

Abstract
NMNAT1 (nicotinamide mononucleotide adenylyltransferase 1) encodes a rate-limiting enzyme that catalyzes the biosynthesis of NAD+ and plays a role in neuroprotection. Mutations in NMNAT1 have been identified to cause a recessive, non-syndromic early form of blindness genetically defined as Leber Congenital Amaurosis 9 (LCA9). One of the most common alleles reported so far in NMNAT1 is the c.769G>A (E257K) missense mutation, which occurs in 70% of all LCA9 cases. However, given its relatively high population frequency and the observation of individuals with homozygous E257K variant without phenotype, the pathogenicity of this allele has been questioned. To address this issue, we have studied the pathogenic effects of this allele by generating a knock-in mouse model. Interestingly, no obvious morphological or functional defects are observed in Nmnat1 E257K homozygous mice up to one year old, even after light-damage. Together with the previous clinical reports, we propose that the E257K allele is a weak hypomorphic allele that has significantly reduced penetrance in the homozygous state. In contrast, compound heterozygous Nmnat1E257K/− mice exhibited photoreceptor defects which were exacerbated upon exposure to light. Furthermore, retina specific Nmnat1 conditional knockout mice exhibit photoreceptor degeneration before the retina has terminally differentiated. These findings suggest that NMNAT1 plays an important role in photoreceptors and is likely involved in both retinal development and maintenance of photoreceptor integrity.
Introduction
Leber Congenital Amaurosis (LCA) is characterized by photoreceptor cell dysfunction and cell death with functional loss of rod and cone cells at an early age of life (den Hollander et al., 2008; Kumaran et al., 2017; Wang et al., 2009). LCA is an inherited form of retinal dystrophy, and more than 20 causative genes had been identified. LCA9 is an autosomal recessive retinal degeneration condition caused by mutations in the NMNAT1 gene, encoding a nicotinamide adenine dinucleotide (NAD+) biosynthetic enzyme, NMNAT1, which synthesizes NAD+ from nicotinamide mononucleotide (NMN) and ATP (Falk et al., 2012; Koenekoop et al., 2012; Sasaki et al., 2015). In patients with LCA9, 38 causative variants have been identified in NMNAT1, and a majority of them are missense mutations (Chiang et al., 2012; Falk et al., 2012; Keen et al., 2003; Koenekoop et al., 2012; Perrault et al., 2012; Thompson et al., 2017). In vitro studies evaluating the functional consequences of human NMNAT1 variants have shown that they result in reduced NMNAT1 enzymatic activity. Previous studies indicate that NMNAT1 plays a role in neuroprotection (Sasaki et al., 2009a; Sasaki et al., 2009b; Williams et al., 2017). In the Golgi complex and mitochondria, two other NMNAT paralogs function similarly to regenerate NAD+, encoded by NMNAT2 and NMNAT3, respectively (Berger et al., 2005; Hassa et al., 2006; Mori et al., 2014; Sasaki et al., 2015). Metabolic profiling showed that NMNAT1 exhibits higher enzymatic activity than other isozymes in mouse tissues (Mori et al., 2014). Despite similar functions, neither paralog can compensate for the loss of NMNAT1 since Nmnat1 homozygous null mice exhibit embryonic lethality (Conforti et al., 2011).
Interestingly, of the variants identified in NMNAT1-LCA9 patients, the E257K (NM_022787: exon 5: c.769G>A: p.E257K) variant is the most frequently observed, accounting for more than 70% of LCA9 cases based on previously published reports (Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012; Siemiatkowska et al., 2014b; Thompson et al., 2017). However, a recent report has identified individuals who are homozygous for the E257K variant and have no ocular phenotypes, and one individual presented with intellectual disability (Siemiatkowska et al., 2014a). In addition, based on the allele frequency in healthy European Americans, the Hardy-Weinberg prediction suggests that a significantly larger portion of LCA patients than observed should be homozygous for this variant. Therefore, it has been postulated that this variant likely has low penetrance in the homozygous state.
To gain insights into the role of Nmnat1 function, two Nmnat1 mutant mice generated by a large-scale N-ethyl-N-nitrosourea mutagenesis screen, Nmnat1V9M and Nmnat1D243G, exhibit retinal degeneration (Greenwald et al., 2016). However, our knowledge of the significance of the E257K NMNAT1 variant still remains limited. Functional studies, in vivo and in vitro, have demonstrated that the NMNAT1 E257K variant results in reduced enzymatic activity based on analysis in red blood cells from LCA9 patients and in HeLa cells overexpressing wildtype and mutant proteins (Koenekoop et al., 2012) and also altered structural stability of NMNAT1 under stress conditions (Sasaki et al., 2015), demonstrating a possible pathogenic effect of this variant on NMNAT1 function.
In this study, to gain insights into the pathogenicity of the NMNAT1 E257K variant, we generated a knock-in Nmnat1 mouse model by introducing the point mutation c.769G>A in exon 4. Nmnat1E257K/E257K homozygous mice did not exhibit a retinal phenotype as assessed by histology and electroretinography (ERG). In addition, we generated compound heterozygous Nmnat1E257K/− mice by crossing Nmant1+/− mice, in which exon 2 is deleted, with Nmnat1E257K/E257K mice. Nmnat1+/− mice are grossly normal and do not exhibit any retinal phenotypes. However, phenotypic characterization of Nmnat1E257K/− mouse retinas revealed significant thinning of the photoreceptor layer beginning at 5 months of age. The retinal degeneration phenotype is progressive and can be accelerated after exposure to strong white light. To test whether the E257K variant affects NMNAT1 localization, we performed immunostaining and observed no change, suggesting that this variant does not alter the protein expression pattern in the retina. We also detected expression of activated ER stress markers in the retina of Nmnat1E257K/− mice after light-induced damage, suggesting that Nmnat1E257K/− mice are more susceptible to ER stress, which likely contributes to photoreceptor degeneration and death.
Furthermore, to overcome the embryonic lethality exhibited by Nmnat1 germline null mice and investigate the role of NMNAT1 in retinal degeneration, we generated Nmnat1 conditional knock-out (Nmnat1 cKO) mice. NMNAT1 protein expression has been reported in the mouse retina (Zhou et al., 2016), but its sub-cellular localization and function in photoreceptors has not been examined. Since photoreceptors are the primary cell types affected in LCA and other inherited retinal dystrophies, we focused on photoreceptor-specific loss of Nmnat1 in conditional knock-out mice. Specifically, we used Chx10-Cre (Rowan and Cepko, 2004), Crx-Cre (Nishida et al., 2003) and Rhodopsin-iCre (Li et al., 2005) transgenic mouse lines, which result in retinal-specific, photoreceptor-specific and rod-specific conditional deletion, respectively. Phenotypic characterization of the retinas of Nmnat1 cKO mice revealed rapid and progressive retinal degeneration in all three conditional mouse lines at an early age, suggesting that Nmnat1 plays a critical role in retinal development. These findings provide insights into the role of NMNAT1 in retinal development and are useful for future therapeutic interventions for LCA9 patients.
Results
1. Expression of NMNAT1 in the retina
To determine the localization of NMNAT1 in mouse retina, we used a NMNAT1 antibody to specifically examine its expression in the wild-type mouse retina. NMNAT1 was detected in the photoreceptor layer of the mouse retina from post-natal day 2 and is consistently expressed throughout post-natal development and throughout adulthood (Fig. 1A). NMNAT1 expression is also detected in the outer plexiform layer (OPL), inner plexiform layer (IPL), and ganglion cell layer (GCL). The specificity of the NMNAT1 antibody was confirmed by a blocking experiment (Fig. 1B). In order to determine its specific expression pattern in photoreceptors, we used antibodies against the ciliary markers pan-Centrin (marker of axoneme) and γ-tubulin (marker of basal body). NMNAT1 localization partially overlaps with γ-tubulin, and its expression is distal to Centrin (Fig. 1C). Furthermore, immunostaining of NMNAT1 in cultured hTERT-RPE1 cells with induced cilia consistently shows that NMNAT1 localizes near the cilia basal body (Fig. 1D), suggesting that NMNAT1 is localized near the basal body of the photoreceptor.
Figure 1. NMNAT1 localizes to the basal body of mouse photoreceptors.
