence of glucose, this equilibrium can shift more favorably towards the charged phenylborates through the PBA-glucose complexation, as the complexation between the uncharged PBA and glucose is very unstable in aqueous solutions.[47] When the concentration of glucose increases in the surrounding of a PBA-functionalized hydrogel, the amount of borate anions increases and that of the uncharged form decreases. Resultantly, as borate ions have higher hydrophilicity than the uncharged form, the solubility of polymer chains increases with the increase of the glucose concentration. Ultimately, the presence and absence of glucose can cause a sharp change in swelling and de-swelling of the PBA-functionalized hydrogels.[46] Con A is a lectin. It can bind to many sugar moieties.[52] In comparison to many other sugar moieties, Con A binds to D-glucose more strongly. Notably, Con A is a homotetramer with four binding sites for sugar moieties.[53] In the active state, Con A can induce the formation of hydrogel when mixed with polymers displaying glucose moieties via polyvalent complexation.[48,54] However, molecular binding is reversible. The presence of free glucose can replace the glucose moieties displayed by polymer chains. It leads to the dissociation of polyvalent Con A-polymer complexes and the gel-solution transition of the hydrogel.[54] Con A can be used by itself in the synthesis of hydrogels. However, the passive dissociation of Con A from the hydrogel system in the absence of glucose can weaken the sensitivity of the hydrogel to the environment. Thus, Con A has been chemically conjugated to the hydrogel component via different chemical reactions such as carbodiimide reaction to immobilize Con A to the hydrogel matrix, which restricts the loss of Con A and improves the sensitivity of hydrogels to glucose.[54,55] Glucose oxidase (GOD) is an oxidoreductase that can catalyze the transformation of glucose to gluconolactone that further hydrolyzes to gluconic acid.[49,50] As the ultimate product of this enzymatic reaction is gluconic acid, the pH microenvironment of a hydrogel with GOD will decrease in the presence of glucose. If the hydrogel has components responsive to low pH, the hydrogel can undergo glucose-triggered pH-responsive changes.[51] Depending on the functional components or groups in the hydrogel matrix, the hydrogel can change its state of protonation, molecular substitution or phase. The pH-responsive hydrogels will be discussed in more details in section 2.2.
Adenosine triphosphate (ATP) and its derivatives are important small molecules in the metabolism as they are not only used as a direct source of energy but also signaling molecules to regulate cell behavior.[56–58] Studies have shown that cells can release ATP or its derivatives,[56] which can interact with the receptors of the same cell releasing this metabolite, neighboring cells, or even both for signal transduction pathways.[56–58] From the viewpoint of biomimicry, it is possible to develop programmable hydrogels that can mimic this natural phenomenon for cell regulation or drug delivery. An important example in this research direction is to apply anti-ATP aptamer to functionalize hydrogels.[16] When the aptamer binds to ATP, its conformational structure is changed. Thus, when a double-stranded DNA helix with this aptamer and its complementary sequence is used to crosslink hydrogel chains, the presence of ATP can cause the dissociation of this helix.[32] As a result, the state of the hydrogel is changed from gel to solution.[16] It is also possible to conjugate the aptamer as a functional side group of the hydrogel matrix.[59] In this scenario, the overall structure of the hydrogel matrix does not change whereas the side functional groups of the hydrogel change their binding state in the presence of ATP or its derivatives. The key difference of these two ATP-responsive hydrogels is that the former hydrogel loses its integrity during the change and the latter does not have to sacrifice the integrity.
2.2. Chemical Stimuli
Redox
Reduction-oxidation reactions (Redox) are chemical reactions involving the change of the oxidation states of atoms. A chemical bond in its reducing state can crosslink polymer chains whereas its reduction leads to the dissociation of the chains. A very typical example of Redox is the conversion of disulfide (S-S) bonds into thiols.[60,61] The Prestwich group developed disulfide cross-linked hyaluronan (HA) hydrogels by mixing thiol-modified HA macromers and exposing the polymer solution to air and/or hydrogen peroxide.[60,61] When the formed hydrogel is in contact with dithiothreitol (DTT), disulfide bonds can be reduced to form thiols. Resultantly, the disulfide cross-linked hydrogels can switch from gel to solution. Disulfide cross-links can also be cleaved using natural reductants such as glutathione (GSH) and cysteine.[62] Notably, the formed thiol groups can be reversibly oxidized to form disulfide bonds. Similar to the disulfide bonds, the diselenide (Se-Se) bonds can be used as crosslinkers for the synthesis of hydrogels.[63] The diselenide bonds are oxidized to seleninic acid in an oxidizing environment with reactive oxygen species (e.g., hydrogen peroxide) and reduced to selenol in a reducing environment with reducing agents (e.g., GSH).[64,65] It is important to point out that the Se-Se bonds have lower bond energy (172 kJ/Mol) than the S-S bonds (240 kJ/Mol).[66] Thus, it is easier to cleave the Se-Se bonds.
Ions
Polymer chains can be crosslinked together by multivalent ions to form hydrogels. A typical example is alginate hydrogels.[67] Alginate consists of guluronic acid (G) and mannuronic acid (M) units, both of which have a carboxyl group. The carboxyl groups and bivalent cations such as Ca2+ can form egg-box-like junctions.[68,69] When alginate hydrogels are incubated in an environment with monovalent cations (e.g., Na+), bivalent cations can be replaced by the monovalent ions.[69] This ion exchange can lead to the partial dissolution of alginate hydrogel or at least significant swelling. Moreover, chelating agents such as citrate can compete against carboxyl groups to acquire bivalent ions from the hydrogel networks, which can cause the complete dissolution of the hydrogels.[70] This procedure is reversible when the swelling alginate hydrogels are incubated in an environment with bivalent cations. Thus, by switching the treatment of alginate hydrogels with different cations or chelating agents, alginate hydrogels can be programmed to change their structures and functions. Pectin is another interesting natural polysaccharide. It has a branched structure. Similar to alginate, pectin forms hydrogels with the egg-box-like junctions in the presence of bivalent or trivalent cations and the functions can be reversed quickly by monovalent cations.[71]
pH
pH-responsive polymer chains change mainly through two mechanisms.[72] One is protonation and the other is the cleavage of acid-labile chemical bonds. Protonation relies on the presence of pendant acidic or basic groups such as carboxyl, sulfonic, phosphate, pyridine and tertiary amines.[72] These functional groups accept protons at low pH whereas releasing protons at high pH. Depending on their sensitivity to the pH range, hydrogels made of these functional groups are classified into pH-responsive acidic hydrogels and pH-responsive basic hydrogels.[72] They undergo ionization with pH change, which will ultimately lead to structural and functional changes of hydrogels. Acidic hydrogels with acidic functional groups are negatively charged at high pH through proton release. Basic hydrogels with basic functional groups are positively charged at low pH through proton acceptance. During the switch of the state of charge, ionization will cause the hydrogels to swell or shrink. In addition to protonation, cleavage is also important to the development of pH-responsive hydrogels. Cleavage depends on the degradation of pH-labile bonds used for the synthesis of the backbones, crosslinkers or pendant groups of hydrogels.[72] Commonly studied pH-labile bonds include acetal/ketal, hydrazone, orthoester, imine, etc.[72] For instance, the Frechet group applied an acetal crosslinker to synthesize a microgel, showing that this microgel was degraded faster at pH 5.0 than at pH 7.4.[73] Different from either protonation or cleavage, polymer-conjugated PBA and salicylhydroxamic acid (SHA) can be used to synthesize programmable hydrogels through reversible covalent bonds that are formed or broken based on the environmental pH values.[74] Another type of pH-responsive hydrogels can be made of i-motif structure that forms with cytosine-rich DNA sequences. The i-motif structure is a four-stranded complex that has two anti-parallel based-paired parallel-stranded duplexes.[75] This structure is stable at low pH but is unstable at high pH. This feature can be applied to develop fast pH-responsive programmable hydrogels.[76,77] I-motif-cross-linked hydrogels undergo reversible transition repeatedly when pH changes between acidic and basic conditions.[76,77]
2.3. Physical Stimuli
Temperature
The solubility of thermo-responsive polymers in aqueous solutions can remarkably change with the variation of temperature. Some of them dissolve in an upper critical solution temperature (UCST) process and others dissolve in a lower critical solution temperature (LCST) process.[78] The change of their solubility depends on the function of temperature versus the concentration. A typical example of thermo-responsive polymers is poly(N-isopropylacrylamide) (PNIPAAm).[79] It exhibits an inverse solubility upon the increase of the temperature above LCST.[80,81] At a temperature lower than LCST, water and the amide groups form H bonds and the ordering of water through isopropyl groups considerably increases. It leads to the significant decrease of both the enthalpy and the entropy of mixing.[79] In contrast, at a temperature higher than LCST, the entropy term of mixing dominates the enthalpy term. It results in a positive free energy, which causes the dehydration and precipitation of PNIPAAm.[79] PNIPAAm can be synthesized in the form of either single chains or cross-linked hydrogels. The methods used for the synthesis of single chains can be simply tuned to developed cross-linked PNIPAAm hydrogels by the addition of a crosslinker into the reaction solution. PNIPAAm can also be functionalized by the addition of other monomers into the reaction solution or physically mixed with other macromolecules without significant interference with its overall thermosensitivity.[79]
Light
Light has been widely studied as an external physical stimulus for controlling the behavior of hydrogels. The irradiation of light is easy and non-invasive to apply. It does not need direct contact and can be delivered remotely. The whole light spectrum has a broad range from UV to near infrared. Depending on the wavelength, the depth of penetration ranges from a few microns to several centimeters.[82–84] UV and visible light have been vastly studied for topical use on the skin and infrared has been employed in the internal tissues or organs. Importantly, light allows stimulation within a defined location at a macroscopic level in general. A variety of functional groups can respond to light through the mechanisms of photocleavage, photoisomerization and photodimerization, including o-nitrobenzylester, azobenzene, coumarin, etc.[85–89] For instance, Luo and Shoichet synthesized a photolabile poly(ethylene glycol) hydrogel with a nitrobenzyl protected moiety;[87] and Fomina and co-workers developed a photodegradable polymer that contained a quinone-methide-based backbone protected with o-nitrobenzyl photo-sensitive groups.[89] Upon the exposure to light, these functional groups can undergo rapid photolysis and degradation.
Mechanical force
The mechanical behavior of hydrogels is determined by the crosslinking density, the molecular weight of polymer chains and the amount of water inside the hydrogel.[90] Nearly all hydrogels exhibit the combinatorial behavior of elasticity, flow and molecular motion. Thus, hydrogels are considered as viscoelastic materials.[90] However, for most hydrogels, when a mechanical force is applied to produce a small strain, the hydrogel exhibits rubber-like elastic behavior, i.e., responding to external mechanical forces with reversible deformation and recovery.[91] The elastic modulus of hydrogels depends on the crosslinking densities of the polymer network and the polymer concentration. However, beyond a small percentage of their initial length, hydrogels can rupture easily.[91] To solve this problem, hydrogels with a semi-interpenetrating network (IPN) have been studied.[92] The IPN consists of two or more networks that are not covalently bonded together. For instance, polyacrylamide and sodium alginate have been integrated to form a hydrogel hybrid with an IPN structure.[92,93] This hydrogel exhibits resilience and rubber-like behavior. The hydrogel hybrid can be stretched to 2 to 17 times of their original length and subsequently recover.[92,93] The hydrogel hybrid can deform and recover their original morphology even after 20 times of cyclic loading and unloading treatment.[92] Thus, mechanical force can be used as a physical stimulus to control the properties of rationally designed hydrogels.
Electric field
It has been widely studied to apply an electric current in vivo in laboratory experiments and clinical applications such as neuropharmacology and transdermal drug delivery.[94] Similar to other physical stimuli, it is easy to control various parameters such as the magnitude of current, duration of each electric pulse and intervals between electric pulses.[94] Thus, using electric fields as a physical stimulus has attracted significant attention in the development of programmable hydrogels. The hydrogels that are responsive to electrical stimulation are mostly prepared from negatively charged polyelectrolytes.[95] The application of an electric field to polyelectrolyte hydrogels can produce a stress gradient in the hydrogel,[96] change local pH values,[97,98] and/or generate electroosmosis of water.[99,100] Moreover, polyelectrolyte hydrogels can erode under an electric field.[94,95] These hydrogels are usually made of two soluble polymers that interact with each other through H-bonds or ionic bonds, or two oppositely-charged polymers.[94,95]
Magnetic field
Magnetic nanoparticles or microparticles can be either physically dispersed or chemically crosslinked in hydrogels.[101] In the presence of external magnetic fields, magnetic particles can produce two important effects. One is hyperthermia that is governed by the transformation of magnetic energy to heat.[102] The other is the macroscopic motion of magnetic particles driven by the magnetic fields.[103,104] As the temperature in a magnetic hydrogel system can increase,[105] any thermo-responsive hydrogels can in principle be developed into magnetic field-responsive hydrogels. The advantage of magnetic field-responsive hydrogels is that the effect of temperature variation on their functional changes can be remotely controlled. Moreover, as magnetic particles can move towards one direction or change the direction of motion in the presence of a fixed or alternating magnetic field, the dimension of hydrogels can be changed to shrink and swell.[101]
3. Functions
When hydrogels are subject to environmental stimulation, their functional structures and properties change in different or coherent ways. As this review focuses on biomedical applications, functional changes will be mainly discussed in the following four aspects including dimension, mechanical support, cell attachment and molecular sequestration (Figure 1), which are more relevant to biomedical applications.
