

Abstract
Background
Currently 20–35% of pregnant women are obese, posing a major health risk for mother and fetus. It is postulated that an abnormal maternal-fetal nutritional environment leads to adverse metabolic programming, resulting in altered substrate metabolism in the offspring and predisposing to risks of obesity and diabetes later in life. Data indicate that oocytes from overweight animals have abnormal mitochondria. We hypothesized that maternal obesity is associated with altered mitochondrial function in healthy neonatal offspring.
Methods
Overweight and obese (Body mass index, (BMI) ≥ 25 kg/m2, n=14) and lean (BMI < 25 kg/m2, n=8), African American pregnant women carrying male fetuses were recruited from the Barnes Jewish Hospital obstetric clinic. Maternal and infant data were extracted from medical records. Infants underwent body composition testing in the first days of life. Circumcision skin was collected for isolation of fibroblasts. Fibroblast cells were evaluated for mitochondrial function, metabolic gene expression, nutrient uptake and oxidative stress.
Results
Skin fibroblasts of infants born to overweight mothers had significantly higher mitochondrial respiration without a concurrent increase in ATP production, indicating mitochondrial inefficiency. These fibroblasts had higher levels of reactive oxygen species and evidence of oxidative stress. Evaluation of gene expression in offspring fibroblasts revealed altered expression of multiple genes involved in fatty acid and glucose metabolism and mitochondrial respiration in infants of overweight mothers.
Conclusion
This study demonstrates altered mitochondrial function and oxidative stress in skin fibroblasts of infants born to overweight mothers. Future studies are needed to determine the long-term impact of this finding on the metabolic health of these children.
Keywords: Obesity, pregnancy, offspring, mitochondria, oxidative stress
Introduction
The obesity epidemic is a major public health concern; it is estimated that by the year 2025, 300 million people worldwide will have metabolic syndrome, with significant secondary health problems contributing to morbidity and mortality, affecting people of all ages.(1) Approximately 25–30% of pregnant women are overweight, defined as a body mass index (BMI) ≥ 25 kg/m2.(2, 3) Increased maternal weight prior and during pregnancy is associated with several complications, including cesarean delivery, fetal growth disorders, preeclampsia, stillbirth, prematurity and congenital anomalies.(4–6) Additionally, data show that offspring born to obese mothers are at a greater risk of development of obesity in childhood, adolescence and adulthood.(7) Elevated prepregnancy BMI correlates positively with higher leptin, inflammatory markers, increased insulin resistance, and increased abdominal and hepatic lipid content in offspring,(8–10) indicating an impact of maternal nutritional status on the metabolic milieu of her offspring. The developmental origins of adult disease hypothesis suggests that a suboptimal nutritional environment in utero, which includes starvation or over-nutrition, can increase susceptibility of the offspring to adult diseases, such as obesity, cardiovascular disease or metabolic syndrome.(11, 12) This theory and the above mentioned observational data is supported by animal models. Studies done in rodents, sheep and non-human primates demonstrate that maternal over-nutrition has been associated with alterations in offspring body composition, such as increased fat mass and reduced muscle mass.(13, 14) These studies also suggest that maternal obesity is poorly correlated with birthweight and that offspring born to obese mothers can have a diverse weight spectrum growth restricted, overweight or have a normal birthweight.(13–15) Data show that a maternal obesogenic diet is associated with metabolic changes including higher glucose, insulin, leptin and triglyceride levels and lower adiponectin secretion in the offspring.(13–16) Evidence also suggests that suboptimal maternal nutrition can alter gene expression and enzyme function of key regulators and mediators of metabolism.(16–18) Taken together, these data indicate an important impact of maternal obesity on the metabolic function of her offspring. It is proposed that these changes in offspring metabolism can subsequently lead to infant obesity and an increased risk for adult diseases.(11, 13) The exact mechanism of this developmental programming is not fully understood.
Mitochondria are at the center of energy metabolism and are strongly implicated in the development of metabolic disease associated with maternal nutrition. Several investigations in mouse models of obesity and diabetes have demonstrated abnormal mitochondria in maternal oocytes.(19–23) Oocytes from mice fed a high fat diet showed signs of altered mitochondrial activity, such as reduced mitochondrial membrane potential and altered mitochondrial morphology on electron microscopy.(20–22) Oocytes of obese female mice had a higher mitochondrial DNA content and increased expression of mitochondrial biogenesis markers.(20, 23) Recent data from mice with metabolic syndrome demonstrated that mitochondrial dysfunction associated with maternal obesity can be passed down to the second and third generation through the female germline.(22) One recent human study noted a correlation between maternal weight and infant size at birth, as well as between mitochondrial DNA and infant size at birth, indicating a possible impact of mitochondrial content on offspring metabolism.(24) However, despite epidemiologic data in humans, there remains minimal data in human infants regarding cellular mitochondrial changes associated with maternal obesity.