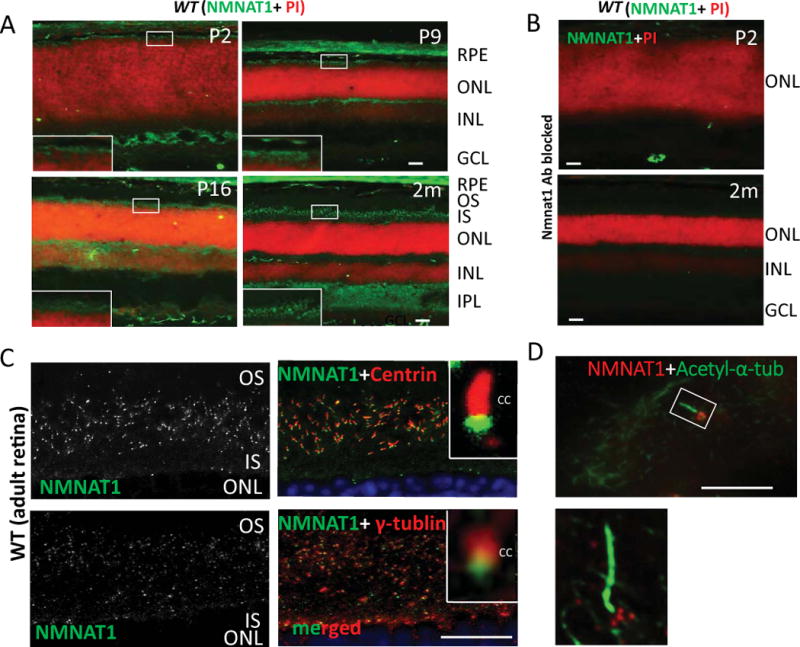
(A) NMNAT1 expression in wild-type frozen sections is detected in the photoreceptor cell layer beginning at P2 and continues throughout adult stages (NMNAT1 shown in green; propidium iodine (PI) shown in red indicating nuclear staining). (B) Cryosections of WT retina treated with a NMNAT1 blocking peptide in combination with NMNAT1 antibody shows lack of NMNAT1 staining, confirming the specificity of the NMNAT1 antibody. (C) Cryosections of WT retina co-stained with anti-NMNAT1, pan-Centrin (an axoneme marker) and γ-tubulin (a marker of the basal body) antibodies. Inserts: high magnification images show NMNAT1 positive signals near the basal body of the photoreceptor. (D) Co-staining of ciliated hTERT-RPE1 cultured cells with NMNAT1 and acetylated α-tubulin (an axoneme marker) shows NMNAT1 positive signal near the basal body. Scale bar: A–B: 20um; C: 10um; D: 2um. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, IPL= inner plexiform layer, GCL= ganglion cell layer, OS= outer segment, IS= inner segment, CC= connecting cilium.
2. Generation of Nmnat1 mutant alleles
Nmnat1E257K/E257K mice harboring a point mutation in exon 4 (Fig. 2A) were generated by BAC recombineering (described in the methods section). Nmnat1 E257K homozygous mice were viable and fertile. We generated a Nmnat1 knockout allele by deleting exon 2 (Fig. 2 B, C) to generate Nmnat1+/− mice. We crossed Nmnat1+/− mice with Nmnat1E257K/E257K mice to generate Nmnat1E257K/− mice. Although Nmnat1−/− mice are embryonic lethal as previously reported (Conforti et al., 2011), Nmnat1+/− mice are viable and fertile, and do not exhibit any readily detectable retinal defects.
Figure 2. Generation of Nmnat1 conditional knockout alleles.
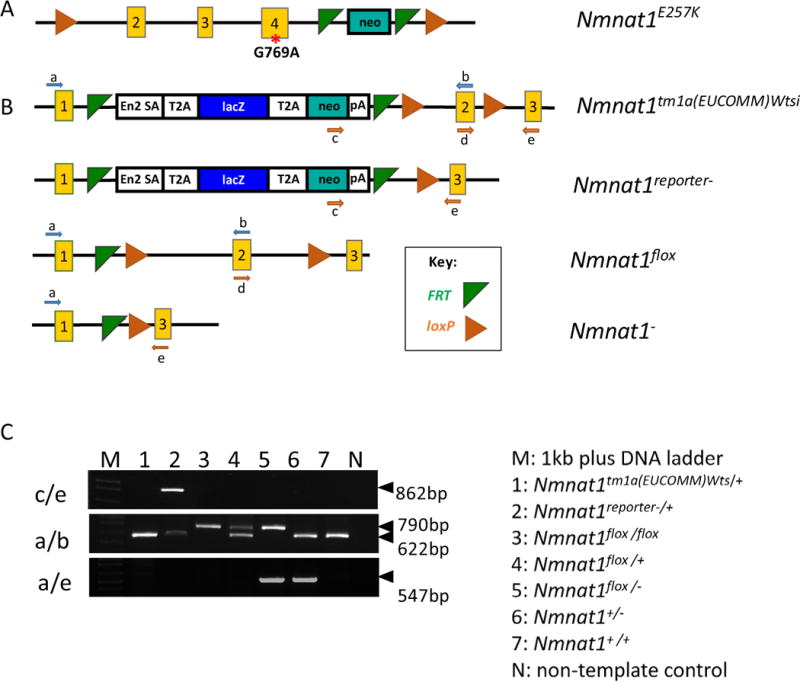
(A) Schematic of a point mutation introduced in exon 4 of Nmnat1 to generate the Nmnat1E257K mutant allele. (B) Generation of Nmnat1 floxed alleles; green triangles indicate the FRT sites flanking the neo cassette, and orange triangles indicate the loxP sites flanking exon 2 (C) PCR performed using 3 primer pairs: c/e, a/b, a/e, flanking the target regions of interest used for genotyping along with legend (right) indicating the appropriate genotypes for lanes 1–7 on the gel (left).
3. Characterization of Nmnat1 E257K Homozygous mouse retina
To study the pathogenicity of the NMNAT1 E257K variant, Nmnat1 mice harboring the E257K allele were generated by creating a point mutation, c.769G>A, in exon 4. Phenotypic characterization of the Nmnat1E257K/E257K homozygous mice showed no detectable retinal degeneration as observed at 5 months of age (Supp. Fig. 1A). Further histological analysis in aged Nmnat1E257K/E257K mice at 8 months also failed to reveal any retinal degeneration phenotypes (Supp. Fig. 1). To test the retinal function in Nmnat1E257K/E257K mice, we performed scotopic and phenotypic full-field electroretinography (ERG) in mice at 12 months of age and observed normal a-wave and b-wave responses to light (data not shown).
Light induced damage is an important factor in accelerating retinal degeneration (Bai and Sheline, 2013). Findings from a previous study show that Nmnat1 Glu257Lys (E257K) mutant enzyme is susceptible to heat induced stress (Sasaki et al., 2015). To examine whether light exposure can increase retinal degeneration in Nmnat1E257K/E257K mice, we subjected Nmnat1E257K/E257K mice to strong white light for 4 hours. Histological examination of mice 2 weeks after exposure to light damage showed that the retinal morphology is unaffected in Nmnat1E257K/E257K mice at 5 months of age (Supp. Fig. 1B).
Nmnat1E257K/− mice exhibit retinal dysfunction and degeneration
To determine whether photoreceptor function is altered in Nmnat1E257K/− mice, we performed ERG on dark-adapted Nmnat1E257K/− mice at different ages under scotopic conditions (Fig. 3 A, B). Starting at 5 months of age, a reduction in the amplitudes of rod-generated a-waves is observed in Nmnat1E257K/− mice and this decline becomes more pronounced with increased age at 12 months of age. Consistently, histological analysis shows evident retinal degeneration in the outer nuclear layer (ONL) at 5 months, and by 12 months, approximately 40% of the photoreceptor cells are degenerated (Fig. 4 A, B), indicating that photoreceptor degeneration is progressive. Additionally, we examined the expression of NMNAT1 in Nmnat1E257K/− retina by immunostaining which showed no significant alteration in NMNAT1 localization or level (Supp. Fig. 2). Immunostaining for rhodopsin (a marker for rod cells) and PNA (a marker for cone cells) showed loss of both rod and cone photoreceptor outer segments of aged Nmnat1E257K/− mice (Fig. 4C, D) at 12 months, demonstrating that both rod and cone photoreceptors are affected.
Figure 3. Nmnat1E257K/− mice exhibit photoreceptor dysfunction.
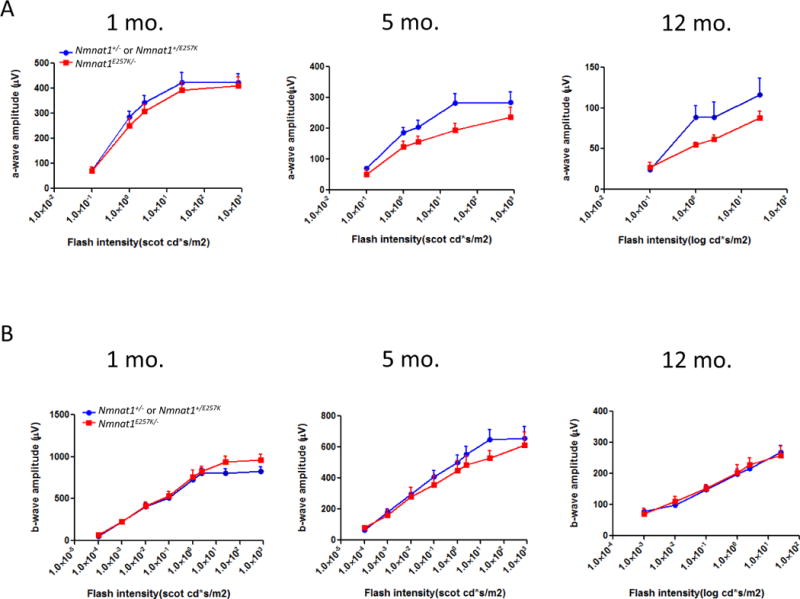
Electroretinogram recordings performed on Nmnat1E257K/− mutant mice and Nmnat1E257K/+ or Nmnat1+/− littermate control mice at 1 month, 5 months, and 12 months of age averaged from n ≥ 5 per group per genotype. (A) A significant reduction in the a-wave amplitude was detected in 5-month-old and 12-month-old Nmnat1E257K/− mutant mice (shown in red) compared to the a-wave amplitude of age-matched control littermates (shown in blue). (B) ERG responses (n≥ 5) of the b-wave show no marked decrease in amplitude in Nmnat1E257K/− mutant mice compared to Nmnat1E257K/+ or Nmnat1+/− littermate control mice at 1 month, 5 months, and 12 months of age.