Figure 1.

Schematic illustration of functional changes. (a) Dimensional change during shrinking and swelling. (b) Change of mechanical support during sequential crosslinking or cleavage. Adapted from Refs. 88 and 143 with permission. (c) Programmable cell attachment and detachment controlled by temperature variation (c1) and the display of functional aptamers (c2-4). Adapted from Refs. 144, 161, 122 and 123 with permission. (d) Comparison between affinity-based molecular recognition and physical entrapment for molecular sequestration.
3.1 Dimension
Hydrogels provide a three-dimensional space for anything that resides inside or interacts with the hydrogels. Thus, the change of the dimension in volume, pore space and/or shape is important to various hydrogel-based applications.[106] As hydrogels contain a high concentration of hydrophilic polymers compared to the surroundings, an osmotic pressure is produced between the inside and the outside of hydrogel.[107] Thus it is thermodynamically favorable for water to enter the network of hydrogel. The penetration of water molecules into the hydrogel network leads to the increase of the hydrogel volume (i.e., swelling) and the decrease of the polymer concentration, which in turn decreases the osmotic pressure. As the polymer chains in the hydrogel network are connected by crosslinkers, the ability of the polymer chains to disperse in the space is restricted by the chemical and/or physical bonds between the chains and the elastic deformation of the polymer chains.[106] With the increase of the swelling, the elastic forces resulting from deformation will increase significantly. The balance between the osmotic pressure and the elastic forces determines the ultimate swelling pressure or degree. When the swelling pressure reaches zero, the swelling reaches equilibrium and the volume does not change. If the swelling pressure is positive, hydrogel will further swell. Otherwise, hydrogel will shrink if the swelling pressure is negative. In addition to the osmotic pressure and the elastic forces, the swelling pressure or degree is determined by the charge of polymer chains and the concentration of electrolytes in the hydrogel system.[108] By modulating the swelling pressure via different parameters such as crosslinking density, charge density, chain length and chain distance, one can modulate the dimensional change of hydrogel. For example, magnetic hydrogels have been developed by dispersing magnetic particles into the hydrogel network.[101] These hydrogels can be triggered by an external magnetic field. When magnetic particles are attracted to magnets, the polymer chains will be driven to aggregate. It produces an attraction force against the swelling pressure, which leads to shrinkage of the hydrogel volume and pore size. The dimensional change of a hydrogel material does not necessarily have to be macroscopically homogeneous. By changing the swelling pressure at different locations of a hydrogel material, one can differently change the dimensions of hydrogel in volume, pore space and/or shape (Figure 1a).[109] With the dimensional change, other functional properties may change simultaneously. For example, the permeability of solutes can either increase or decrease, depending on whether the hydrogel swells or shrinks.
3.2 Mechanical Support
Mechanical support is an important function of hydrogels. The ability to control the mechanical behavior of hydrogels has received great attention. The mechanical behavior can be characterized in shear rheometry, tension, compression and local indentation.[110] For instance, shear rheometry is often measured to examine the change of the storage and loss moduli of hydrogels since hydrogels are intrinsically viscoelastic with time-dependent deformation owing to fluid flow in the interstitial space of hydrogels.
A cross-linking PEG diacrylate macromer with two nitrobenzyl groups has been applied as a crosslinker to synthesize a photocleavable hydrogel.[88] Upon light exposure, the crosslinkers underwent cleavage (Figure 1b). It led to a significant decrease in the storage modulus. Depending on whether light exposure was continuous or alternate, the storage modulus decreased continuously or sequentially.[88] Resultantly, the hydrogel was programmed by light to soften in functional support of the live cells. Hydrogels can also be stiffened via sequential crosslinking with the principles of Michael addition reaction and free radical polymerization (Figure 1b).[111] In addition to the unidirectional softening or stiffening, hydrogels can be developed to switch alternately between the soft and stiff states. For instance, in a hybrid collagen/alginate hydrogel system, the alginate component was used to manipulate the mechanical property of the hybrid hydrogel by the alternate exposure to Ca2+ and citrate.[112]
3.3 Cell Attachment
It is easier for cells to adhere and grow on hydrophobic substrates than on hydrophilic surfaces. Thus, controlling the hydrophobicity and hydrophilicity at the substrate-cell interface is an effective method to regulate cell binding.[113,114] Takezawa et al. and Okano et al. independently developed a thin PNIPAAm hydrogel film on the culture dishes to regulate the state of cell attachment (Figure 1c).[113,114] At 37 °C, PNIPAAm dehydrates to become hydrophobic, which allows for cell attachment and growth. At a temperature lower than LCST, PNIPAAm fully hydrates and become hydrophilic, which leads to cell detachment.
Most hydrogels are biologically inert without cell binding sites. Thus, it is often important to functionalize hydrogels with functional moieties for cell attachment. Natural adhesion proteins (e.g., collagen) found in the extracellular matrix (ECM) can be used alone or incorporated into hydrogel for cell binding.[115] However, while these adhesion proteins provide cells with the binding sites similar to the natural ECM, their uses may be limited by denaturation during the synthesis of hydrogels or fast degradation.[116,117] Small adhesion peptide sequences derived from the adhesion proteins have also been widely applied to functionalize hydrogels for cell adhesion. The small peptides have advantages such as easy synthesis through chemical routes, easy conjugation to polymer chains and little immunogenicity. However, peptides often require circularization and multimerization to achieve sufficient stability and binding affinity.[116] It is particularly important to emphasize that most peptides bind to target cell receptors with very high dissociation constants at the μM levels that indicate very low binding affinities.[118] In contrast, nucleic acid aptamers can bind to target molecules with very low dissociation constants from nM to pM.[119] When these biomolecules are used to functionalize hydrogels, their states can be modulated to regulate the outcomes of cell binding. A cell-binding resistant hydrogel can be activated by light to display reactive functional groups that can further react with cell adhesion biomolecules for cell binding.[87] Using this method, the immobilization of biomolecules in the hydrogel can be spatially controlled for cell binding. This method can be tuned to conjugate a caged biomolecule into the hydrogel.[85,86] Once triggered by light, the caging groups are removed and the cell adhesion molecules are exposed to the cells for binding. This method can be further tuned by using sequential and/or reversible chemical reactions to present and release cell adhesion molecules.[120,121]
Different from those strategies that are based on chemical conjugation and bond cleavage, specific and competitive molecular recognition can be applied to control the display of cell binding biomolecules. One example is the display of the RGD peptide via peptide-peptide interactions. By using a pair of acidic and basic leucine zipper domains, the cell-binding RGD peptide and the shielding PEG can be reversibly displayed on cell culture dishes to regulate cell binding and release.[35] Compared to this peptide-based competition, nucleic acid-based hybridization has more diversity in binding control.[122–125] Wang and co-workers applied nucleic acid aptamers to develop three types of hydrogels for the control of cell adhesion (Figure 1c).[122,123,125] The forms of aptamers include conjugated active aptamer,[125] conjugated inert aptamer,[122] and hybridized active aptamer.[123] In the first two forms, the conjugated aptamers can reversibly change their active and inert conformational structures through hybridization with complementary sequences.[122,125] In the third form, the hybridized aptamer is not subject to activation or inactivation but it is released and replenished for cell binding to the hydrogel.[123] Similar to this third form, the DNA-antibody conjugate can also be displayed and released on the hydrogel through DNA hybridization to regulate cell binding and release.[124]
3.4 Molecular Sequestration
Hydrogels are inherently a porous material. On one hand, the pores allow molecules to be absorbed into the hydrogel networks; on the other hand, the absorbed molecules may leave from the pores easily. [126–128] To stably sequester molecules of interest, the pore size needs to be tuned to decrease after molecule absorption. It can in principle be realized by controlling the density of hydrogel chains and the length of crosslinkers. However, there is a limit for the decrease of the pore size.[129] Moreover, in numerous biomedical applications such as tissue engineering and regenerative medicine, the small size will negatively affect other properties such as cell infiltration and nutrient transport. Instead of decreasing the pore size, researchers have developed another strategy for molecular sequestration based on molecular recognition (Figure 1d).
Heparin is a highly sulfated linear polysaccharide that can bind proteins with cationic residues (e.g., lysine) with medium to high affinity (as indicated by KD of 10-6 to 10−9 M). Great effort has been made to apply heparin to functionalize hydrogel for protein sequestration based on electrostatic heparin-protein interactions, which is similar to heparan sulfate that is an important component discovered in the natural extracellular matrix (ECM) in sequestration of soluble signaling molecules.[130] While heparin is very powerful in developing protein-sequestering hydrogels, a few issues need to be borne in mind for using it to develop programmable hydrogels. Firstly, heparin binds to a diverse array of proteins without high specificity. Secondly, as heparin-mediated protein binding is mainly limited to positively charged molecules owing to the dependence on electrostatic interactions, heparin may not be able to sequester neutral or negatively charged proteins in the hydrogels. Thirdly, as a natural polymer, heparin has batch-to-batch variation in molecular weight and molecular weight distribution, which is often overlooked but important to determine the reproducibility of manufacturing high-quality hydrogels. Fourthly, when heparin binds to a certain protein, it is hard to regulate this molecular sequestration except the use of heparinase for the potential of developing programmable hydrogels.
Peptide-mediated molecular sequestration may solve some of the issues associated with heparin such as binding specificity and reproducibility.[118] Based on crystallographic analysis, site-directed mutagenesis studies and phage display selection, functional peptides can be identified for recognition of target proteins.[118] Once peptides are defined, they can be chemically synthesized. It limits the issues related to reproducibility or endotoxin contamination that is often a problem for biologics purified from animal tissues. Many peptides interact with their target proteins mainly through hydrophobic interactions. For instance, a single16-mer peptide identified from α2-Macroglobulin in binding transforming growth factor-β (TGF-β) includes 12 hydrophobic amino acids.[131] However, as peptides are smaller and their binding to target proteins are mainly based on hydrophobic interactions, the binding affinities of most peptides against proteins are low at the μM level.[118] Moreover, while peptides are not the same as heparin in binding to a variety of molecules promiscuously, the binding specificity of peptides can still be an issue. For instance, the single16-mer peptide identified from α2-Macroglobulin binds both (TGF-β) and platelet-derived growth factor (PDGF-BB) is virtually identical while these two proteins do not share any significant sequence identity.[131] Moreover, the regulation of peptide-protein binding would be hard except the use of an identical peptide sequence at a high concentration to compete the initially bound peptide.[35]
Nucleic acid aptamers may offer an opportunity to address the aforementioned issues of molecular sequestration for the following reasons. Firstly, in principle, aptamers can be selected against target proteins with binding affinities and specificities comparable to or even higher than antibodies.[24,132–134] It is particularly important to emphasize that aptamer selection has been significantly advanced. An important advance is the development of aptamers with modified nucleotides at the 5-position with hydrophobic aromatic moieties (i.e., SOMAmers).[132,133,135] As these aptamers have protein-like side chains, their binding affinities have been tremendously increased from the nM to pM level.[132,135] It is also important to note that as aptamers bind targets with high specificities, multiple aptamers can be integrated into one hydrogel for independent sequestration of multiple proteins simultaneously. Secondly, truncated aptamers have lengths mainly ranging from 20 to 40 nt. Thus, it is easy to synthesize aptamers with the standard solid-state oligonucleotide synthesis method without batch-to-batch variation or biological contamination. Thirdly, aptamers can be chemically integrated into hydrogels without the concern about possible denaturation as by nature nucleic acids are stable in various harsh chemical conditions. Fourthly, as single-stranded oligonucleotides, nucleic acid aptamers can bind to not only target proteins but also their complementary sequences by nature. Worthy of emphasizing is that it is easy to add a non-essential nucleotide tail to either 3’, 5’ or even both ends of an aptamer, with which the complementary sequences can bind the aptamer more easily to compete against the target protein.[27,28,36] It will allow for the convenient regulation of aptamer-mediated molecular sequestration.
Wang and co-workers have systematically studied the functionality of aptamers as pendent groups of hydrogels for protein sequestration.[25–29,36,136] In the first study, PDGF-BB and its aptamer were incorporated into a polyacrylamide hydrogel with free radical polymerization.[25] The sequestration efficiency of the aptamer-functionalized hydrogel was ~90% whereas the native hydrogel was ~30%. While the data clearly showed that nucleic acid aptamers could sequester PDGF-BB in the hydrogel, it was also found that the efficiency of aptamer incorporation into the hydrogel was ~90%. Increasing the initial aptamer amount further decreased the incorporation efficiency.[136] This issue is important as the free aptamers will neutralize the freed proteins. It is also important to note that proteins have different sensitivity to free radicals. To avoid protein denaturation resulting from free radicals, they applied aptamers to functionalize particles, washed the particles and incorporated the aptamer-functionalized particles into an agarose or poloxamer hydrogel.[27–29] However, both methods regardless of the involvement of free radicals or particles require the simultaneous incorporation of proteins and aptamers (or aptamer-functionalized particles) into the hydrogels during the synthesis of the hydrogels. To develop an off-the-shelf protein-sequestering biomaterial that would be ideal in real-world applications, it is necessary to decouple the synthesis of biomaterials from the loading of proteins. To achieve this goal, the same research group developed an aptamer-functionalized superporous hydrogel for protein sequestration.[26,59,136,137] In this method, aptamers and monomers were first polymerized to form the superporous hydrogel without proteins. After the formation of the superporous hydrogel, the hydrogel was thoroughly washed to remove unreacted molecules and catalysts. After the washing, the hydrogels were loaded with fresh proteins, which could be facilitated by the dehydration of hydrogels.[26,136] As protein loading was decoupled from hydrogel synthesis, the procedure of hydrogel synthesis did not affect the protein bioactivity. Importantly, the results showed that the hydrogel functionalized with high-affinity aptamers could sequester 99.7% of loaded PDGF-BB, which means that only 0.3% of loaded PDGF-BB was washed away from the superporous hydrogel during the 24-h washing incubation.[26] It clearly shows the power of nucleic acid aptamers in molecular sequestration in highly permeable porous hydrogels.