Figure 4. Nmnat1E257K/− retinas show progressive photoreceptor degeneration.
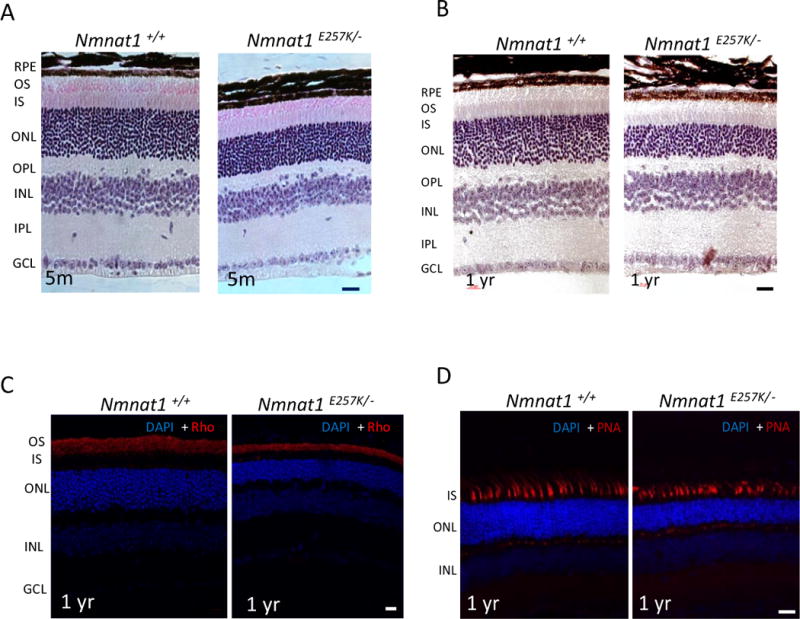
(A) Nmnat1E257K/− mutant mice exhibit mild photoreceptor degeneration at 5 months of age, which is progressive and more severe by 12 months (B) in comparison to Nmnat1 WT mice. (C) Immunostaining with Rhodopsin (Rho) antibody (shown in red) shows thinning of the outer segment (OS) in Nmnat1E257K/− mutant mice compared to Nmnat1 WT mice. (D) PNA staining shows attenuated cones in Nmnat1E257K/− retina compared to Nmnat1 WT retina. Scale bar: 20 μm.
4. Light-induced damage triggers ER stress and rapid photoreceptor degeneration in Nmnat1E257K/− mice
To examine whether light exposure can increase retinal degeneration in Nmnat1E257K/− mice, we subjected Nmnat1E257K/− mice, along with control littermates, to strong white light for 4h. Histological examination reveals marked photoreceptor degeneration two weeks after light exposure of Nmnat1E257K/− mice at 8 weeks of age (Fig. 5C), consistently throughout the retina (Fig. 5D). ERG results show noticeable reduction in scotopic a-wave (rod cells) and b-wave (bipolar cells) responses in light-damaged Nmnat1E257K/− mouse retinas (Fig. 5 A, B), consistent with changes in histology and retinal morphometry.
Figure 5. Light induced damage triggers rapid degeneration of photoreceptors of Nmnat1E257K/− mutant mice.
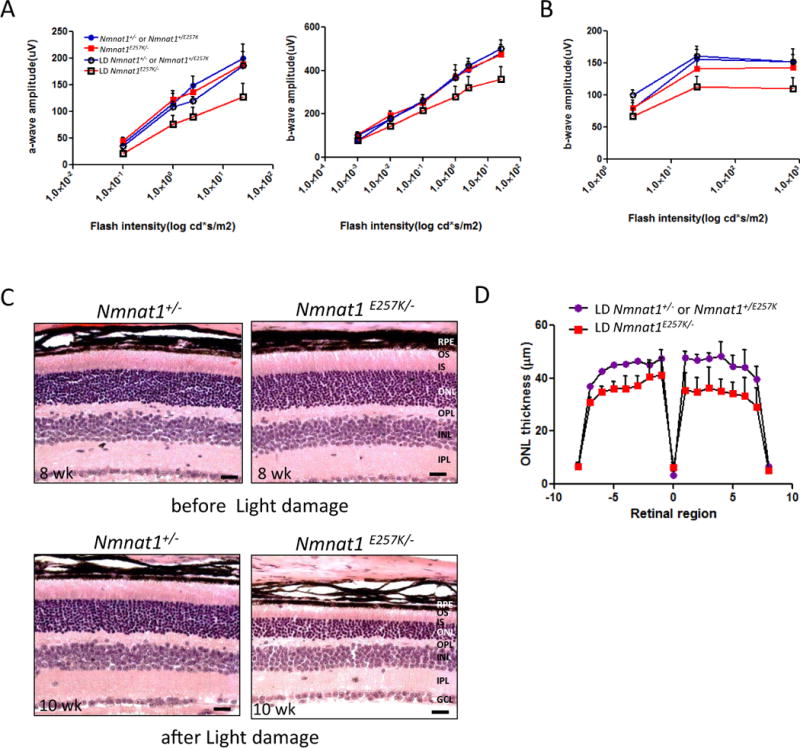
(A, B) Scotopic and photopic ERG were recorded to evaluate the before and after effects of light damage (LD) on Nmnat1E257K/− mutant mice compared to control age-matched littermates (n ≥ 5 per genotype). (C, D) H&E staining and retinal morphometry show marked photoreceptor degeneration in 10 weeks-old mutant mice after light exposure at 8 weeks of age compared to controls. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, OPL= outer plexiform layer, IPL= inner plexiform layer, GCL= ganglion cell layer, OS= outer segment, IS= inner segment.
Furthermore, since Nmnat1E257K/− mice showed retinal degeneration upon light-induced damage, we examined whether Nmnat1E257K/− mice exhibited ER stress under these conditions. Immunostaining with markers of ER stress showed positive staining for BIP and Caspase-12 at one, two, and four days (Supp. Fig. 3–5) after light exposure. CHOP positive signals were observed in Nmnat1E257K/− retinas examined both two days and four days after light exposure (Supp. Fig. 4–5); Caspase 12 staining (Fig. 6 A, B) and TUNEL positive signals (Fig. 6 C) were observed in retinal sections of Nmnat1E257K/− mice both 7 and 14 days after light exposure.
Figure 6. Light damage induces cell death in Nmnat1E257K/− retinas at 7 and 14 days after light exposure.
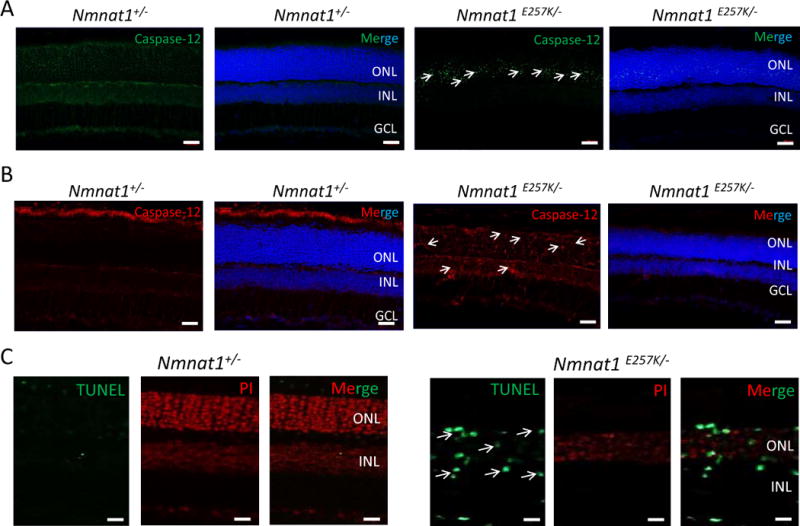
Cryosections of 8 weeks-old Nmnat1E257K/− retinas stained for Caspase-12, a marker of ER stress, after light exposure (A, B). Caspase-12 is not detected in the ONL of Nmnat1+/− retinas while strong staining is observed in the ONL of Nmnat1E257K/− retinas at 7 days (A) and 14 days (B) after light exposure. A subset of positive signals is marked by arrowheads. (C) TUNEL-positive cells (green) are detected mostly in the INL and ONL of paraffin embedded Nmnat1E257K/− retinas at 14 days after light exposure (right), while no signal is observed in control retina after light exposure (left). Scale bar: 20 μm. DAPI (blue) was used to counterstain nuclei in Panels A and B. Propidium Iodide (PI) was used to counterstain nuclei in Panel C. ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
5. Generation of Nmnat1 conditional knockout alleles
Since Nmnat1 null mice are embryonic lethal, this presents a limitation in further understanding the function of NMNAT1 in retinal disease pathogenesis. Therefore, we generated conditional knockout alleles using the Cre/loxP recombination system to delete Nmnat1 specifically in the eye. To generate an Nmnat1 floxed allele, mice carrying the Nmnat1tm1a (EUCOMM) Wtsi allele were crossed to Rosa-FLPe mice to remove the reporter tagged insertion, and two loxP sites flanking exon 2 were introduced into the targeted locus (Fig. 2B, C). Nmnat1flox/flox homozygous mice are viable, fertile and lack gross morphological defects. Using Nmnat1flox/flox and Nmnat1+/− mice in specific Cre backgrounds, we generated Nmnat1-Chx10-Cre-cKO (affecting all retina cells), Nmnat1-Crx-Cre-cKO (affecting only photoreceptor cells) and Nmnat1-iCre-cKO (affecting only rod photoreceptors) mice.
6. Conditional loss of Nmnat1 causes early onset retinal degeneration
Most Patients with bi-allelic Nmnat1 variants have severe, early onset vision loss presented as LCA. To examine if retina-specific conditional deletion of Nmnat1 recapitulates the phenotypes observed in LCA patients, we used Chx10-Cre mice expressing Cre recombinase in retinal progenitor cells to produce pan-retinal deletion. Histological analysis at post-natal day 2, 9, and 15 (P2, P9, P15) shows that Nmnat1-Chx10-Cre-cKO mice exhibit retinal degeneration marked by reduced thickness of inner nuclear and outer nuclear layers compared to littermate control mice at P9 (Fig. 7B), and a drastic reduction of retinal thickness is observed by P15 (Fig. 7C). No morphological changes were observed in the P2 retina of Nmnat1-Chx10-Cre-cKO mice (Fig. 7A).
Figure 7. Nmnat1Flox/−; Chx10-Cre conditional knockout mice exhibit rapid photoreceptor degeneration.
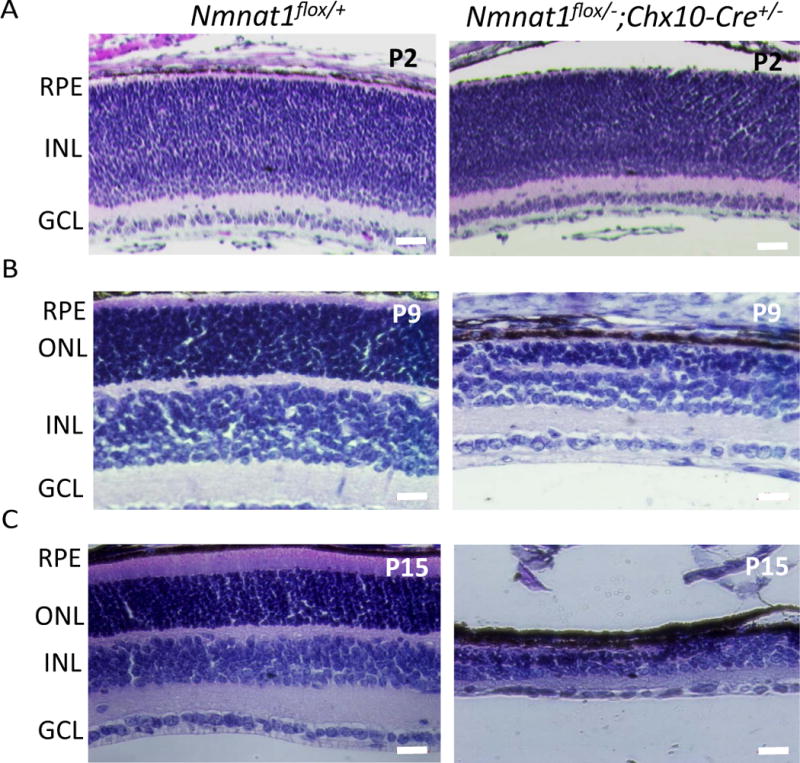
(A) Conditional deletion of Nmnat1 in the entire retina using Chx10-Cre did not show any readily detectable phenotype at P2 compared to control Nmnat1flox/+ mice by H&E staining. (B) At P9, severe reduction in the thickness of the ONL is observed in Nmnat1 cKO mice compared to control Nmnat1flox/+ mice. (C) At P15, severe thinning of the ONL and overall retina is observed in Nmnat1 cKO mice compared to control Nmnat1flox/+ mice. Scale bar: 20 μm. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
NMNAT1 is expressed in the retina in various cell types, including photoreceptors and cells of the OPL, IPL and GCL(Zhou et al., 2016) (Fig. 1). Since photoreceptors are highly metabolically active cells with high energy demands to regulate and sustain their function, and NMNAT1 regulates NAD+ biosynthesis, we hypothesized that selective depletion of NMNAT1 in photoreceptors is sufficient to cause retinal dysfunction. We generated Nmnat1 conditional knockout mice in photoreceptors by crossing Nmnat1+/−; Crx-Cre mice to Nmnat1flox/flox mice. Crx-driven Cre is detectable in photoreceptors as early as E11.5, allowing the deletion to occur early during development. Immunostaining of NMNAT1 in Nmnat1-Crx-Cre-cKO and control retina at P15 confirmed that NMNAT1 expression is strongly reduced throughout the Nmnat1-Crx-Cre-cKO retina as expected (Fig. 8B). RHO is undetectable due to significantly shortened outer segments (Fig. 8B). We performed histological analysis of the Nmnat1-Crx-Cre-cKO mouse retinas at several post-natal stages, including P3, P6, P15 and P28 (Fig. 8A). Although the histology appeared normal at P6 and only retinal lamination defects were observed in Nmnat1-Crx-Cre-cKO mice, progressive thinning of the outer nuclear layer, a hallmark of retinal degeneration, and reduced overall retinal thickness was apparent by P15. Furthermore, by P28, most photoreceptor cells are degenerated in Nmnat1-Crx-Cre-cKO mice as the outer nuclear layer is essentially absent in the retina (Fig. 8A).
Figure 8. Conditional ablation of Nmnat1 in photoreceptors using Crx-Cre causes rapid retinal degeneration.
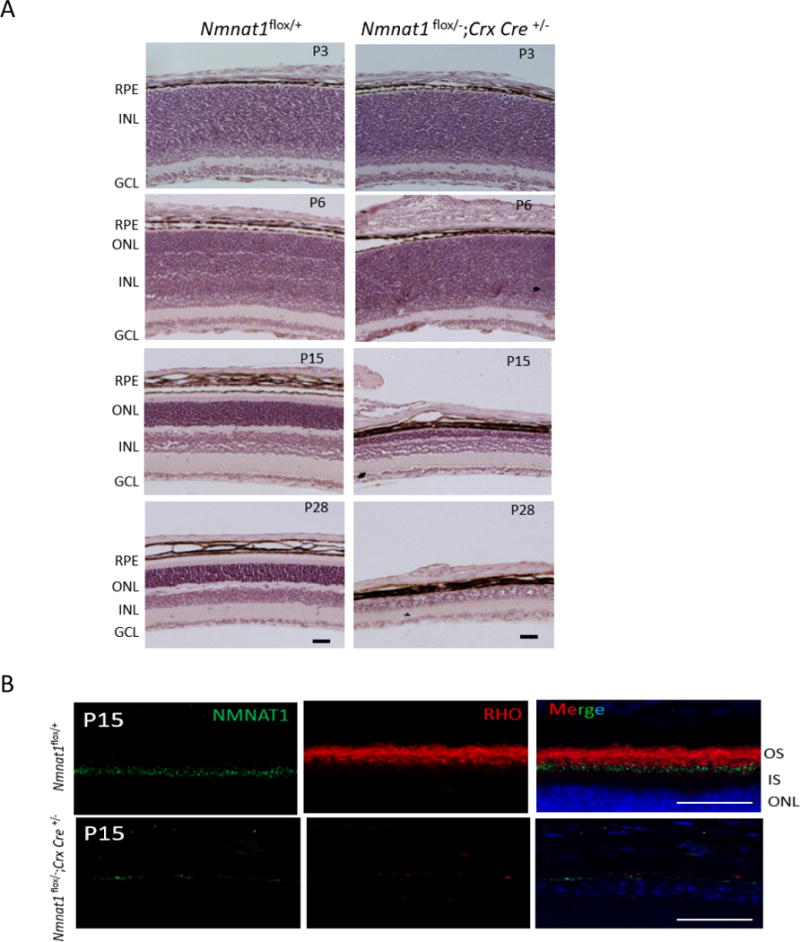
(A) H&E staining on Nmnat1 Crx-Cre cKO (Nmnat1Flox/−; Crx-Cre) mice reveals defects in lamination at P6 and strong loss of retinal tissue by P15. At P28, there is a significant reduction in the overall thickness of the retina as well as the photoreceptor ONL layer in Nmnat1Flox/−; Crx-Cre mutant retina compared to Nmnat1Flox/+ control retina. (B) Rhodopsin (red) staining shows complete loss of outer segments in Nmnat1Flox/−; Crx-Cre retina at P15, consistent with strong loss of NMNAT1 immunoreactivity (green). In comparison, the expression of Rho in the OS and NMNAT1 in control Nmnat1Flox/+ retina appears to be normal as expected. Scale bar: 20 μm. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
We further dissected the effect of NMNAT1 in photoreceptors by selectively ablating it only in rod cells. We generated Nmnat1-Rhodopsin-iCre-cKO (Nmnat1-iCre-cKO) mice by crossing Nmnat1+/−; iCre mice to Nmnat1flox/flox mice. As expected, Nmnat1-iCre-cKO mice exhibited substantial loss of NMNAT1 in rod photoreceptors and fewer NMNAT1-positive cells remained compared to the expression levels in Nmnat1flox/+ controls (Supp. Fig. 6B). At P7, no differences are observed in the thickness of the ONL between control and Nmnat1iCre-cKO retinas. At P28, the ONL of Nmnat1-iCre-cKO mice retina was markedly reduced compared to littermate control mice, indicating that most rods are lost (Supp. Fig. 6A). We also performed immunostaining with a rod outer segment marker, Rhodopsin, and show that no rod outer segments are detectable in Nmnat1-iCre-CKO retinas by P15 (Supp. Fig. 6B). Our data suggests that loss of Nmnat1 in rod photoreceptors alone is sufficient to cause retinal degeneration in mice, although less severe than the phenotype of Nmnat1-Chx10-Cre-Cko or Nmnat1-Crx-Cre-cKO mice. Cumulatively, our findings provide strong evidence to support the essential involvement of a NAD+ biosynthetic enzyme, NMNAT1, in the development and survival of photoreceptors and visual function.