4. Applications
Programmable hydrogels could be developed with the aforementioned principles and stimulated to undergo functional changes for various promising applications. To limit the scope of the following discussion, four applications will be presented and certain case studies closely relevant to these applications will be discussed, including mechanistic studies in mechanobiology, tissue engineering, cell separation and protein delivery.
4.1 Mechanistic Studies in Mechanobiology
Hydrogels have been widely studied to understand fundamental mechanisms in mechanobiology, particularly cell-ECM interactions, primarily because hydrogels exhibit structural and mechanical similarities to human tissues.[138–140] However, most hydrogels were historically developed as static substrates to study cell-ECM interactions. This characteristic is different from the nature of dynamic changes that are an inherent characteristic of the ECM and can be easily found during many developmental and disease processes such as embryo development, inflammation and cancer progression.[141,142] Therefore, there is a great need to develop programmable hydrogels whose mechanical properties can be changed under control to mimic these biological processes for basic studies.
A hydrogel can change its stiffness from a stiff state to a soft state and vice versa. These changes can be programmed using various stimuli as described in section 2 such as ions, pH, light, temperature and biomolecules (e.g., DNA). The convenient way of changing stiffness is to control the crosslinking density of hydrogels. Photolabile crosslinkers have been incorporated into the hydrogel network as the backbone[88] in addition to being used as pendent groups.[87] Upon sequential on-and-off light exposure, the degradation was triggered to start and stop (Figure 1b). As a result, the storage modulus of the hydrogel decreased sequentially (Figure 2a).[88] Moreover, as expected, the degree of decrease in storage modulus could be controlled by the variation of exposure time and light intensity.[88] By inducing the degradation of the hydrogel, the cell morphology could be changed from a rounded shape to an elongated shape (Figure 2a),[88] which also indicates the possible change of mechanotransduction pathways. Mechanotransduction can also be achieved by regulating the density of RGD peptides conjugated to the hydrogel. Anseth and co-workers further showed that upon the removal of RGD peptides from the hydrogel in the presence of human mesenchymal stem cells (MSCs), the production of glycosaminoglycans (GAGs) could be increased by four times within approximately 10 days.[88] These studies demonstrated that programmable hydrogels are a promising tool for understanding cell interaction with dynamic microenvironment and cell differentiation in a three-dimensional space.
Figure 2.

Sequential cleavage and crosslinking of hydrogels. (a) Light-mediated sequential cleavage for hydrogel softening. F: rounded shape of a cell in the hydrogel without light treatment; G: elongated shape of a cell in the hydrogel with light treatment (from ref. 88 with permission). (b) Sequential polymerization for hydrogel stiffening (from ref. 143 with permission).
In addition to reducing the stiffness of a hydrogel sequentially, it is possible to increase the stiffness of hydrogels via sequential crosslinking. Burdick and co-workers applied a Michael addition reaction and photo-initiated free radical polymerization to regulate the stiffness of hyaluronic acid hydrogels (Figure 2b).[111,143] Following these two reactions, the value of the Young’s modulus was doubled quickly.[111] Later studies further showed that step-wise light exposure led to the sequential increase of stiffness.[143] The traction force could be increased by one order of magnitude within 4h after hydrogel stiffening, during which the average cell area was increased by nearly three times.[143] In addition to examining this short-term cell response to the change of the stiffness, the same research group also examined the long-term cell response, finding that the responses of differentiating and undifferentiated human MSCs to mechanical changes were different. Undifferentiated rather than differentiating cells responded to mechanical changes more favorably.[143] Moreover, the results suggest that later stiffening is beneficial for adipogenic differentiation and earlier stiffening is beneficial for osteogenic differentiation.[143]
These aforementioned studies have demonstrated that programmable stiffening or softening of hydrogels can be achieved for basic understanding of cell response to the microenvironment by integrating reactive functional moieties into pendent groups or crosslinkers in the presence of live cells. However, during the softening or stiffening procedure, other properties of hydrogels are changed simultaneously. For instance, during the secondary crosslinking or triggered degradation, it is inevitable to change the pore size of the hydrogel. When cells are cultured in a three-dimensional space, molecular transport will definitely change with the change of the pore size. It may be another factor that may affect cell response to the microenvironment during the change of matrix stiffness. Moreover, while sequential and unidirectional softening or stiffening can be programmed to understand cell behavior in a dynamic microenvironment,[88,143] a further in-depth understanding of cell behavior may be achieved by using reversible programmable hydrogels.[112]
4.2 Tissue Engineering
Programmable hydrogels hold great potential for tissue engineering applications. A typical example is to develop a temperature-programmed hydrogel surface using PNIPAAm to engineer functional cell sheets (Figure 1C). This seminal work was initially started in 1990 by two research groups.[113,114]
Takezawa et al. mixed PNIPAAm and collagen to prepare a homogenous solution, which was uniformly casted on the dish surface to form a thin hydrogel film for the culture of human dermal fibroblasts at 37 °C.[113] In the presence of collagen, the PNIPAAm hydrogel coating allowed the cells to attach and grow on the PNIPAAm-coated culture surface. After the cells reached the confluency, the cell sheet was detached and floated from the surface in 15 min without cell-cell dissociation when the temperature was shifted from 37 °C to 15 °C.
Instead of coating pre-synthesized PNIPAAm on the dish surface, Okano and co-workers directly grafted the PNIPAAm coating onto the dish surface.[114] (Strictly speaking, this hydrogel coating is more similar to a polymer brush system.) The measurement of contact angles showed that the hydrophobicity of the grafted surface reversibly changed from the hydrophilic state to the hydrophobic state when the temperature was shifted from 10 °C to 37 °C and vice versa. While they did not use any specific cell adhesion molecules (e.g., collagen), the degree of cell attachment and growth on the PNIPAAm-grafted surface was the same as that on the traditional dishes. Moreover, nearly 100% of the cultured cells were detached from PNIPAAm-grafted surface when the temperature was shifted from 37°C to 4 °C whereas less than 10% of the cells were detached from the control dish.
These two pioneering studies clearly show that it is promising to regulate a programmable hydrogel for the reversible cell attachment-detachment control in tissue engineering, while there is a difference between these two studies in terms of the necessity of using collagen, which presumably resulted from the release of free PNIPAAm in Takezawa’s work. Following these two studies, a variety of thermo-responsive hydrogel/coating systems have been developed.[144] Basic mechanisms for cell detachment have also been studied, showing that the change of cell morphology is important during the cell detachment procedure.[144] More importantly, after over ten years of effort, the engineered cell sheets were developed into grafts for in vivo implantation.[145,146]
Nishida, Okano and co-workers harvested human corneal epithelial stem cells and seeded these cells onto a PNIPAAm surface.[146] Different from their initial study,[114] before seeding the epithelial stem cells, a mitomycin C-treated 3T3 feeder layer was plated on the PNIPAAm surface.[146] After the epithelial cells grew to confluency on the feeder layer, they were able to further grow into a multilayered epithelial sheet that had three to five cell layers with basal cells, flattened middle cells and polygonal superficial cells.[146] This cell sheet had a much higher light transmission in comparison to the tissue-engineered constructs growing with fibrin gels and amniotic membranes. Importantly, when the multilayered cell sheet was placed over the bare corneal stroma in a rabbit model, it could quickly and stably adhere to the stroma without the need of suturing. Moreover, the implanted corneal sheet stayed at the initial location throughout the entire experiment up to six months.[146]
Based on this compelling animal study, Nishida, Okano and co-workers initiated the first clinical application of cell sheets for corneal surface regeneration (Figure 3a).[145] The cell source of the cell sheet was the patient’s own oral mucosal epithelia cells. The cell sheet was directly implanted onto the stromal bed without sutures. Following the surgery, the cell sheet was covered with a contact lens for protection and frequently treated for hydration with artificial tears. Within one week post transplantation, the corneal surface was completely reepithelialized. Stromal vascularization gradually appeared on the peripheral region of the cornea. Importantly, corneal transparency could be maintained during the 14 month of observation.[145]
Figure 3.
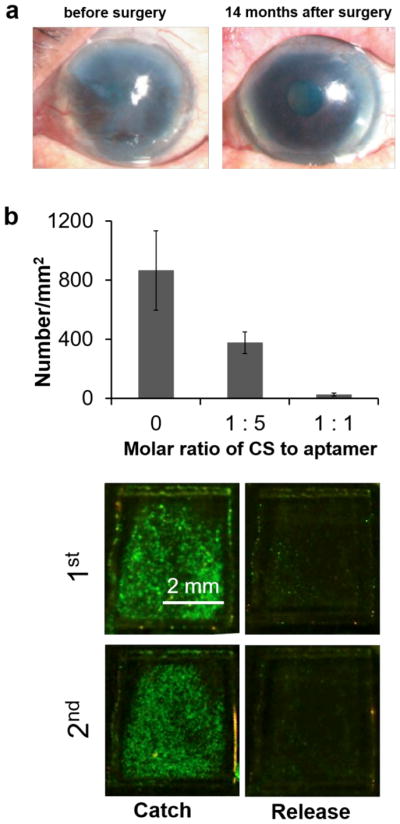
Reversible cell attachment and detachment for tissue engineering and cell separation. (a) Corneal reconstruction with tissue-engineered epithelial sheets cultured on the PNIPAAm surface. Pictures of corneal surface before surgery and covered by the tissue engineered cell sheet 14 months after surgery (from ref. 145 with permission). (b) CS-programmed aptamer-functionalized surface for reversible cell attachment and detachment (from refs. 161 and 123 with permission).
In addition to corneal tissue engineering, this PNIPAAm-based cell sheet engineering approach was also studied for ex vivo engineering of many other tissues such as heart and liver.[147,148] This approach has a key advantage over other tissue engineering strategies. It is scaffold-free, involving no synthetic polymers that may cause acute or chronic inflammation in vivo before full degradation. However, the growth of the cell layers in the cell sheet was not uniform.[146] Some locations had more layers than others. The cells in different locations exhibited different morphology.[146] Moreover, the same research group later claimed that it is difficult to isolate autologous stem cells from an adult tissue because of their low number and capacity to differentiate into target cells, which affects the long-term success of implantation.[149] The use of induced pluripotent stem cells may offer a promising method to solve the problem of cell source.[149,150] Besides the cell source issue, whether other mechanisms may negatively affect the long-term success of implantation remains unclear. An in-depth understanding and discussion of the function of the cell sheets after implantation will advance the future design of novel cell sheets with better quality. It is also important to note that when multiple cell sheets are constructed together to form thick three-dimensional tissues, the cells would face the problem of hypoxia and limited transport of nutrients and wastes. It is a common challenge currently faced by all tissue engineering methods, i.e., how to vascularize the large-dimension engineered tissues.[151]
4.3 Cell Separation
Advances in proteomics and bioinformatics have demonstrated great potential for the discovery of more and more specific cell biomarkers.[152,153] A cell type would be identified with a specific affinity ligand that binds the corresponding cell surface biomarker. This specific ligand-biomarker recognition can be applied to the separation of a defined cell population (e.g., circulating cancer cells, cancer stem cells, or differentiated stem cells) from a heterogeneous cell mixture, which is an essential part of clinical diagnosis and basic research.[154,155]
Ligand-functionalized surface has already been studied for cell separation. For example, antibody-conjugated microbeads have been applied to magnetic-activated cell sorting.[156] Antibody-functionalized microfluidic systems have also been studied for the isolation of rare circulating cancer cells in a single step from the whole blood without pre-labeling, pre-dilution, or any other processing steps.[157] However, while strong and specific antibody-cell binding is beneficial to cell separation, it may turn on intracellular signaling cascades or even induce the death of cells,[158,159] causing the failure of the downstream cell analysis or cell-based tissue engineering or implantation. Different from these studies, the Murthy Group developed an alginate-based hydrogel surface to catch and release cardiac fibroblasts for potential tissue engineering applications.[160] The cell-catching function was regulated by alternating Ca2+ and chelating reagents (e.g., citrate). One concern with this hydrogel system is that alginate molecules may be continuously attached on the cell surface after cell separation owing to the multivalent ligand-receptor interactions. Thus, it is critical to develop novel programmable hydrogels and methods for the recovery of viable and ideally intact cells.