Discussion
NMNAT1 encodes a rate-limiting enzyme that generates NAD+ from nicotinic acid mononucleotide (NaMN) and nicotinamide mononucleotide (NMN) (Berger et al., 2005), and is involved in nuclear NAD+ homeostasis. Although NMNAT1 is ubiquitously expressed in humans, mutations in NMNAT1 have been identified to cause a non-syndromic early form of blindness genetically defined as LCA9 in which both rod and cone photoreceptors are degenerated. In this report, we studied the pathogenic effects of the E257K variant, which occurs in 70% of all LCA9 cases, using a knock-in mouse model. Characterization of the mouse retina morphology and function revealed an absence of rapid photoreceptor cell degeneration in Nmnat1 E257K homozygous mice in 129 SvEv/C57BL6 mixed background. Even upon light-induced damage, Nmnat1E257K/E257K mice did not exhibit any detectable retinal degeneration in adulthood. Together, these data show that the lack of retinal phenotype observed in Nmnat1E257K/E257K mice is consistent with individuals harboring the E257K allele in the homozygous state, suggesting that the E257K variant alone is not sufficient to cause rapid retinal degeneration. However, compound heterozygous Nmnat1E257K/− mice, genotypically similar to LCA9 patients with compound heterozygous NMNAT1 mutations, exhibited characteristic morphological and functional photoreceptor defects as measured by electrophysiology, which were exacerbated upon exposure to light damage. Thus, our data indicates that Nmnat1E257K/− mice are more susceptible to light-induced damage.
Based on the observations in NMNAT1-LCA9 patients, together with findings from both Nmnat1 mutant mouse models described in this study, one possibility is that the severity of the retinal pathology depends on the level of reduction in NMNAT1 enzymatic activity. For instance, the E257K homozygous individuals exhibiting no retinal phenotype (Siemiatkowska et al., 2014a) could be explained by the fact that the NMNAT1 enzymatic activity is not sufficiently affected to trigger retinal degeneration. In vitro studies have demonstrated that the ATP catalytic activity of the E257K mutation on NMNAT1 is comparable with wild type, while the NMN activity is only moderately reduced (Sasaki et al., 2015). This is also consistent with the findings obtained from Nmnat1 E257K homozygous mutant mice which also exhibit no retinal phenotypes. Thus, our findings strengthen the previously reported claim that the E257K variant is not fully penetrant. However, combining the E257K allele with a loss of function allele means that the enzymatic activity is severely reduced to levels (i.e. less than 50%) that result in LCA9 in patients, as in the case reported by us and other previously (Chiang et al., 2012; Falk et al., 2012; Keen et al., 2003; Koenekoop et al., 2012; Perrault et al., 2012; Thompson et al., 2017). This is consistent with the findings obtained from compound heterozygous Nmnat1E257K/− mice, which also exhibit retinal degeneration. Furthermore, other small nucleotide variants (SNVs), that could alter expression levels of NMNAT1 in cis or in trans with the E257K mutation could explain why some NMNAT1 E257K homozygotes have disease whereas others do not. This possibility has also been suggested previously (Siemiatkowska et al., 2014a), and our current findings from the Nmnat1 mouse models provide further validity to its basis.
Interestingly, the phenotypes observed in compound heterozygous Nmnat1E257K/− mice is relatively mild compared to the LCA pathology presented in patients. This may be attributed to cross-species differences and/or the modification from the genetic background of the mice strain. To further understand the mechanisms involved in photoreceptor degeneration observed in Nmnat1E257K/− mice, we exposed Nmnat1E257K/− mice to strong light, which has been shown to accelerate photoreceptor cell death (Bai and Sheline, 2013). Upon prolonged exposure to light, the retinal degeneration phenotype in Nmnat1E257K/− mice significantly worsened, and triggered activation of the ER stress response pathway. These findings provide evidence supporting the role of NMNAT1 in protecting post-mitotic photoreceptor cells against stress induced-instability in the retina. Previous studies have shown that NAD+ plays a protective role in photoreceptor survival in Zn2+-mediated light induced conditions in mice, resulting in retinal degeneration (Bai and Sheline, 2013). When NMNAT1 function is impaired in the Nmnat1E257K/− mouse retinas, the decrease in NAD+ likely results in an increased susceptibility to light, contributing to the retinal degeneration observed, which is enhanced under light-stress conditions. These findings demonstrate that the presumed inhibition of NAD+ biosynthesis resulting from Nmnat1 mutations in mice affects ER stress pathways, causing visual impairment and ultimate photoreceptor death.
Furthermore, analysis of NMNAT1 localization in the mouse retina shows its expression in the photoreceptor layer as early as P2 and is continuously expressed throughout postnatal development. Consistent with previously findings (Greenwald et al., 2016), we have shown that NMNAT1 deficiency affects both rod and cone cells functionally in Nmnat1 mutant mice (Fig. 4C and D, Fig. 5B). This suggests that NMNAT1 is likely expressed in both photoreceptor cell types. Specifically, in the photoreceptor layer, it partially co-localizes with γ-tubulin, a marker for the basal body, and is distal to Centrin, a marker for the axoneme. These findings show that NMNAT1 localizes to the cilia basal body in photoreceptor cells in the mouse retina. Based on this localization pattern, an alternative possibility is that NMNAT1 has novel, unidentified roles in the photoreceptor connecting cilium. Structural and functional defects in the connecting cilium have been associated with a spectrum of ciliopathies, including LCA and RP (Eblimit et al., 2015; Rachel et al., 2012; Soens et al., 2016; Xu et al., 2016). Additionally, we and others have shown that NMNAT1 is also expressed in the OPL, IPL and GCL of the mouse retina (Zhou et al., 2016). In RGC-5 cells, NMNAT1 is localized in both the nuclei and cytoplasm of ganglion cells where it plays essential roles in protecting RGCs from high glucose-induced injury, serving as a neuroprotective agent. Further evidence of this protective role is shown in a mouse model of glaucomatous neurodegeneration, where viral based gene therapy (AAV2-Nmnat1) alleviates the axon and soma loss as well as ganglion cell function (Williams et al., 2017). Furthermore, the OPL and IPL form a dense network of synapses and fibrils, respectively, between photoreceptor cells, horizontal cells, etc., where NMNAT1 is detected; however, the precise expression of NMNAT1 in these cell types remains unexplored and warrants further investigation. Therefore, our findings provide a strong foundation for future studies to further investigate the function of NMNAT1 in the retina.
NMNAT1 encodes a rate-limiting enzyme that generates NAD+ from nicotinic acid mononucleotide (NaMN) and nicotinamide mononucleotide (NMN). NAD+ synthesis can also occur using other pathways, such as tryptophan-driven synthesis and from nicotinamide riboside (NR) (Imai and Guarente, 2014; Imai and Yoshino, 2013). NAD+ is an essential coenzyme involved in several metabolic processes and is required for energy production. Since photoreceptors are highly metabolically active cells that require high levels of ATP production to function properly (Fu and Yau, 2007; Yau and Hardie, 2009), we hypothesized that depletion of Nmnat1 in photoreceptors may result in retinal degeneration. In this study, we probed the function of Nmnat1 specifically in the mouse retina by generating conditional loss-of-function alleles and deleting them in a tissue- and cell type-specific manner in the eye. All Nmnat1 cKO mice (driven by Chx10-Cre, Crx-Cre, and Rhodopsin-iCre) exhibit severe, rapid and progressive retinal degeneration and blindness. Importantly, most photoreceptors in the Nmnat1 Chx10-Cre cKO retina are lost before the retina has terminally differentiated, which suggests that Nmnat1 is likely to be functionally important during the retinal developmental stages as well as in maintaining photoreceptor integrity post-developmentally. Interestingly, in patients, although NMNAT1 mutations are present ubiquitously in all cell types, their disease manifestation is uniquely restricted to the retina (Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012), where their rod and cone cell functions are severely impaired. In our Nmnat1 conditional deletion mouse models, we provide strong evidence that ablation of Nmnat1 function in the retina results in photoreceptor degeneration and cell death.
Taken together, our current findings provide evidence that the E257K NMNAT1 allele has significantly low penetrance, though it contributes to retinal degeneration phenotypes observed both in LCA9 patients and Nmnat1 mutant mice when present in the compound heterozygous state with a homozygous loss of function allele. Additionally, there is a likelihood of other SNVs that are present in LCA9 patients that could alter expression levels of NMNAT1 in concert with the E257K mutation, that could result in variable disease phenotypes. Clinically, this finding is of relevance since it helps us better evaluate the molecular diagnosis of LCA9 patients with the E257K variant. Furthermore, we show that loss of NMNAT1 in specifically in the eye causes severe early-onset retinal degeneration prior to terminal differentiation of photoreceptor cells, implicating its importance in photoceptor development and survival. Interestingly, we found that NMNAT1 expression is restricted to the basal body of the photoreceptor connecting cilium. Although it is well known that NMNAT1 is an enzyme that functions in synthesizing NAD+ and plays a role in neuroprotection (Sasaki et al., 2009a; Sasaki et al., 2009b; Williams et al., 2017), it is possible that it may have additional unknown functions in the connecting cilium which contribute to the severe retinal pathologies shown in this study. Functional analysis of NMNAT1 in the photoreceptor connecting cilium may provide insights into its specific role in retinal development. Additionally, Nmnat1 conditional knock-out mice may serve as a useful tool for further gene-therapy based studies to test therapeutic interventions, which can ultimately benefit in development of treatments for LCA9 patients.
Methods and Materials
Generation of Nmnat1 mutant mice
To generate Nmnat1 mutant mice, C57BL/6NTac-Nmnat1tm1a (EUCOMM)Wtsi embryonic stem cells (strain ID EM:04346, purchased from EUCOMM Mutant mice), were microinjected into C57BL6 blastocysts to generate chimeras. Male chimeras were bred to C57BL/6J females to test for germline transmission of the Nmnat1tm1a (EUCOMM)Wtsi allele. They were also bred to FLPeR mice (Gt (ROSA) 26Sortm1(FLP1)Dym, obtained from Jackson Labs) to remove the FRT flanked lacZ/neomycin sequence and generate a Nmnat1flox allele. Nmnat1flox mice were crossed to Hprt-Cre (129S1/Sv-Hprttm1 (Cre)Mnn/J, Jackson Labs) to generate the Nmnat1−/− allele. To generate the c.769G>A point mutation, we used a two-step method requiring a counter selection marker (sensitivity to sucrose with SacB). Briefly, we first used recombineering technology to introduce a Cat-SacB cassette into the Nmnat1-BAC DNA which is cloned in the P[acman] vector. We then constructed a DNA fragment containing a point mutation in exon 4 (c.769G>A) along with a Neo cassette to replace the Cat-SacB cassette in the Nmnat1-BAC. AB 2.2 embryonic stem cells (Mouse ES Cell Core Facility at Baylor College of Medicine) derived from the 129 SvEv strain were electroporated with linearized targeting vector. DNA from embryonic stem cell lines was digested and analyzed by Southern blot using 5′ and 3′ probes (flanking the recombination arms and not included in the targeting vector) and genomic PCR. Targeted cell lines were selected and microinjected into the C57BL6 blastocysts to generate chimeras. One chimera underwent germline transmission. To genotype subsequent F2 and F3 generations of c.769G>A mice, we used a genomic PCR assay with a set of primers from the targeted region. The primers for the point mutation are: Nmnat1-PL451-reco-R 5′-TAGGCCAGTGCTCTACCAACT-3′ and 1LoxpcheckF 5′-ATCGCATTGTCTGAGTAGGTGTC-3′.
Homozygous Nmnat1flox females were bred with Nmnat1flox/− mice expressing different Cre recombinases to generate Nmnat1 conditional knockout mice. Crx-Cre (Tg (Crx-Cre/ERT2)1Tfur), Chx10-Cre (Tg (Chx10-EGFP/Cre-ALPP) 2Clc/J) mice were obtained from Jackson Labs. iCre mice were a gift from Dr. Ching-Kang (Jason) Chen. To genotype subsequent F2 and F3 generations of mice, we used a genomic PCR assay with a set of primers from the targeted region. These primers are: a (5′-TCATGTAGGGAACTCAGAGCTGGT-3), b (5′-TTTCCCACGTATCCACTTCC-3′), c (5′-GCCATCACGAGATTTCGATT-3′) and d (5′-TTGCCACCAAGAACTCACAC-3′) and e (5′-TGAAAGAGGCAAGGGCTTAG).
Immunocytochemistry
hTERT-RPE1 cells were cultured as previously described (Gorden et al., 2008). To induce ciliogenesis, cells were seeded on coverslips and serum starved for 48 hrs to detect endogenous Nmnat1 expression. Ciliated cells were fixed in 2% PFA for 20 min, treated with 1% Triton X-100 in PBS for 5 min, and blocked in 2% bovine serum albumin (BSA)\in PBS for 30 minutes. Fixed cells were stained for 1 hr with the corresponding primary antibodies: rabbit-Nmnat1 polyclonal antibody (1:1000, Abcam ab45652), mouse monoclonal α-acetylated tubulin (1:1000, Sigma-Aldrich). Coverslips were then washed in PBS and stained for 45 min with secondary antibodies (anti-mouse Alexa Fluor 488, and anti-rabbit Cy3, 1:500, Invitrogen/Molecular Probes). Coverslips were washed again with PBS and briefly with mQ water before mounting in Vectashield (Vector Laboratories, Burlingame, CA, USA). Samples for immunocytochemistry were analyzed on a Zeiss Axio Imager ZI fluorescence microscope (Zeiss), equipped with a 63x objective oil lens. Optical sections were generated through structured processed using Axiovision 4.3 (Zeiss), Adobe Photoshop CS4, and FIJI software.
Electroretinographic Analysis
Prior to ERG testing, mice were allowed to dark-adapt overnight. Under dim red illumination, mice were anesthetized with an intraperitoneal injection of ketamine (70 mg/kg), xylazine (14 mg/kg), and acepromazine (1.2 mg/kg). A single drop of 0.5% proparacaine was applied to each eye for corneal anesthesia and drops of 1% tropicamide and 2.5% phenylephrine were used to dilate the pupils. Mice were placed on a heating pad maintained at 39 °C inside a Ganzfeld dome. Platinum electrodes mounted on micromanipulators were positioned on each cornea and a small amount of 2.5% methylcellulose was applied to each eye. Platinum subdermal needle electrodes were inserted into the tail and forehead to serve as ground and reference, respectively. Signals were bandpass filtered from 0.1 – 1,000 Hz and digitally sampled at 10 kHz. Flashes in the 30 kHz scotopic range were generated by a pair of cyan Luxeon K2 LEDs (λpeak = 505 nm, ∆λ1/2 = 30 nm; Phillips Lumileds, San Jose, CA) wired in series. Square pulses of 0.5 ms duration and varying currents were driven through the LEDs to create flashes of different intensities. Bright flashes were generated with 1500 W xenon flash bulbs (Novatron, Dallas, TX). At the lowest intensity, 25 responses were averaged with a delay of 4 seconds between each flash. As flash intensity increased, fewer responses were averaged with a longer delay between flashes. For analysis of ERG waveforms, the a-wave was measured from baseline to the trough of the initial negative deflection (unfiltered) and the b-wave was measured from the a-wave trough to the peak of the subsequent positive deflection (low-pass filtered, fc = 60 Hz).
Immunohistochemistry
Eyes were enucleated from mutant and wild-type mice as previously described (Agrawal et al., 2017; Eblimit et al., 2018). For evaluation of Nmnat1 localization in photoreceptor cells of wild-type, unfixed mouse eyes were harvested and frozen in OCT. 10–14 μm cryosections were cut and treated with 0.1% Triton X100 in PBS for 10 minutes and subsequently blocked in 5% normal goat serum in PBS (blocking buffer) for 30 minutes. Cryosections were then incubated overnight at 4°C with corresponding primary antibodies and followed by incubation of secondary antibodies at room temperature for 1 hr in blocking buffer. Modified Davidson’s Fixative was used to fix eyes overnight for paraffin embedding. Seven micrometer eye sections were cut (Microtome, Leica). Slides were deparaffinized and antigen retrieval was performed as described previously (Agrawal et al., 2017; Arno et al., 2016). Slides were washed in PBS, incubated for 1 hr at room temperature in hybridization buffer (10% normal goat serum, 0.1% Triton X-100, PBS), then incubated overnight in primary antibody diluted in hybridization buffer. Slides were then washed in PBS, incubated with secondary antibody diluted in hybridization buffer at room temperature for 2 hr, washed in PBS, mounted with anti-fade medium (Prolong; Invitrogen) to reduce bleaching, and cover-slipped.