Nucleic acid aptamers hold great potential for the development of programmable hydrogels for reversible catch and release of live cells. Before studying the function of the hydrogels, Wang and co-workers examined nonspecific cell binding on a solid glass surface.[44,161] The cells were found to nonspecifically bind the naked glass and silanized glass with a very high density.[44] This observation is consistent with some studies[162–165] but different from others showing that only aptamer-functionalized metal and glass surfaces induced cell attachment whereas naked control surface did not.[166–168] The difference of the results from those studies is unclear. Wang and co-workers further found that when the glass surface was coated with polyacrylamide or PEG hydrogels, the nonspecific cell binding was significantly inhibited with ~5–10 cells/mm2.[44,161] In contrast, when aptamer-functionalized hydrogel was coated on the glass surface, aptamers could induce specific cell binding to the surface with over 1000 cells/mm2.[44,161] These results demonstrate that rationally designed hydrogels could inhibit nonspecific cell binding and that aptamers could allow for the specific binding of target cells to hydrogels.
During cell separation, it is equally important to release target cells noninvasively after the cells are captured on the surface of a device. Two methods can be applied to treat aptamer-functionalized hydrogels. The first method is to treat the hydrogels with nucleases.[44] In the presence of endonucleases, 99% of the captured cells could be released within half an hour and 98% of them remained viable.[44] However, while nucleases can be used to trigger the cleavage of aptamers for cell release, it is important to note that aptamers need to be chemically modified against nucleases in real-world cell separation applications since aptamers need to maintain high stability during the procedure of cell separation. In this case, nuclease-mediated degradation may not be an appropriate principle for cell separation. The second method is to treat the hydrogels with triggering sequences complementary to aptamers (Figure 1C).[161] Wang and co-workers initiated the first study of using complementary sequences (CS) to inactivate the binding functionality of aptamers in the study of a hydrogel-based artificial extracellular matrix in 2011 (Figure 3b).[161] The cell density decreased significantly from 800 to 30 cells/mm2 when the molar ratio of CS to aptamer was 1:1. Alternatively, the same research group applied primary and secondary CSs to program the hydrogel surface for cyclic aptamer display and displacement.[123] In this work, the binding function of the aptamer was not inactivated whereas the aptamer changed its hybridization partner to leave from the hydrogel surface. As a result, the captured cells were released from the hydrogel surface with 99% efficiency and 99% viability. Importantly, the hydrogel could be programmed to repeatedly catch and release the cells (Figure 3b).[123] These concepts have been confirmed by later studies.[122,124,169]
While the aforementioned studies have suggested that cells could be captured and released noninvasively from an aptamer-functionalized programmable surface for cell separation, all of them were focused on the monovalent display of aptamers on the hydrogel surface and cell catch and release in a static condition. To enhance cell catch particularly in a dynamic flow condition, it is important to develop polyvalent aptamers on the hydrogel surface. Wang and co-workers applied hybridization chain reaction[170] to tether polyvalent aptamers on the hydrogel surface.[171,172] The results showed that the display of polyvalent aptamers on the hydrogel surface led to the catch of more cells within a shorter period of time in both static and dynamic flow conditions.[171,172] Moreover, the cells could be released from the polyvalent aptamer-functionalized hydrogel surface with triggering CS.[172]
It is important to note that while significant progress has been made in developing aptamer-functionalized hydrogels as programmable materials for reversible cell catch and release, all of the previous studies were carried out in a clean environment without biofluids such as the blood, cell culture media or liquids with a complicated cell population. Moreover, cells would have the inclination to spread and secret ECM with membranous protrusions after binding to a surface.[171,122] It could cause nonspecific cell binding to any surface at a later stage, which was seldom reported and discussed in the literature but could be a significant issue for cell separation if the cells reside on a surface for a long period of time before the treatment with the cell releasing solution. It is also important to note that aptamer-mediated cell attachment and detachment on programmable hydrogels hold potential for not only cell separation but also tissue engineering applications.[123,161]
4.4 Protein Delivery
Proteins are promising drugs for the treatment of various human diseases. Numerous antibodies have recently been approved by FDA for systemic drug delivery in treating human diseases.[173] However, many proteins (e.g., growth factors) would cause severe side effects due to their broad biodistribution in non-target tissues and high potency of cell stimulation.[174] Therefore, polymeric delivery systems, particularly hydrogels, have been extensively studied to achieve localized and controlled protein release.[175–177]
Davis and Langer pioneered the development of hydrogels for protein delivery in the 1970s. [126–128] However, owing to the high permeability of hydrogels, these initial attempts failed to demonstrate effective protein sequestration for controlled protein delivery. Decrease of the pore size has been proposed and studied to reduce the fast release of proteins,[128] which remains a major strategy today. However, while the concentration of monomers or crosslinkers can be increased, the threshold pore size of normal hydrogel networks is at the level of tens nanometers.[178] Thus, attention has been paid to the development of affinity-based hydrogels with peptides, heparin and metal ions for the delivery of proteins or proteins with tags, which has been well discussed in the literature.[118,179–182] To avoid significant overlap with the literature and redundancy, the following discussion will be focused on the application of nucleic acid aptamers as binding, sensing and triggering elements to develop programmable hydrogels in sustained and triggered protein release (Figure 4).
Figure 4.

Aptamers as pendent groups for sustained and programmed protein release from hydrogels. (a) Protein dissociation for sustained release. a1: schematic illustration; a2: experimental results. Adapted from refs. 25 and 29 with permission. (b) Sequential release of single or multiple proteins. CS: complementary sequence. Aptamers can not only bind target proteins but also sense their triggering CSs. b1: schematic illustration; b2: Release of one protein for two times (green arrow: CS triggering); b3: programmed release of two proteins. Adapted from refs. 27 and 28 with permission. (c) Integration of external (e.g., light) and internal triggers for self-programmed protein release. Adapted from ref. 137 with permission. (d) Integration of two internal triggers for metabolite-regulated self-programmed protein release. Two aptamers are used in this hydrogel system. One (green arrow) binds the target protein and senses the triggering CS (blue arrow). The other (purple arrow) senses the metabolite for triggering initiation of the protein-bound aptamer. Adapted from ref. 59 with permission.
The first study showing sustained protein release from aptamer-functionalized hydrogels was reported in 2010 by Wang and co-workers (Figure 4a).[25] Polyacrylamide hydrogel and anti-PDGF-BB aptamer were used as a model in this work. In the presence of the aptamer, the initial 24 h burst release was decreased from ~70% to ~10%. After this fast release, ~6% of PDGF-BB was slowly released in the next 6 days. On average, 1% was released each day. A first-order diffusion-reaction model was also developed. The experimental results were consistent with the modeling analysis. Following this work, the same research group developed aptamer-functionalized in situ injectable hydrogels[29] that can be delivered in vivo to fill tissue defects of any geometry with minimal surgery. Three aptamer derivatives were developed with different binding affinities by sequence mutation. The cumulative release exhibited a pseudo-linear relationship with the log scale of KD of these aptamers. These early studies clearly demonstrate that aptamers can mediate the sustained release of proteins from hydrogels. However, those hydrogel systems have numerous shortcomings such as the production of free radicals during the formation of hydrogels, the low loading amount of proteins, the low bioactivity of proteins and the inability of incorporating live cells with proteins.
To solve these problems, Wang and co-workers synthesized off-the-shelf aptamer-functionalized superporous hydrogels with the average pore size of 50 to 100 μm.[136,183] Because the superb capability of solution absorption, the loading amount of proteins was increased from the initial picogram level to nanogram and microgram levels. Even with the increase of the protein loading amounts and pore sizes, aptamers could mediate the sustained release of proteins. In the presence of high-affinity aptamer, only 3% of 2 μg PDGF-BB was released in two weeks.[183] In the presence of low-affinity aptamer, 60% was gradually released during the same period of time. The ability of aptamer in mediating the sustained protein release was also confirmed in the VEGF release study showing that 25% of VEGF was released in two weeks.[136] Many protein release studies reported in the literature did not show protein bioactivity before the animal experiments. While animal studies are important to evaluate the functionality of a protein delivery system, a simple in vitro examination of protein bioactivity with intact proteins as control would help design and understand protein delivery systems. Moreover, as biomaterials can more or less cause inflammation that can stimulate a variety of bioactivity in the implantation location,[184] failure to demonstrate the bioactivity of released proteins increases uncertainty about whether the biological effects come from inflammation or the delivered proteins. In comparison to intact stock VEGF (10 ng/mL), the released VEGF from the aptamer-functionalized superporous hydrogels at day 14 could maintain approximately 60% bioactivity based on the cell tube formation assay.[136,185] VEGF is very fragile and often difficult to maintain its bioactivity in traditional polymeric systems.[186] For instance, Gu and coworkers showed that VEGF bioactivity was decreased to ~15% after 10 days of in vitro release;[187] and Ekaputra and coworkers showed that VEGF bioactivity was decreased to ~5% after 7 days of in vitro release.[188] This big difference between the reported data demonstrates that aptamer-functionalized superporous hydrogels are a promising protein delivery system to load a large amount of proteins with high protein bioactivity. In addition, the superporous hydrogels can be used as a cell carrier owing to their large pore size and the loaded proteins can enhance the survival of the carried cells in the hydrogels. [136,185]
While aptamer-functionalized hydrogels can release proteins in a sustained manner for various therapeutic applications, it is also desirable to develop protein delivery systems that can release the right protein drug at the right time in the right dose. To satisfy this requirement, it is important to program the releasing function of aptamer-functionalized hydrogels. As aptamers can hybridize with their CSs and change the functional conformations, CS can be used as a triggering element to program protein-bound aptamers in hydrogels.[27,28,36,183]
Sullenger and co-workers for the first time applied CS as an antidote to control the function of an anticoagulant aptamer in the blood through the aptamer-CS hybridization,[30,31] which was confirmed by a later study using the anti-thrombin aptamer in an aqueous solution.[189] Mi and Tan and their co-workers applied nucleic acid aptamers as a crosslinker to synthesize a responsive gel-sol transition system, which could be converted into solutions once triggered.[16,17] However, none of these elegant studies showed a protein release profile. Notably, as the gel-sol transition would cause an indiscriminate release of loaded cargoes, the gel-sol transition system may not satisfy the release control of multiple proteins. It may also cause the abrupt loss of mechanical integrity of hydrogels, which is beneficial for biosensing applications[190] but problematic for biomedical applications such as protein delivery and tissue engineering.
Wang and co-workers applied aptamers as pendent groups rather than crosslinkers to develop programmable hydrogels and provided the direct evidence of pulsatile protein release from the hydrogels (Figure 4b).[27,28,36,183] Since aptamers are pendent groups, protein release is independent of other properties (e.g., mechanical integrity) of the programmable hydrogels. It is also important to note that while aptamers can be used as both crosslinkers and functional side groups, the amounts of aptamers needed to synthesize or functionalize a hydrogel is tremendously different in these two different strategies. The amount of aptamers for crosslinking has to be overwhelmingly high to ensure the sufficient crosslinking density and to resist high swelling of hydrogels. In contrast, aptamers used as functional side groups may only need a small amount to fulfill a requirement. Thus, the amount of triggering CSs used for these two different situations would be different. In their initial attempts, aptamers were applied to functionalize the microparticle surface and the microparticles were dispersed in the hydrogel.[27] The treatment with CS led to periodically pulsatile PDGF-BB release during a 25 day release test.[27] The efficiency of triggered PDGF-BB release could be tuned by varying the length and composition of CS.[36] The study also showed that while a longer CS is supposed to favor protein release thermodynamically, it is not the case kinetically.[36] It may result from the slower diffusion of the longer CS in the hydrogel and the higher steric hindrance, which is different from the triggering situation in aqueous solutions. These factors may decrease the sensitivity of triggered release. With this concern in mind, the same research group directly conjugated aptamers to the superporous hydrogels. With the presence of the large pores, the CS-triggered hydrogel could release ~5% of loaded PDGF-BB within 30 min whereas ~02.% was released in 24 h from the untriggered hydrogel.[183] Moreover, the periodic exposure of the hydrogel to the CS treatment led to sequential PDGF-BB release at pre-determined time points.[183] However, all of these studies focused on the sequential release of single proteins, which can also be achieved by using other chemical, physical or biological stimuli. To further demonstrate the advantage of aptamer-functionalized hydrogels, two aptamers were integrated into one hydrogel system.[28] VEGF and PDGF-BB could be sequentially and specifically released from the dual aptamer-functionalized hydrogel,[28] which is challenging to achieve using traditional responsive hydrogels that would simultaneously release multiple proteins without differentiation. However, while the concept of sequential protein release was approved, it is important to note that CS was used as an external molecular trigger. To develop a self-programmed hydrogel, it is desirable to incorporate aptamer and CS into one hydrogel.