For NMNAT1 blocking experiments, 5 ul of anti-rabbit NMNAT1 antibody was mixed with 5 ul of blocking peptide (Abcam) and incubated in 500ul of hybridization buffer at 4°C overnight followed by immunostaining as described above. Dilutions and sources of antibodies were as follows: rabbit-Nmnat1 polyclonal antibody (1:100, Abcam ab45652), mouse γ-tubulin antibody (1:200, Sigma), mouse anti-pan Centrin (Millipore, 20H-5, 1:1000), mouse anti-rhodopsin (B6-30N) (1:200, a generous gift from W. Clay Smith), Cy3 anti-rabbit (1:500, Jackson Immunochemicals), secondary antibodies were conjugated with Alexa fluor 488 (1:500, Molecular Probes), and DAPI staining reagent (dilution 1:8000). Fluorescent images were captured with a Zeiss LSM 510 confocal microscope and processed with Image J and Adobe Photoshop software. Images were processed using Axiovision 4.3 (Zeiss) and Adobe Photoshop CS4.
Light damage studies
Light damage (LD) studies were carried out as previously published (Wenzel et al., 2000) but with some modifications. Specifically, mice were dark-adapted for 48 h, and LD was induced after pupil dilation with 0.1% atropine (Sigma) by exposure to 10,000 lux diffuse white fluorescent light (150 watt spiral lamp, Commercial Electric) (lights on at 11:00 a.m.) for 4 hours. Mice were then reared in a normal light-dark cycle until being sacrificed for histology.
Measurements of Outer Nuclear Layer (ONL) Thickness
Mouse eyes were dissected, embedded in paraffin, and sectioned along the vertical meridian. The thickness of the outer nuclear layer (ONL) was measured at 18 positions equally spaced along the retina (9 positions each in the superior and inferior hemispheres) in retinal tissue from at least 3 biological replicates. For each position, three measurements were taken and the average value of these three measurements was calculated. Measurements were made using a camera lucida connected to a light microscope, a WACOM graphics tablet (WACOM, Vancouver, WA), and AxioVision LE Rel. 4.1 software (Zeiss, Goettingen, Germany). Before each measurement session, the setup was calibrated using a stage micrometer (Klarmann Rulings, Litchfield, NH).
Detection of Apoptotic Cells in the Retina
Apoptotic cell death was detected by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay according to the manufacturer’s manual (Roche, In Situ Cell Death Detection it, 11684795910, Roche Diagnostics, Indianapolis, IN). Paraffin embedded retinal sections were used for the TUNEL assay.
Supplementary Material
Supplemental Figure 1. Characterization of Nmnat1E257K/E257K retina.
(A) H&E staining of Nmnat1E257K/E257K mice at 5 months and 8 months of age shows no detectable phenotype in the retina in comparison to WT mice. (B) Retinal morphology of Nmnat1E257K/E257K mutant mice at 5 months of age shows no difference in phenotype before and after light damage for 4 hours. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
Supplemental Figure 2. Expression of NMNAT1 in Nmnat1E257K/− retinas is unaffected.
Cryosections of 6 weeks-old Nmnat1E257K/− mouse retinas were stained with NMNAT1 antibody (green) (lower panel). The localization pattern of NMNAT1 is unaffected in Nmnat1E257K/− retina compared to wild-type retina (upper panel). Co-staining of Centrin, a marker of axoneme (red), shows that NMNAT1 is distal to Centrin, indicating its localization to the basal body of the connecting cilium. Scale bar: 5 μm. OS= outer segment, IS= inner segment, CC= connecting cilium.
Supplemental Figure 3. Light-induced damage causes cell death and ER stress in Nmnat1E257K/− retinas within one day after light exposure.
Frozen sections of 8 weeks-old Nmnat1E257K/− retinas were stained for markers of cell death and ER stress one day after light exposure. (A) Caspase-12 positive signals are observed in the ONL of Nmnat1E257K/− mutant retina while no staining is detected in control Nmnat1+/− retina. (B) Positive BIP staining is observed in the ONL and IS of Nmnat1E257K/− mutant retina compared to Nmnat1 heterozygous control retina. DAPI (blue) was used to counterstain nuclei. Arrowhead indicates selected positive signals. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 4. Cell death and ER stress are increased in Nmnat1E257K/− retinas two days after light exposure.
Frozen sections of 8 weeks-old Nmnat1E257K/− mouse retina were stained for markers of ER stress two days after light exposure. (A) Caspase-12 positive signals are observed in the ONL of Nmnat1E257K/− retinas while no staining is detected in control Nmnat1E257K/+ retinas. (B) Positive BIP staining is observed in the ONL of Nmnat1E257K/− retinas and absent in the control Nmnat1E257K/+ retina. (C) Positive CHOP staining is observed in the ONL of Nmnat1E257K/− retinas (arrowheads indicate a subset of positive signals), which is absent in the Nmnat1E257K/+ control. DAPI (blue) shows nuclei staining. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 5. Cell death and ER stress are increased in Nmnat1E257K/− retinas four days after light exposure.
Cryosections of 8 weeks-old Nmnat1E257K/− retinas were stained for markers of cell death and ER stress four days after light exposure., Caspase-12 positive signals are observed in the ONL (panel A top, arrowheads) of Nmnat1E257K/− retinas while no staining is detected in control Nmnat1+/− retinas. BIP (Panel A bottom, arrowheads) staining shows positive signals in the ONL of Nmnat1E257K/− retinas and CHOP staining (panel B, arrowheads) is also positive in the ONL of Nmnat1E257K/− retinas, absent in the control Nmnat1+/− sections. DAPI (blue) was used to counterstain nuclei. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 6. Rod-specific deletion of Nmnat1 results in marked degeneration of photoreceptors.
(A) H&E staining on paraffin embedded retinal sections revealed no obvious phenotype at P7, but a drastic reduction in the thickness of the ONL and overall retinal thickness was observed at P28 in Nmnat1Flox/−;iCre retinal sections compared to Nmnat1flox/+ retina. (B) Rho immunostaining shows no positive signal in the outer segments of photoreceptors and strong loss of NMNAT1 in Nmnat1Flox/−; iCre retina compared to controls which exhibit normal expression of both NMNAT1 and Rho. Scale bar: 20 μm. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
HIGHLIGHTS.
The manuscript describes detailed characterization of the most common NMNAT1 variant, E257K, associated with LCA9, using a knock-in mouse model for the first time, and delineation of the cell type specific function of NMNAT1 in the retina using conditional knock-out mice.
Acknowledgments
We thank Dr. Yiyun Chen and Huidan Xu for their help in making mouse constructs. We thank Shangyi Fu for help with histology. This work was supported by the Retina Research Foundation and the National Eye Institute [R01EY020540] to G.M. and R.C., 5T32EY007102-23 to S.A. (PI: G.M.). Nmnat1 mutant mice were generated by the Mouse ES Cell Core at BCM that is partially supported by the BCM IDDRC [Grant Number 5P30HD024064-23] from the Eunice Kennedy Shriver National Institute of Child Health & Human Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal SA, Burgoyne T, Eblimit A, Bellingham J, Parfitt DA, Lane A, Nichols R, Asomugha C, Hayes MJ, Munro PM, Xu M, Wang K, Futter CE, Li Y, Chen R, Cheetham ME. REEP6 Deficiency Leads to Retinal Degeneration through Disruption of ER Homeostasis and Protein Trafficking. Hum Mol Genet. 2017 doi: 10.1093/hmg/ddx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arno G, Agrawal SA, Eblimit A, Bellingham J, Xu M, Wang F, Chakarova C, Parfitt DA, Lane A, Burgoyne T, Hull S, Carss KJ, Fiorentino A, Hayes MJ, Munro PM, Nicols R, Pontikos N, Holder GE, Ukirdc Asomugha C, Raymond FL, Moore AT, Plagnol V, Michaelides M, Hardcastle AJ, Li Y, Cukras C, Webster AR, Cheetham ME, Chen R. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am J Hum Genet. 2016;99:1305–1315. doi: 10.1016/j.ajhg.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Sheline CT. NAD(+) maintenance attenuates light induced photoreceptor degeneration. Experimental eye research. 2013;108:76–83. doi: 10.1016/j.exer.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Chen Y, Yi X, Wu R, Gan H, Shi Y, Chen Y, Barnett C, Wheaton D, Day M, Sutherland J, Heon E, Weleber RG, Gabriel LA, Cong P, Chuang K, Ye S, Sallum JM, Qi M. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet. 2012;44:972–974. doi: 10.1038/ng.2370. [DOI] [PubMed] [Google Scholar]