Wang and co-workers further integrated aptamer and CS into one hydrogel that was developed to mimic step-wise molecular recognition pathways observed in live cells or tissues.[59,137] As CS is initially incorporated into the hydrogel, CS functions as an internal molecular trigger and has the ability to get access to neighboring aptamers for self-programmed protein release control. The first attempt was made by using sequential photoreaction and hybridization (Figure 4c).[137] CS was conjugated to the hydrogel with a photolabile group. After the hydrogel was exposed to light, the immobilized CS was freed to approach the protein-bound aptamer. Subsequently, the CS-aptamer hybridization led to the release of the aptamer-bound protein. Periodic light exposure led to sequential protein release and cell migration at pre-determined time points.[137] As an ideal protein release system is able to release protein drugs in response to the variation of metabolic activity, a second attempt was made to recapitulate the procedure of cellular signal transduction to control protein release in response to a small metabolite through sequential displacement and hybridization reactions (Figure 4d).[59] Totally four biomolecules including two aptamers, a dual-functional CS and a protein were incorporated into a two-compartment hydrogel. In the presence of adenosine, the anti-adenosine aptamer changed its conformation to free the aptamer-bound CS. The freed CS subsequently hybridized with the protein-bound aptamer to form a new nucleic acid duplex, which led to protein release. Protein release from the same hydrogel was periodically stimulated for four times.[59] While adenosine and PDGF-BB were used as a model in this work, the concept is in principle suitable for any metabolites (e.g., glucose) and proteins (e.g., insulin).
There are a few advantages of using aptamers to develop programmable hydrogels. Firstly, aptamers have high affinities and specificities in binding target proteins, which cannot be easily compensated with any other ligands except antibodies. Thus, in principle, it is possible to incorporate many aptamers into a hydrogel based on a certain need for sustained release, triggered release or their combinations. Secondly, aptamers can change their functional conformations in the presence of rationally designed CSs that are either used as external triggering molecules or incorporated into the hydrogel together with aptamers as a biomimetic internal triggering intermediate. This function is the key for the development of programmable hydrogels. Based on the currently available knowledge, it is challenging or virtually impossible for antibodies to fulfill this requirement. Thirdly, aptamers bind both proteins and CSs with high affinities and specificities. Thus, the entire programmable system has strong and specific aptamer-CS and aptamer-protein interactions. This dual binding fidelity ensures the high accuracy of control. Fourthly, in addition to the incorporation of multiple aptamers for controlling the release of multiple proteins, programmable hydrogels can be further functionalized with multiple cell-binding aptamers to expand the diversity of programmable hydrogels. It is important to note that there is a common concern with the stability of nucleic acid aptamers in the body fluids that contain various nucleases. While this concern is reasonable, the nuclease degradation of aptamers within the hydrogel is significantly different from that in aqueous solutions or on the hydrogel surface even if aptamers are not chemically modified. Aptamers exhibit reasonable stability once chemically incorporated into the hydrogel network. Moreover, even if their stability does not reach the expectation, they can be chemically modified to achieve high stability.[191] Because of these advantages, aptamer-functionalized hydrogels have recently attracted more and more attention in the field of biomaterials.[192,193]
5. Conclusion and Outlook
The definition of programmable hydrogels is given in this review. Moreover, programmable hydrogels are introduced from the viewpoints of principles, functions and biomedical applications. While it is obvious that great progress has been made in this research area, there are several issues that need to be considered for future advances.
The ability to achieve spatiotemporal control of a change would be ideal, which has been proposed and discussed in the literature for many years. However, most if not all programmable hydrogels undergo changes at the macroscopic level. That is the change of the entire bulk of the hydrogel. One may suggest applying high-focus stimuli such as light to achieve spatial control to spare the unstimulated regions of the hydrogel. Indeed two-photon techniques have high spatial resolution; however, it would be challenging to apply such a stimulus for most real-world applications (e.g., protein delivery) by triggering a three-dimensional space via a few microns by a few microns. It would be particularly challenging to differentiate the extracellular space from the randomly distributed cells in a three-dimensional hydrogel in vitro or tissue in vivo. Thus, the development of programmable hydrogels that can be spatiotemporally controlled remains a challenge. The integration of multiple external and internal triggering events for one functional change has provided a potential solution to address this question.[59,137] However, more work is needed to enrich such a new research direction for the advanced development of programmable hydrogels that are responsive to coherent or sequential stimuli at both macroscopic and microscopic levels. As a result, a local small-scale change in a bulk hydrogel will not affect the overall properties of the hydrogel. This requirement for spatiotemporal control also suggests a need to explore novel methods for real-time and in-situ examination of temporal and local variation of properties and functions of programmable hydrogels.
Functional changes are often coupled together. For instance, when the pore size of a hydrogel is increased or decreased, it will cause the change of not only the permeability but also mechanical properties and ligand density. These changes will further affect cell attachment and protein release. The ability to decouple these simultaneous changes would help better understand basic behavior of cells or develop more effective strategies for treatment of diseases. Moreover, the effectiveness of functional changes is often a non-linear function of time. It decays with the times of stimulation. This situation is reasonable as the change in a hydrogel is either directly or indirectly related to concentration. For instance, the protein amount in the hydrogel decreases after each pulsatile release.[59] As the release is a function of the concentration gradient, the following release would decrease with the triggering times and cycles. This issue may also hold true to other functional changes such as the decrease or increase of stiffness. These concerns with functional changes need to be considered when programmable hydrogels are designed for biomedical applications, particularly those requiring very frequent and periodic changes (e.g., insulin release for treatment of diabetes).
Programmable hydrogels have been studied for various biomedical applications as exemplified herein. These promising applications may be tuned for other areas. For instance, the principle for programmable catch and release of proteins and cells can be applied to reversible catch of nano- or micro-particles.[194] It is also possible to integrate two or more than two programmable hydrogels into one multifunctional programmable hydrogel for one application such as the development of cancer cell homing, killing and releasing hydrogel[195], stem cell homing and differentiating hydrogel, and 4-D cell culture hydrogels.[196,197] However, it is important to note that most programmable hydrogels are currently studied in an in vitro environment. When they are used to help understand certain basic biological mechanisms or to separate target cells from a cell mixture in vitro, their current functions may fulfill the requirements. However, if they are developed for in vivo applications such as protein delivery, cell homing and tissue engineering, numerous technical challenges need to be considered. For instance, the penetration of light, triggering molecules or metabolites into hydrogels through the dense tissues or the circulation needs to be examined systematically, which needs to be addressed in the future.
Acknowledgments
Support from the U.S. National Science Foundation (DMR-1332351 and DMR-1322332) and the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01HL122311) is greatly acknowledged. Dr. Jinping Lai, Dr. Peng Shi, Mr. James Coyne and Ms. Lidya Abune are also acknowledged for drawing the schemes and reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd GT, Davis KE, Pisani D, Tarver JE, Ruta M, Sakamoto M, Hone DW, Jennings R, Benton MJ. Dinosaurs and the Cretaceous Terrestrial Revolution. Proc R Soc B Biol Sci. 2008;275:2483–2490. doi: 10.1098/rspb.2008.0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang W. How do squid and octopuses change color? Sci Am. 2001;284:100. [Google Scholar]
- 3.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible Hydrogels Based on Photopolymerized Poly(ethylene glycol)-co-poly (α-hydroxy acid) Diacrylate Macromers. Macromolecules. 1993;26:581–587. doi: 10.1021/ma00056a005. [DOI] [Google Scholar]
- 4.Lu S, Anseth KS. Release behavior of high molecular weight solutes from poly(ethylene glycol)-based degradable networks. Macromolecules. 2000;33:2509–2515. doi: 10.1021/ma9915024. [DOI] [Google Scholar]
- 5.Zustiak SP, Leach JB. Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties. 2010:1348–1357. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohn D, Younes H. Biodegradable PEO/PLA block copolymers. J Biomed Mater Res. 1988;22:993–1009. doi: 10.1002/jbm.820221104. [DOI] [PubMed] [Google Scholar]
- 7.Jonker AM, Löwik DWPM, Van Hest JCM. Peptide- and protein-based hydrogels. Chem Mater. 2012;24:759–773. doi: 10.1021/cm202640w. [DOI] [Google Scholar]
- 8.Coviello T, Matricardi P, Marianecci C, Alhaique F. Polysaccharide hydrogels for modified release formulations. J Control Release. 2007;119:5–24. doi: 10.1016/j.jconrel.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Roh YH, Ruiz RCH, Peng S, Lee JB, Luo D. Engineering DNA-based functional materials. Chem Soc Rev. 2011;40:5730. doi: 10.1039/c1cs15162b. [DOI] [PubMed] [Google Scholar]
- 10.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 11.Dumortier G, Grossiord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23:2709–2728. doi: 10.1007/s11095-006-9104-4. [DOI] [PubMed] [Google Scholar]
- 12.Nagahara S, Matsuda T. Hydrogel formation via hybridization of oligonucleotides derivatized in water-soluble vinyl polymers. Polym Gels Networks. 1996;4:111–127. doi: 10.1016/0966-7822(96)00001-9. [DOI] [Google Scholar]
- 13.Um SH, Lee JB, Park N, Kwon SY, Umbach CC, Luo D. Enzyme-catalysed assembly of DNA hydrogel. Nat Mater. 2006;5:797–801. doi: 10.1038/nmat1741. [DOI] [PubMed] [Google Scholar]
- 14.Yan L, Zhu Z, Zou Y, Huang Y, Liu D, Jia S, Xu D, Wu M, Zhou Y, Zhou S, Yang CJ. Target-responsive “sweet” hydrogel with glucometer readout for portable and quantitative detection of non-glucose targets. J Am Chem Soc. 2013;135:3748–3751. doi: 10.1021/ja3114714. [DOI] [PubMed] [Google Scholar]
- 15.Dave N, Chan MY, Huang PJJ, Smith BD, Liu J. Regenerable DNA-functionalized hydrogels for ultrasensitive, instrument-free mercury(II) detection and removal in water. J Am Chem Soc. 2010;132:12668–12673. doi: 10.1021/ja106098j. [DOI] [PubMed] [Google Scholar]
- 16.Yang H, Liu H, Kang H, Tan W. Engineering target-responsive hydrogels based on aptamer-target interactions. J Am Chem Soc. 2008;130:6320–6321. doi: 10.1021/ja801339w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei B, Cheng I, Luo KQ, Mi Y. Capture and release of protein by a reversible DNA-induced sol-gel transition system. Angew Chemie - Int Ed. 2008;47:331–333. doi: 10.1002/anie.200704143. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Faulkner-Jones A, Dun AR, Jin J, Chen P, Xing Y, Yang Z, Li Z, Shu W, Liu D, Duncan RR. Rapid formation of a supramolecular polypeptide-DNA Hydrogel for in situ three-dimensional multilayer bioprinting. Angew Chemie - Int Ed. 2015;54:3957–3961. doi: 10.1002/anie.201411383. [DOI] [PubMed] [Google Scholar]