- Conforti L, Janeckova L, Wagner D, Mazzola F, Cialabrini L, Di Stefano M, Orsomando G, Magni G, Bendotti C, Smyth N, Coleman M. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J. 2011;278:2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Eblimit A, Agrawal SA, Thomas K, Anastassov IA, Abulikemu T, Mardon G, Chen R. Conditional loss of Spata7 in photoreceptors causes progressive retinal degeneration in mice. Experimental eye research. 2018;166:120–130. doi: 10.1016/j.exer.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblimit A, Nguyen TM, Chen Y, Esteve-Rudd J, Zhong H, Letteboer S, Van Reeuwijk J, Simons DL, Ding Q, Wu KM, Li Y, Van Beersum S, Moayedi Y, Xu H, Pickard P, Wang K, Gan L, Wu SM, Williams DS, Mardon G, Roepman R, Chen R. Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Human molecular genetics. 2015;24:1584–1601. doi: 10.1093/hmg/ddu573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, Staniszewska M, Shukla R, Palavalli L, Mohand-Said S, Waseem NH, Jalali S, Perin JC, Place E, Ostrovsky J, Xiao R, Bhattacharya SS, Consugar M, Webster AR, Sahel JA, Moore AT, Berson EL, Liu Q, Gai X, Pierce EA. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Yau KW. Phototransduction in mouse rods and cones. Pflugers Arch. 2007;454:805–819. doi: 10.1007/s00424-006-0194-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topcu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald SH, Charette JR, Staniszewska M, Shi LY, Brown SDM, Stone L, Liu Q, Hicks WL, Collin GB, Bowl MR, Krebs MP, Nishina PM, Pierce EA. Mouse Models of NMNAT1-Leber Congenital Amaurosis (LCA9) Recapitulate Key Features of the Human Disease. Am J Pathol. 2016;186:1925–1938. doi: 10.1016/j.ajpath.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab 15 Suppl. 2013;3:26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen TJ, Mohamed MD, McKibbin M, Rashid Y, Jafri H, Maumenee IH, Inglehearn CF. Identification of a locus (LCA9) for Leber’s congenital amaurosis on chromosome 1p36. Eur J Hum Genet. 2003;11:420–423. doi: 10.1038/sj.ejhg.5200981. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, Schwartzentruber J, Solanki N, Traboulsi EI, Cheng J, Logan CV, McKibbin M, Hayward BE, Parry DA, Johnson CA, Nageeb M, Finding of Rare Disease Genes Canada C. Poulter JA, Mohamed MD, Jafri H, Rashid Y, Taylor GR, Keser V, Mardon G, Xu H, Inglehearn CF, Fu Q, Toomes C, Chen R. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017 doi: 10.1136/bjophthalmol-2016-309975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chen D, Sauve Y, McCandless J, Chen YJ, Chen CK. Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis. 2005;41:73–80. doi: 10.1002/gene.20097. [DOI] [PubMed] [Google Scholar]
- Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, Orsomando G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One. 2014;9:e113939. doi: 10.1371/journal.pone.0113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, Furukawa T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nature neuroscience. 2003;6:1255–1263. doi: 10.1038/nn1155. [DOI] [PubMed] [Google Scholar]
- Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort-Delhemmes S, Delphin N, Fares-Taie L, Gerber S, Xerri O, Edelson C, Goldenberg A, Duncombe A, Le Meur G, Hamel C, Silva E, Nitschke P, Calvas P, Munnich A, Roche O, Dollfus H, Kaplan J, Rozet JM. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012;44:975–977. doi: 10.1038/ng.2357. [DOI] [PubMed] [Google Scholar]
- Rachel RA, Li T, Swaroop A. Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia. 2012;1:22. doi: 10.1186/2046-2530-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Developmental biology. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Margolin Z, Borgo B, Havranek JJ, Milbrandt J. Characterization of Leber Congenital Amaurosis-associated NMNAT1 Mutants. J Biol Chem. 2015;290:17228–17238. doi: 10.1074/jbc.M115.637850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009a;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009b;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemiatkowska AM, Schuurs-Hoeijmakers JH, Bosch DG, Boonstra FN, Riemslag FC, Ruiter M, de Vries BB, den Hollander AI, Collin RW, Cremers FP. Nonpenetrance of the most frequent autosomal recessive leber congenital amaurosis mutation in NMNAT1. JAMA Ophthalmol. 2014a;132:1002–1004. doi: 10.1001/jamaophthalmol.2014.983. [DOI] [PubMed] [Google Scholar]
- Siemiatkowska AM, van den Born LI, van Genderen MM, Bertelsen M, Zobor D, Rohrschneider K, van Huet RA, Nurohmah S, Klevering BJ, Kohl S, Faradz SM, Rosenberg T, den Hollander AI, Collin RW, Cremers FP. Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis. Mol Vis. 2014b;20:753–759. [PMC free article] [PubMed] [Google Scholar]
- Soens ZT, Li Y, Zhao L, Eblimit A, Dharmat R, Li Y, Chen Y, Naqeeb M, Fajardo N, Lopez I, Sun Z, Koenekoop RK, Chen R. Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet Med. 2016;18:1044–1051. doi: 10.1038/gim.2015.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, De Roach JN, McLaren TL, Montgomery HE, Hoffmann LH, Campbell IR, Chen FK, Mackey DA, Lamey TM. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol Genet Genomic Med. 2017;5:652–667. doi: 10.1002/mgg3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al-Rajhi AA, Bray M, Lewis RA, Lupski JR, Mardon G, Koenekoop RK, Chen R. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84:380–387. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, Reme CE. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:81–88. doi: 10.1523/JNEUROSCI.20-01-00081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756–760. doi: 10.1126/science.aal0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yamada T, Sun Z, Eblimit A, Lopez I, Wang F, Manya H, Xu S, Zhao L, Li Y, Kimchi A, Sharon D, Sui R, Endo T, Koenekoop RK, Chen R. Mutations in POMGNT1 cause non-syndromic retinitis pigmentosa. Hum Mol Genet. 2016;25:1479–1488. doi: 10.1093/hmg/ddw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, Hardie RC. Phototransduction motifs and variations. Cell. 2009;139:246–264. doi: 10.1016/j.cell.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou RM, Shen Y, Yao J, Yang H, Shan K, Li XM, Jiang Q, Yan B. Nmnat 1: a Security Guard of Retinal Ganglion Cells (RGCs) in Response to High Glucose Stress. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;38:2207–2218. doi: 10.1159/000445576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Characterization of Nmnat1E257K/E257K retina.
(A) H&E staining of Nmnat1E257K/E257K mice at 5 months and 8 months of age shows no detectable phenotype in the retina in comparison to WT mice. (B) Retinal morphology of Nmnat1E257K/E257K mutant mice at 5 months of age shows no difference in phenotype before and after light damage for 4 hours. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.
Supplemental Figure 2. Expression of NMNAT1 in Nmnat1E257K/− retinas is unaffected.
Cryosections of 6 weeks-old Nmnat1E257K/− mouse retinas were stained with NMNAT1 antibody (green) (lower panel). The localization pattern of NMNAT1 is unaffected in Nmnat1E257K/− retina compared to wild-type retina (upper panel). Co-staining of Centrin, a marker of axoneme (red), shows that NMNAT1 is distal to Centrin, indicating its localization to the basal body of the connecting cilium. Scale bar: 5 μm. OS= outer segment, IS= inner segment, CC= connecting cilium.
Supplemental Figure 3. Light-induced damage causes cell death and ER stress in Nmnat1E257K/− retinas within one day after light exposure.
Frozen sections of 8 weeks-old Nmnat1E257K/− retinas were stained for markers of cell death and ER stress one day after light exposure. (A) Caspase-12 positive signals are observed in the ONL of Nmnat1E257K/− mutant retina while no staining is detected in control Nmnat1+/− retina. (B) Positive BIP staining is observed in the ONL and IS of Nmnat1E257K/− mutant retina compared to Nmnat1 heterozygous control retina. DAPI (blue) was used to counterstain nuclei. Arrowhead indicates selected positive signals. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 4. Cell death and ER stress are increased in Nmnat1E257K/− retinas two days after light exposure.
Frozen sections of 8 weeks-old Nmnat1E257K/− mouse retina were stained for markers of ER stress two days after light exposure. (A) Caspase-12 positive signals are observed in the ONL of Nmnat1E257K/− retinas while no staining is detected in control Nmnat1E257K/+ retinas. (B) Positive BIP staining is observed in the ONL of Nmnat1E257K/− retinas and absent in the control Nmnat1E257K/+ retina. (C) Positive CHOP staining is observed in the ONL of Nmnat1E257K/− retinas (arrowheads indicate a subset of positive signals), which is absent in the Nmnat1E257K/+ control. DAPI (blue) shows nuclei staining. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 5. Cell death and ER stress are increased in Nmnat1E257K/− retinas four days after light exposure.
Cryosections of 8 weeks-old Nmnat1E257K/− retinas were stained for markers of cell death and ER stress four days after light exposure., Caspase-12 positive signals are observed in the ONL (panel A top, arrowheads) of Nmnat1E257K/− retinas while no staining is detected in control Nmnat1+/− retinas. BIP (Panel A bottom, arrowheads) staining shows positive signals in the ONL of Nmnat1E257K/− retinas and CHOP staining (panel B, arrowheads) is also positive in the ONL of Nmnat1E257K/− retinas, absent in the control Nmnat1+/− sections. DAPI (blue) was used to counterstain nuclei. Scale bar: 20 μm. ONL= outer nuclear layer, INL= inner nuclear layer.
Supplemental Figure 6. Rod-specific deletion of Nmnat1 results in marked degeneration of photoreceptors.
(A) H&E staining on paraffin embedded retinal sections revealed no obvious phenotype at P7, but a drastic reduction in the thickness of the ONL and overall retinal thickness was observed at P28 in Nmnat1Flox/−;iCre retinal sections compared to Nmnat1flox/+ retina. (B) Rho immunostaining shows no positive signal in the outer segments of photoreceptors and strong loss of NMNAT1 in Nmnat1Flox/−; iCre retina compared to controls which exhibit normal expression of both NMNAT1 and Rho. Scale bar: 20 μm. RPE= retinal pigment epithelium, ONL= outer nuclear layer, INL= inner nuclear layer, GCL= ganglion cell layer.