- 19.Lin DC, Yurke B, Langrana NA. Mechanical Properties of a Reversible, DNA-Crosslinked Polyacrylamide Hydrogel. J Biomech Eng. 2004;126:104–110. doi: 10.1115/1.1645529. [DOI] [PubMed] [Google Scholar]
- 20.Sigal B, Alberts N. Genetic recombination: the nature of a crossed strand-exchange between two homologous DNA molecules. J Mol Biol. 1972;71:789–792. doi: 10.1016/s0022-2836(72)80039-1. [DOI] [PubMed] [Google Scholar]
- 21.Richardson CC. Strand Displacement during Deoxyribonucleic Acid Synthesis at Single Strand Breaks. J Biol Chem. 1971;246:2692–2701. [PubMed] [Google Scholar]
- 22.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 23.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 24.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soontornworajit B, Zhou J, Shaw MT, Fan T-H, Wang Y. Hydrogel functionalization with DNA aptamers for sustained PDGF-BB release. Chem Commun. 2010;46:1857. doi: 10.1039/b924909e. [DOI] [PubMed] [Google Scholar]
- 26.Battig MR, Huang Y, Chen N, Wang Y. Aptamer-functionalized superporous hydrogels for sequestration and release of growth factors regulated via molecular recognition. Biomaterials. 2014;35:8040–8048. doi: 10.1016/j.biomaterials.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Soontornworajit B, Zhou J, Wang Y. A hybrid particle–hydrogel composite for oligonucleotide-mediated pulsatile protein release. Soft Matter. 2010;6:4255. doi: 10.1039/c0sm00206b. [DOI] [Google Scholar]
- 28.Battig MR, Soontornworajit B, Wang Y. Programmable release of multiple protein drugs from aptamer-functionalized hydrogels via nucleic acid hybridization. J Am Chem Soc. 2012;134:12410–12413. doi: 10.1021/ja305238a. [DOI] [PubMed] [Google Scholar]
- 29.Soontornworajit B, Zhou J, Zhang Z, Wang Y. Aptamer-functionalized in situ injectable hydrogel for controlled protein release. Biomacromolecules. 2010;11:2724–2730. doi: 10.1021/bm100774t. [DOI] [PubMed] [Google Scholar]
- 30.Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TL, Monroe D, Sullenger BA. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90–94. doi: 10.1038/nature00963. [DOI] [PubMed] [Google Scholar]
- 31.Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G, Scardino E, Fay WP, Sullenger BA. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol. 2004;22:1423–1428. doi: 10.1038/nbt1023. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Lu Y. Non-base pairing DNA provides a new dimension for controlling aptamer-linked nanoparticles and sensors. J Am Chem Soc. 2007;129:8634–8643. doi: 10.1021/ja072075+. [DOI] [PubMed] [Google Scholar]
- 33.Yang CJ, Jockusch S, Vicens M, Turro NJ, Tan W. Light-switching excimer probes for rapid protein monitoring in complex biological fluids. Proc Natl Acad Sci U S A. 2005;102:17278–83. doi: 10.1073/pnas.0508821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyata T, Asami N, Uragani T. A reversibly antigen-responsive hydrogel. Nature. 1999;399:766–768. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Liu Y, Riesberg JJ, Shen W. Dynamic presentation of immobilized ligands regulated through biomolecular recognition. J Am Chem Soc. 2010;132:13630–13632. doi: 10.1021/ja1054669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soontornworajit B, Zhou J, Snipes MP, Battig MR, Wang Y. Affinity hydrogels for controlled protein release using nucleic acid aptamers and complementary oligonucleotides. Biomaterials. 2011;32:6839–6849. doi: 10.1016/j.biomaterials.2011.05.074. [DOI] [PubMed] [Google Scholar]
- 37.Ulijn Rein V, Bibi Nurguse, Jayawarna Vineetha, Thornton Paul D, Todd Simon J, Mart Robert J, Smith Andrew M, Gough JE. Bioresponsive hydrogels. Mater Today. 2007;10:40–48. doi: 10.1016/S1369-7021(07)70049-4. [DOI] [Google Scholar]
- 38.Krishna OD, Kiick KL. Protein- and peptide-modified synthetic polymeric biomaterials. Biopolymers. 2010;94:32–48. doi: 10.1002/bip.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleiner DE, Stetler-Stevenson WG. Structural biochemistry and activation of matrix metalloproteases. Curr Opin Cell Biol. 1993;5:891–897. doi: 10.1016/0955-0674(93)90040-W. [DOI] [PubMed] [Google Scholar]
- 40.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc Natl Acad Sci. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Park Y, Tae G, Lee KB, Hwang SJ, Kim IS, Noh I, Sun K. Synthesis and characterization of matrix metalloprotease sensitive-low molecular weight hyaluronic acid based hydrogels. J Mater Sci Mater Med. 2008;19:3311–3318. doi: 10.1007/s10856-008-3469-3. [DOI] [PubMed] [Google Scholar]
- 42.Roberts RJ. Restriction endonucleases. CRC Crit Rev Biochem. 1976;4:123–164. doi: 10.3109/10409237609105456. [DOI] [PubMed] [Google Scholar]
- 43.Shevelev IV, Hubscher U. The 3’-5’ exonucleases. Nat Rev Mol Cell Biol. 2002;3:364–376. doi: 10.1038/nrm804. [DOI] [PubMed] [Google Scholar]
- 44.Li S, Chen N, Zhang Z, Wang Y. Endonuclease-responsive aptamer-functionalized hydrogel coating for sequential catch and release of cancer cells. Biomaterials. 2013;34:460–469. doi: 10.1016/j.biomaterials.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 45.Liu J. Oligonucleotide-functionalized hydrogels as stimuli responsive materials and biosensors. Soft Matter. 2011;7:6757. doi: 10.1039/c1sm05284e. [DOI] [Google Scholar]
- 46.Kataoka K, Miyazaki H, Bunya M, Okano T, Sakurai Y. Totally synthetic polymer gels responding to external glucose concentration: Their preparation and application to on-off regulation of insulin release. J Am Chem Soc. 1998;120:12694–12695. doi: 10.1021/ja982975d. [DOI] [Google Scholar]
- 47.Matsumoto A, Yoshida R, Kataoka K. Glucose-responsive polymer gel bearing phenylborate derivative as a glucose-sensing moiety operating at the physiological pH. Biomacromolecules. 2004;5:1038–1045. doi: 10.1021/bm0345413. [DOI] [PubMed] [Google Scholar]
- 48.Brownlee M, Cerami A. A glucose-controlled insulin-delivery system: semisynthetic insulin bound to lectin. Science. 1979;206:1190–1191. doi: 10.1126/science.505005. [DOI] [PubMed] [Google Scholar]
- 49.Ishihara K, Kobayashi M, Ishimaru N, Shinohara I. Glucose Induced Permeation Control of Insulin through a Complex Membrane Consisting of Immobilized Glucose Oxidase and a Poly(amine) Polym J. 1984;16:625–631. doi: 10.1295/polymj.16.625. [DOI] [Google Scholar]
- 50.Kost J, Horbett TA, Ratner BD, Singh M. Glucose-sensitive membranes containing glucose oxidase: Activity, swelling, and permeability studies. J Biomed Mater Res. 1985;19:1117–1133. doi: 10.1002/jbm.820190920. [DOI] [PubMed] [Google Scholar]
- 51.Fischel-Ghodsian F, Brown L, Mathiowitz E, Brandenburg D, Langer R. Enzymatically controlled drug delivery. Proc Natl Acad Sci. 1988;85:2403–2406. doi: 10.1073/pnas.85.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker G, Reeke JW, Edelman GN. Location of the saccharide binding site of concanavalin A. J Biol Chem. 1971;246:6123–6125. [PubMed] [Google Scholar]
- 53.Edelman GM, Cunningham Ba, Reeke GN, Becker JW, Waxdal MJ, Wang JL. The covalent and three-dimensional structure of concanavalin A. Proc Natl Acad Sci U S A. 1972;69:2580–2584. doi: 10.1073/pnas.69.9.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JJ, Park K. Modulated insulin delivery from glucose-sensitive hydrogel dosage forms. J Control Release. 2001;77:39–47. doi: 10.1016/S0168-3659(01)00447-3. [DOI] [PubMed] [Google Scholar]
- 55.Kim Sung Wan, Paii Chaul Min, Kimiko M, Seminoff LA, Holmberg DL, Gleeson JM, Wilson DE, Mack EJ. Self-regulated glycosylated insulin delivery. J Control Release. 1990;11:193–201. doi: 10.1016/0168-3659(90)90132-D. [DOI] [Google Scholar]
- 56.Burnstock G. Historical review: ATP as a neurotransmitter. Trends Pharmacol Sci. 2006;27:166–176. doi: 10.1016/j.tips.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Drury AN, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68:213–237. doi: 10.1183/09031936.00137511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corriden R, Insel PA. Basal Release of ATP: An Autocrine-Paracrine Mechanism for Cell Regulation. Sci Signal. 2010;3:re1-re1. doi: 10.1126/scisignal.3104re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai J, Li S, Shi X, Coyne J, Zhao N, Dong F, Mao Y, Wang Y. Displacement and hybridization reactions in aptamer-functionalized hydrogels for biomimetic protein release and signal transduction. Chem Sci. 2017 doi: 10.1039/C7SC03023A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shu XZ, Liu Y, Luo Y, Roberts MC, Prestwich GD. Disulfide cross-linked hyaluronan hydrogels. Biomacromolecules. 2002;3:1304–1311. doi: 10.1021/bm025603c. [DOI] [PubMed] [Google Scholar]
- 61.Shu XZ, Liu Y, Palumbo F, Prestwich GD. Disulfide-crosslinked hyaluronan-gelatin hydrogel films: A covalent mimic of the extracellular matrix for in vitro cell growth. Biomaterials. 2003;24:3825–3834. doi: 10.1016/S0142-9612(03)00267-9. [DOI] [PubMed] [Google Scholar]
- 62.Choh S-Y, Cross D, Wang C. Facile Synthesis and Characterization of Disulfide-Cross-Linked Hyaluronic Acid Hydrogels for Protein Delivery and Cell Encapsulation. Biomacromolecules. 2011;12:1126–1136. doi: 10.1021/bm101451k. [DOI] [PubMed] [Google Scholar]
- 63.Cheng X, Jin Y, Sun T, Qi R, Fan B, Li H. Oxidation- and thermo-responsive poly(N-isopropylacrylamide-co-2-hydroxyethyl acrylate) hydrogels cross-linked via diselenides for controlled drug delivery. RSC Adv. 2015;5:4162–4170. doi: 10.1039/C4RA13500H. [DOI] [Google Scholar]
- 64.Kloc K, Mlochowski J, Syper L. Oxidation of Organic Diselenides with Hydrogen Peroxide to Alkane- and Areneseleninic Acids and Selenium-Containing Heterocycles. 1989;8:811–813. [Google Scholar]
- 65.Hassan W, Teixeira Rocha JB. Interaction profile of diphenyl diselenide with pharmacologically significant thiols. Molecules. 2012;17:12287–12296. doi: 10.3390/molecules171012287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. doi:14988435. [DOI] [PubMed] [Google Scholar]
- 67.Lee KY, Mooney DJ. Alginate: Properties and biomedical applications. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grant GT, Morris ER, Rees DA, Smith PJC, Thom D. Biological interactions between polysaccharides and divalent cations: The egg-box model. FEBS Lett. 1973;32:195–198. doi: 10.1016/0014-5793(73)80770-7. [DOI] [Google Scholar]
- 69.Mørch ÝA, Donati I, Strand BL, Skjåk-Bræk G. Effect of Ca2+, Ba2+, and Sr2+ on alginate microbeads. Biomacromolecules. 2006;7:1471–1480. doi: 10.1021/bm060010d. [DOI] [PubMed] [Google Scholar]
- 70.Shoichet MS, Li RH, White ML, Winn SR. Stability of hydrogels used in cell encapsulation: An in vitro comparison of alginate and agarose. Biotechnol Bioeng. 1996;50:374–381. doi: 10.1002/(SICI)1097-0290(19960520)50:4<374::AID-BIT4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 71.Munarin F, Tanzi MC, Petrini P. Advances in biomedical applications of pectin gels. Int J Biol Macromol. 2012;51:681–689. doi: 10.1016/j.ijbiomac.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 72.Kocak G, Tuncer C, Bütün V. pH-Responsive polymers. Polym Chem. 2017;8:144–176. doi: 10.1039/C6PY01872F. [DOI] [Google Scholar]
- 73.Murthy N, Xu M, Schuck S, Kunisawa J, Shastri N, Frechet JM. A macromolecular delivery vehicle for protein-based vaccines: acid-degradable protein-loaded microgels. Proc Natl Acad Sci U S A. 2003;100:4995–5000. doi: 10.1073/pnas.0930644100\r0930644100. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roberts MC, Hanson MC, Massey AP, Karren EA, Kiser PF. Dynamically restructuring hydrogel networks formed with reversible covalent crosslinks. Adv Mater. 2007;19:2503–2507. doi: 10.1002/adma.200602649. [DOI] [Google Scholar]
- 75.Gueron KG, Leroy J-L, Maurice A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature. 1993;366:461–464. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 76.Guo W, Lu CH, Qi XJ, Orbach R, Fadeev M, Yang HH, Willner I. Switchable bifunctional stimuli-triggered poly-N-isopropylacrylamide/DNA hydrogels. Angew Chemie - Int Ed. 2014;53:10134–10138. doi: 10.1002/anie.201405692. [DOI] [PubMed] [Google Scholar]
- 77.Cheng E, Xing Y, Chen P, Sun Yang Y, Zhou D, Xu T, Fan Q, Liu D. A pH-triggered, fast-responding DNA hydrogel. Angew Chemie - Int Ed. 2009;48:7660–7663. doi: 10.1002/anie.200902538. [DOI] [PubMed] [Google Scholar]
- 78.Niskanen J, Tenhu H. How to manipulate the upper critical solution temperature (UCST)? Polym Chem. 2017;8:220–232. doi: 10.1039/C6PY01612J. [DOI] [Google Scholar]
- 79.Schild HG. Poly ( N-Isopropylacrylamide ): Experiment , Theory and Application. Prog Polym Sci. 1992;17:163–249. doi: 10.1016/0079-6700(92)90023-R. [DOI] [Google Scholar]
- 80.Scarpa JS, Mueller DD, Klotz IM. Slow hydrogen-deuterium exchange in a non-.alpha.-helical polyamide. J Am Chem Soc. 1967;89:6024–6030. doi: 10.1021/ja01000a006. [DOI] [Google Scholar]
- 81.Heskins M, Guillet JE. Solution Properties of Poly(N-isopropylacrylamide) J Macromol Sci Part A - Chem. 1968;2:1441–1455. doi: 10.1080/10601326808051910. [DOI] [Google Scholar]
- 82.Ku G, Wang LV. Deeply penetrating photoacoustic tomography in biological tissues enhanced with an optical contrast agent. Opt Lett. 2005;30:507. doi: 10.1364/OL.30.000507. [DOI] [PubMed] [Google Scholar]
- 83.Eichler J, Knof J, Lenz H. Measurements on the depth of penetration of light (0.35-1.0 μm) in tissue. Radiat Environ Biophys. 1977;14:239–242. doi: 10.1007/BF01323942. [DOI] [PubMed] [Google Scholar]
- 84.Padalkar MV, Pleshko N. Wavelength-dependent penetration depth of near infrared radiation into cartilage. Analyst. 2015;140:2093–2100. doi: 10.1039/C4AN01987C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petersen S, Alonso JM, Specht A, Duodu P, Goeldner M, Del Campo A. Phototriggering of cell adhesion by caged cyclic RGD peptides. Angew Chemie - Int Ed. 2008;47:3192–3195. doi: 10.1002/anie.200704857. [DOI] [PubMed] [Google Scholar]
- 86.Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, Del Campo A, García AJ. Light-triggered in vivo activation of adhesive peptides regulates cell adhesion, inflammation and vascularization of biomaterials. Nat Mater. 2015;14:352–360. doi: 10.1038/nmat4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo Y, Shoichet MS. A photolabile hydrogel for guided three-dimensional cell growth and migration. Nat Mater. 2004;3:249–253. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 88.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fomina N, McFearin C, Sermsakdi M, Edigin O, Almutairi A. UV and near-IR triggered release from polymeric nanoparticles. J Am Chem Soc. 2010;132:9540–9542. doi: 10.1021/ja102595j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anseth KS, Bowman CN, Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996;17:1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 91.Chandran PL, Barocas VH. Microstructural Mechanics of Collagen Gels in Confined Compression: Poroelasticity, Viscoelasticity, and Collapse. J Biomech Eng. 2004;126:152. doi: 10.1115/1.1688774. [DOI] [PubMed] [Google Scholar]
- 92.Omidian H, Rocca JG, Park K. Elastic, superporous hydrogel hybrids of polyacrylamide and sodium alginate. Macromol Biosci. 2006;6:703–710. doi: 10.1002/mabi.200600062. [DOI] [PubMed] [Google Scholar]
- 93.Sun JY, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z. Highly stretchable and tough hydrogels. Nature. 2012;489:133–136. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Murdan S. E lectro-responsive drug delivery from hydrogels. J Control Release. 2003;92:1–17. doi: 10.1016/s0168-3659(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 95.Sawahata K, Hara M, Yasunaga H, Osada Y. Electrically controlled drug delivery system using polyelectrolyte gels. J Control Release. 1990;14:253–262. doi: 10.1016/0168-3659(90)90165-P. [DOI] [Google Scholar]
- 96.TANAKA T, NISHIO I, SUN S-T, UENO-NISHIO S. Collapse of Gels in an Electric Field. Science. 1982;218:467–469. doi: 10.1126/science.218.4571.467. [DOI] [PubMed] [Google Scholar]
- 97.De Rossi D, Chiarelli P, Buzzigoli G, Domenici C, Lazzeri L. Contractile behaviour of electrically activated mechanochemical polymer actuators. ASAIO Trans. 1986;32:157–162. [PubMed] [Google Scholar]
- 98.Osada Y, Hasebe M. Electrically activated mechanochemical devices using polyelectrolyte gels. Chem Lett. 1985;14:1285–1288. doi: 10.1246/cl.1985.1285. [DOI] [Google Scholar]
- 99.Kwon IC, Bae YH, Kim SW. Characteristics of charged networks under an electric stimulus. J Polym Sci Part B Polym Phys. 1994;32:1085–1092. doi: 10.1002/polb.1994.090320613. [DOI] [Google Scholar]
- 100.Kwon IC, Bae YH, Okano T, Kim SW. Drug release from electric current sensitive polymers. J Control Release. 1991;17 doi: 10.1016/0168-3659(91)90054-H. [DOI] [Google Scholar]
- 101.Li Y, Huang G, Zhang X, Li B, Chen Y, Lu T, Lu TJ, Xu F. Magnetic hydrogels and their potential biomedical applications. Adv Funct Mater. 2013;23:660–672. doi: 10.1002/adfm.201201708. [DOI] [Google Scholar]
- 102.Rosensweig REE. Heating magnetic fluid with alternating magnetic field. J Magn Magn Mater. 2002;252:370–374. doi: 10.1016/S0304-8853(02)00706-0. [DOI] [Google Scholar]
- 103.Hsieh DS, Langer R, Folkman J. Magnetic modulation of release of macromolecules from polymers. Proc Natl Acad Sci U S A. 1981;78:1863–7. doi: 10.1073/pnas.78.3.1863. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=319235&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li Y, Huang G, Gao B, Li M, Genin GM, Lu TJ, Xu F. Magnetically actuated cell-laden microscale hydrogels for probing strain-induced cell responses in three dimensions. NPG Asia Mater. 2016;8:e238. doi: 10.1038/am.2015.148. [DOI] [Google Scholar]
- 105.Satarkar NS, Hilt JZ. Magnetic hydrogel nanocomposites for remote controlled pulsatile drug release. J Control Release. 2008;130:246–251. doi: 10.1016/j.jconrel.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 106.Peppas J, Huang NA, Torres-Lugo Y, Ward M, Zhang JH. Physicochemical Foundations and Structural Design of Hydrogels in Medicine and Biology. Annu Rev Biomed Eng. 2000:9–29. doi: 10.1146/annurev.bioeng.2.1.9. doi:1523-9829/00/0825-0009. [DOI] [PubMed] [Google Scholar]
- 107.De KS, Aluru NR, Johnson B, Crone WC, Beebe DJ, Moore J. Equilibrium swelling and kinetics of pH-responsive hydrogels: Models, experiments, and simulations. J Microelectromechanical Syst. 2002;11:544–555. doi: 10.1109/JMEMS.2002.803281. [DOI] [Google Scholar]
- 108.Gupta P, Vermani K, Garg S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov Today. 2002;7:569–579. doi: 10.1016/S1359-6446(02)02255-9. [DOI] [PubMed] [Google Scholar]
- 109.Löwenberg C, Balk M, Wischke C, Behl M, Lendlein A. Shape-Memory Hydrogels: Evolution of Structural Principles To Enable Shape Switching of Hydrophilic Polymer Networks. Acc Chem Res. 2017;50:723–732. doi: 10.1021/acs.accounts.6b00584. [DOI] [PubMed] [Google Scholar]
- 110.Oyen ML. Mechanical characterisation of hydrogel materials. Int Mater Rev. 2014;59:44–59. doi: 10.1179/1743280413Y.0000000022. [DOI] [Google Scholar]
- 111.Khetan S, Katz JS, Burdick JA. Sequential crosslinking to control cellular spreading in 3-dimensional hydrogels. Soft Matter. 2009;5:1601. doi: 10.1039/b820385g. [DOI] [Google Scholar]
- 112.Gillette BM, Jensen JA, Wang M, Tchao J, Sia SK. Dynamic hydrogels: Switching of 3D microenvironments using two-component naturally derived extracellular matrices. Adv Mater. 2010;22:686–691. doi: 10.1002/adma.200902265. [DOI] [PubMed] [Google Scholar]
- 113.Takezawa T, Mori Y, Yoshizato K. Cell culture on a thermo-responsive polymer surface. Nat Biotechnol. 1990;8:854–856. doi: 10.1038/nbt0990-854. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1366797. [DOI] [PubMed] [Google Scholar]
- 114.Yamada N, Okano T, Sakai H, Karikusa F, Sawasaki Y, Sakurai Y. Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Makromol Chemie, Rapid Commun. 1990;11:571–576. doi: 10.1002/marc.1990.030111109. [DOI] [Google Scholar]
- 115.Antoine EE, Vlachos PP, Rylander MN. Review of Collagen I Hydrogels for Bioengineered Tissue Microenvironments: Characterization of Mechanics, Structure, and Transport. Tissue Eng Part B Rev. 2014;20:683–696. doi: 10.1089/ten.teb.2014.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hersel U, Dahmen C, Kessler H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials. 2003;24:4385–4415. doi: 10.1016/S0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 117.Schussler O, Coirault C, Louis-Tisserand M, Al-Chare W, Oliviero P, Menard C, Michelot RJ, Bochet P, Salomon DR, Chachques JC, Carpentier A, Lecarpentier Y. Use of arginine-glycine-aspartic acid adhesion peptides coupled with a new collagen scaffold to engineer a myocardium-like tissue graft. Nat Clin Pract Cardiovasc Med. 2009;6:240–249. doi: 10.1038/ncpcardio1451. [DOI] [PubMed] [Google Scholar]
- 118.Belair DG, Le NN, Murphy WL. Design of growth factor sequestering biomaterials. Chem Commun. 2014;50:15651–15668. doi: 10.1039/C4CC04317K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gandavarapu NR, Azagarsamy MA, Anseth KS. Photo-click living strategy for controlled, reversible exchange of biochemical ligands. Adv Mater. 2014;26:2521–2526. doi: 10.1002/adma.201304847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deforest CA, Tirrell DA. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 122.Li S, Gaddes ER, Chen N, Wang Y. Molecular encryption and reconfiguration for remodeling of dynamic hydrogels. Angew Chemie - Int Ed. 2015;54:5957–5961. doi: 10.1002/anie.201500397. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Z, Chen N, Li S, Battig MR, Wang Y. Programmable hydrogels for controlled cell catch and release using hybridized aptamers and complementary sequences. J Am Chem Soc. 2012;134:15716–15719. doi: 10.1021/ja307717w. [DOI] [PubMed] [Google Scholar]
- 124.Zhang Z, Li S, Chen N, Yang C, Wang Y. Programmable display of DNA-protein chimeras for controlling cell-hydrogel interactions via reversible intermolecular hybridization. Biomacromolecules. 2013;14:1174–1180. doi: 10.1021/bm400096z. [DOI] [PubMed] [Google Scholar]
- 125.Chen N, Zhang Z, Soontornworajit B, Zhou J, Wang Y. Cell adhesion on an artificial extracellular matrix using aptamer-functionalized PEG hydrogels. Biomaterials. 2012;33:1353–1362. doi: 10.1016/j.biomaterials.2011.10.062. [DOI] [PubMed] [Google Scholar]
- 126.Davis BK. Control of diabetes with polyacrylamide implants containing insulin. Experientia. 1972;28:348. doi: 10.1007/BF01928732. [DOI] [PubMed] [Google Scholar]
- 127.Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 128.Davis BK. Diffusion of polymer gel implants. Proc Natl Acad Sci U S A. 1974;71:3120–3. doi: 10.1073/pnas.71.8.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Okay O. Macroporous copolymer networks. Prog Polym Sci. 2000;25:711–779. doi: 10.1016/S0079-6700(00)00015-0. [DOI] [Google Scholar]
- 130.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3:1–33. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Webb DJ, Roadcap DW, Dhakephalkar A, Gonias SL. A 16-amino acid peptide from human a 2 -macroglobulin binds transforming growth factor-b and platelet-derived growth factor-BB. Protein Sci. 2000;9:1986–1992. doi: 10.1110/ps.9.10.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kraemer S, Vaught JD, Bock C, Gold L, Katilius E, Keeney TR, Kim N, Saccomano NA, Wilcox SK, Zichi D, Sanders GM. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS One. 2011;6 doi: 10.1371/journal.pone.0026332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vaught JD, Bock C, Carter J, Fitzwater T, Otis M, Schneider D, Rolando J, Waugh S, Wilcox SK, Eaton BE. Expanding the chemistry of DNA for in vitro selection. J Am Chem Soc. 2010;132:4141–4151. doi: 10.1021/ja908035g. [DOI] [PubMed] [Google Scholar]
- 134.Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, Gold L, Janjic N. Nucleic acid ligands with protein-like side chains: Modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther - Nucleic Acids. 2014;3:e201. doi: 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, Gold L, Janjic N. Nucleic acid ligands with protein-like side chains: Modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther - Nucleic Acids. 2014;3:e201. doi: 10.1038/mtna.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang X, Battig MR, Chen N, Gaddes ER, Duncan KL, Wang Y. Chimeric Aptamer-Gelatin Hydrogels as an Extracellular Matrix Mimic for Loading Cells and Growth Factors. Biomacromolecules. 2016;17:778–787. doi: 10.1021/acs.biomac.5b01511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lai J, Jiang P, Gaddes ER, Zhao N, Abune L, Wang Y. Aptamer-Functionalized Hydrogel for Self-Programmed Protein Release via Sequential Photoreaction and Hybridization. Chem Mater. 2017;29:5850–5857. doi: 10.1021/acs.chemmater.7b00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Discher DE. Tissue Cells Feel and Respon to the Stiffness of Their Substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 139.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 140.Beningo KA, Lo CM, Wang YL. Flexible polyacrylamide substrata for the analysis of mechanical interactions at cell-substratum adhesions. Methods Cell Biol. 2002;2002:325–339. doi: 10.1016/S0091-679X(02)69021-1. [DOI] [PubMed] [Google Scholar]
- 141.Mierke CT. The fundamental role of mechanical properties in the progression of cancer disease and inflammation. Reports Prog Phys. 2014;77 doi: 10.1088/0034-4885/77/7/076602. [DOI] [PubMed] [Google Scholar]
- 142.Yanez LZ, Han J, Behr BB, Pera RAR, Camarillo DB. Human oocyte developmental potential is predicted by mechanical properties within hours after fertilization. Nat Commun. 2016;7 doi: 10.1038/ncomms10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guvendiren M, Burdick JA. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat Commun. 2012;3:792–799. doi: 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- 144.Kumashiro Y, Yamato M, Okano T. Cell attachment-detachment control on temperature-responsive thin surfaces for novel tissue engineering. Ann Biomed Eng. 2010;38:1977–1988. doi: 10.1007/s10439-010-0035-1. [DOI] [PubMed] [Google Scholar]
- 145.Nishida K, Yamato M, Hayashida Y, Watanabe K, Yamamoto K, Adachi E, Nagai S, Kikuchi A, Maeda N, Watanabe H, Okano T, Tano Y. Corneal Reconstruction with Tissue-Engineered Cell Sheets Composed of Autologous Oral Mucosal Epithelium. N Engl J Med. 2004;351:1187–1196. doi: 10.1056/NEJMoa040455. [DOI] [PubMed] [Google Scholar]
- 146.Nishida K, Yamato M, Hayashida Y, Watanabe K, Maeda N, Watanabe H, Yamamoto K, Nagai S, Kikuchi A, Tano Y, Okano T. Functional bioengineered corneal epithellial sheet grafts from corneal stem cells expanded ex vivo on a temperature-responsive cell culture surface. Transplantation. 2004;77:379–385. doi: 10.1097/01.TP.0000110320.45678.30. [DOI] [PubMed] [Google Scholar]
- 147.Shimizu T. Fabrication of Pulsatile Cardiac Tissue Grafts Using a Novel 3-Dimensional Cell Sheet Manipulation Technique and Temperature-Responsive Cell Culture Surfaces. Circ Res. 2002;90:40e–48. doi: 10.1161/hh0302.105722. [DOI] [PubMed] [Google Scholar]
- 148.Ohashi K, Koyama F, Tatsumi K, Shima M, Park F, Nakajima Y, Okano T. Functional life-long maintenance of engineered liver tissue in mice following transplantation under the kidney capsule. J Tissue Eng Regen Med. 2010;4:141–148. doi: 10.1002/term.225. [DOI] [PubMed] [Google Scholar]
- 149.Hayashi R, Ishikawa Y, Sasamoto Y, Katori R, Nomura N, Ichikawa T, Araki S, Soma T, Kawasaki S, Sekiguchi K, Quantock AJ, Tsujikawa M, Nishida K. Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature. 2016;531:376–380. doi: 10.1038/nature17000. [DOI] [PubMed] [Google Scholar]
- 150.Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 151.Rouwkema J, Rivron NC, van Blitterswijk CA. Vascularization in tissue engineering. Trends Biotechnol. 2008;26:434–441. doi: 10.1016/j.tibtech.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 152.Simpson RJ, Bernhard OK, Greening DW, Moritz RL. Proteomics-driven cancer biomarker discovery: looking to the future. Curr Opin Chem Biol. 2008;12:72–77. doi: 10.1016/j.cbpa.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 153.Boguskl MS, McIntosh MW. Biomedical informatics for proteomics. Nature. 2003;422:233–237. doi: 10.1038/nature01515. [DOI] [PubMed] [Google Scholar]
- 154.Huang D, Xiang N, Tang W, Zhang X, Ni Z. Microfluidics-based circulating tumor cells separation. Prog Chem. 2015;27:882–912. doi: 10.7536/PC150121. [DOI] [Google Scholar]
- 155.Nicodemou A, Danisovic L. Mesenchymal stromal/stem cell separation methods: concise review. Cell Tissue Bank. 2017;18:443–460. doi: 10.1007/s10561-017-9658-x. [DOI] [PubMed] [Google Scholar]
- 156.Haukanes BI, Kvam C. Application of magnetic beads in bioassays. Biotechnology. 1993;11:60–63. doi: 10.1038/nbt0193-60. [DOI] [PubMed] [Google Scholar]
- 157.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Henke C, Bitterman P, Roongta U, Ingbar D, Polunovsky V. Induction of fibroblast apoptosis by anti-CD44 antibody: implications for the treatment of fibroproliferative lung disease. Am J Pathol. 1996;149:1639–50. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1865279&tool=pmcentrez&rendertype=abstract. [PMC free article] [PubMed] [Google Scholar]
- 159.Smith JA, Bluestone JA. T cell inactivation and cytokine deviation promoted by anti-CD3 mAbs. Curr Opin Immunol. 1997;9:648–654. doi: 10.1016/S0952-7915(97)80044-1. [DOI] [PubMed] [Google Scholar]
- 160.Plouffe BD, Brown MA, Iyer RK, Radisic M, Murthy SK. Controlled capture and release of cardiac fibroblasts using peptide-functionalized alginate gels in microfluidic channels. Lab Chip. 2009;9:1507. doi: 10.1039/b823523f. [DOI] [PubMed] [Google Scholar]
- 161.Chen N, Zhang Z, Soontornworajit B, Zhou J, Wang Y. Cell adhesion on an artificial extracellular matrix using aptamer-functionalized PEG hydrogels. Biomaterials. 2012;33:1353–1362. doi: 10.1016/j.biomaterials.2011.10.062. [DOI] [PubMed] [Google Scholar]
- 162.Lee MH, Brass DA, Morris R, Composto RJ, Ducheyne P. The effect of non-specific interactions on cellular adhesion using model surfaces. Biomaterials. 2005;26:1721–1730. doi: 10.1016/j.biomaterials.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 163.Kairdolf BA, Mancini MC, Smith AM, Nie S. Minimizing nonspecific cellular binding of quantum dots with hydroxyl-derivatized surface coatings. Anal Chem. 2008;80:3029–3034. doi: 10.1021/ac800068q. [DOI] [PubMed] [Google Scholar]
- 164.Yang CY, Huang LY, Shen TL, Andrew Yeh J. Cell adhesion, morphology and biochemistry on nanotopographic oxidized silicon surfaces. Eur Cells Mater. 2010;20:415–430. doi: 10.22203/eCM.v020a34. [DOI] [PubMed] [Google Scholar]
- 165.Harbers GM, Emoto K, Greef C, Metzger SW, Woodward HN, Mascali JJ, Grainger DW, Lochhead MJ. Functionalized poly(ethylene glycol)-based bioassay surface chemistry that facilitates bio-immobilization and inhibits nonspecific protein, bacterial, and mammalian cell adhesion. Chem Mater. 2007;19:4405–4414. doi: 10.1021/cm070509u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xu Y, Phillips JA, Yan J, Li Q, Fan ZH, Tan W. Aptamer-based microfluidic device for enrichment, sorting, and detection of multiple cancer cells. Anal Chem. 2009;81:7436–7442. doi: 10.1021/ac9012072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guo KT, Scharnweber D, Schwenzer B, Ziemer G, Wendel HP. The effect of electrochemical functionalization of Ti-alloy surfaces by aptamer-based capture molecules on cell adhesion. Biomaterials. 2007;28:468–474. doi: 10.1016/j.biomaterials.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 168.Chen L, Liu X, Su B, Li J, Jiang L, Han D, Wang S. Aptamer-mediated efficient capture and release of T lymphocytes on nanostructured surfaces. Adv Mater. 2011;23:4376–4380. doi: 10.1002/adma.201102435. [DOI] [PubMed] [Google Scholar]
- 169.Wan Y, Liu Y, Allen PB, Asghar W, Mahmood MAI, Tan J, Duhon H, Kim Y, Ellington AD, Iqbal SM. Capture, isolation and release of cancer cells with aptamer-functionalized glass bead array. Lab Chip. 2012;12:4693. doi: 10.1039/c2lc21251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dirks RM, Pierce NA. Triggered amplification by hybridization chain reaction. Proc Natl Acad Sci U S A. 2004;101:15275–15278. doi: 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Richards E, Li S, Chen N, Battig MR, Wang Y. Polymerization of affinity ligands on a surface for enhanced ligand display and cell binding. Biomacromolecules. 2014;15:4561–4569. doi: 10.1021/bm501347s. [DOI] [PubMed] [Google Scholar]
- 172.Gaddes ER, Gydush G, Li S, Chen N, Dong C, Wang Y. Aptamer-Based Polyvalent Ligands for Regulated Cell Attachment on the Hydrogel Surface. Biomacromolecules. 2015;16:1382–1389. doi: 10.1021/acs.biomac.5b00165. [DOI] [PubMed] [Google Scholar]
- 173.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, Stegmann TJ, Udelson JE, Rosengart TK. Clinical Trials in Coronary Angiogenesis: Issues, Problems, Consensus : An Expert Panel Summary. Circulation. 2000;102:e73–e86. doi: 10.1161/01.CIR.102.11.e73. [DOI] [PubMed] [Google Scholar]
- 175.Chen RR, Mooney DJ. Polymeric growth factor delivery strategies for tissue engineering. Pharm Res. 2003;20:1103–1112. doi: 10.1023/A:1025034925152. [DOI] [PubMed] [Google Scholar]
- 176.Hoare TR, Kohane DS. Hydrogels in drug delivery: Progress and challenges. Polymer (Guildf) 2008;49:1993–2007. doi: 10.1016/j.polymer.2008.01.027. [DOI] [Google Scholar]
- 177.Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853–2888. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 178.Okay O. Macroporous copolymer networks. 2000 doi: 10.1016/S0079-6700(00)00015-0. [DOI] [Google Scholar]
- 179.Vulic K, Shoichet MS. Affinity-based drug delivery systems for tissue repair and regeneration. Biomacromolecules. 2014;15:3867–3880. doi: 10.1021/bm501084u. [DOI] [PubMed] [Google Scholar]
- 180.Sakiyama-Elbert SE. Incorporation of heparin into biomaterials. Acta Biomater. 2014;10:1581–1587. doi: 10.1016/j.actbio.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liang Y, Kiick KL. Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 2014;10:1588–1600. doi: 10.1016/j.actbio.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lin CC, Metters AT. Bifunctional monolithic affinity hydrogels for dual-protein delivery. Biomacromolecules. 2008;9:789–795. doi: 10.1021/bm700940w. [DOI] [PubMed] [Google Scholar]
- 183.Battig MR, Huang Y, Chen N, Wang Y. Aptamer-functionalized superporous hydrogels for sequestration and release of growth factors regulated via molecular recognition. Biomaterials. 2014;35:8040–8048. doi: 10.1016/j.biomaterials.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 184.Franz S, Rammelt S, Scharnweber D, Simon JC. Immune responses to implants - A review of the implications for the design of immunomodulatory biomaterials. Biomaterials. 2011;32:6692–6709. doi: 10.1016/j.biomaterials.2011.05.078. [DOI] [PubMed] [Google Scholar]
- 185.Zhao N, Battig MR, Xu M, Wang X, Xiong N, Wang Y. Development of a Dual-Functional Hydrogel Using RGD and Anti-VEGF Aptamer. Macromol Biosci. 2017;17:1–8. doi: 10.1002/mabi.201700201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simón-Yarza T, Formiga FR, Tamayo E, Pelacho B, Prosper F, Blanco-Prieto MJ. Vascular endothelial growth factor-delivery systems for cardiac repair: An overview. Theranostics. 2012;2:541–552. doi: 10.7150/thno.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gu F, Neufeld R, Amsden B. Sustained release of bioactive therapeutic proteins from a biodegradable elastomeric device. J Control Release. 2007;117:80–89. doi: 10.1016/j.jconrel.2006.09.077. [DOI] [PubMed] [Google Scholar]
- 188.Ekaputra AK, Prestwich GD, Cool SM, Hutmacher DW. The three-dimensional vascularization of growth factor-releasing hybrid scaffold of poly (e{open}-caprolactone)/collagen fibers and hyaluronic acid hydrogel. Biomaterials. 2011;32:8108–8117. doi: 10.1016/j.biomaterials.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 189.Dittmer WU, Reuter A, Simmel FC. A DNA-based machine that can cyclically bind and release thrombin. Angew Chemie - Int Ed. 2004;43:3550–3553. doi: 10.1002/anie.200353537. [DOI] [PubMed] [Google Scholar]
- 190.Xiong X, Wu C, Zhou C, Zhu G, Chen Z, Tan W. Responsive DNA-based hydrogels and their applications. Macromol Rapid Commun. 2013;34:1271–1283. doi: 10.1002/marc.201300411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ni X, Castanares M, Mukherjee A, Lupold SE. Nucleic acid aptamers: clinical applications and promising new horizons. Curr Med Chem. 2011;18:4206–14. doi: 10.2174/092986711797189600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang Z, Du J, Li Y, Wu J, Yu F, Chen Y. An aptamer-patterned hydrogel for the controlled capture and release of proteins via biorthogonal click chemistry and DNA hybridization. J Mater Chem B. 2017;5:5974–5982. doi: 10.1039/C7TB00883J. [DOI] [PubMed] [Google Scholar]
- 193.Enam SF, Krieger JR, Saxena T, Watts BE, Olingy CE, Botchwey EA, Bellamkonda RV. Enrichment of endogenous fractalkine and anti-inflammatory cells via aptamer-functionalized hydrogels. Biomaterials. 2017;142:52–61. doi: 10.1016/j.biomaterials.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 194.Jiang P, Li S, Lai J, Zheng H, Lin C, Shi P, Wang Y. Nanoparticle-Programmed Surface for Drug Release and Cell Regulation via Reversible Hybridization Reaction. ACS Appl Mater Interfaces. 2017;9:4467–4474. doi: 10.1021/acsami.6b14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Li S, Chen N, Gaddes ER, Zhang X, Dong C, Wang Y. A Drosera-bioinspired hydrogel for catching and killing cancer cells. Sci Rep. 2015;5 doi: 10.1038/srep14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gao B, Yang Q, Zhao X, Jin G, Ma Y, Xu F. 4D Bioprinting for Biomedical Applications. Trends Biotechnol. 2016;34:746–756. doi: 10.1016/j.tibtech.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 197.Ruskowitz ER, DeForest CA. Photoresponsive biomaterials for targeted drug delivery and 4D cell culture. Nat Rev Mater. 2018;3:17087. doi: 10.1038/natrevmats.2017.87. [DOI] [Google Scholar]