We sought to evaluate metabolism and mitochondrial function in healthy human neonates. To our knowledge, ours is the first study to analyze the impact of maternal obesity during pregnancy on mitochondrial function in human offspring. In order to evaluate metabolism and mitochondrial function in human cells, we sought to obtain tissue from healthy neonates that would otherwise be discarded. To that end, this study focused on male infants with planned circumcision. We hypothesized that infants born to overweight mothers would have altered skin fibroblast mitochondrial function compared to infants born to lean mothers.
Methods
Study Design
This prospective observational cohort study included mother-infant pairs who delivered at Barnes-Jewish Hospital in St. Louis, MO between August 2014 and June 2015. This study was an ancillary study to an existing study of overweight pregnant African American women (“LIFE-MOMS”, NCT101768793). Approval for this study was obtained from the Institutional Review Board at Washington University School of Medicine. Women were approached for enrollment with informed consent during a prenatal visit and consented if they met inclusion criteria, which included: maternal age between 18–35 years, singleton gestation less than 34 weeks at time of enrollment, normal fetal anatomy, and carrying a male fetus with plans for circumcision. Women were excluded for any of the following: baseline Diabetes Mellitus Type 1 and 2, congenital abnormalities, history of gestational diabetes, active substance abuse or treatment with medications known to impact metabolism or body weight. Maternal demographic and clinical data were extracted from the electronic medical records (including age, pre-pregnancy BMI – defined based on self-reported weight at first prenatal visit), gestational weight gain (GWG – determined as difference between pre-pregnancy weight and weight at delivery), mode of delivery, pregnancy complications, and socioeconomic status). Gestational age (GA) was confirmed by first trimester ultrasound. Expecting mothers were categorized as lean (BMI < 25 kg/m2) or overweight/obese (BMI ≥ 25 kg/m2). Overweight mothers did not have gestational diabetes. Women had a visit to the Clinical Research Unit (CRU) at Washington University between 32 and 34 weeks gestation to have their body fat percentage estimated via skin fold measurement (25, 26) and fasting blood work including: HDL, LDL, total cholesterol, glucose and insulin.
Infant demographic and clinical data were extracted from the chart following delivery (including gestational age, APGAR scores, birth weight, length and head circumference, and delivery complications). Newborns underwent a visit in the CRU between 24–72 hours of life for body composition, measured by the Peapod (Cosmed USA Inc) and for measurement of resting metabolic rate via indirect calorimetry, skin fold testing, and standard length and height. At the time of circumcision, which was done 18–72 hours after birth, skin was collected in culture media for isolation of fibroblasts.
Isolation and culture of skin fibroblasts
Skin fibroblasts were isolated by the Genome Engineering & Induced Pluripotent Stem Cell Center at Washington University School of Medicine. Cells were received frozen at passage 3. Fibroblasts were cultured in alpha minimal essential media (MEM) (Gibco) supplemented with 50ml FBS, 1ml Penicillin/Streptomycin (1%) and 2ml L-glutamine (0.4%) and incubated at 37°C in 5% CO2 atmosphere. All analyses described throughout this study were carried out at passage 4–7.
Mitochondrial Function Testing
Fibroblasts were grown in T75 flasks to 70–80% confluence and then gently detached by trypsinization, washed in phosphate-buffered saline (PBS) and counted. Oxygen consumption rate (OCR) was measured in adherent fibroblasts with a XF96 Extracellular Flux Analyzer (Agilent Technologies). Each cell line was seeded in 8 wells of a 96 well plate at a density of 4 × 103 cells/well in 80 ul of alpha MEM and incubated for 12–18 hours at 37°C in 5% CO2 atmosphere. After replacing the growth medium with unbuffered Dulbecco modified eagle medium (DMEM) (pH 7.4, 1000mg/L glucose) supplemented with 1 mM pyruvate and 10 mM D-glucose, cells were incubated for 30–60 minutes in a CO2 free incubator. Baseline measurements of OCR were determined, followed by the addition of oligomycin (1uM), FCCP (carbonilcyanide p-triflouromethoxyphenylhydrazone 1uM), and Antimycin/Rotenton (1uM combined). Determinations of baseline, maximal respiratory rate, ATP production and spare capacity were made as previously described.(27)
RNA extraction and quantitative real-time PCR
Fibroblasts were rinsed in PBS, and homogenized in TRIzol reagent (Invitrogen) followed by subsequent RNA isolation as per manufacturer’s instructions. RNA was quantified and used to generated cDNA via the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Synthesized cDNA was used for quantitative PCR (qPCR) using SYBR Green (Applied Biosystems) with appropriate primers, and performed in triplicate on a Stratagene Mx3005P qPCR machine, as previously described. (28) Relative quantification after normalization to 36B4 expression was calculated using the 2− ΔCT formula. Primer sequences are available upon request.
Mitochondrial Superoxide staining
Fibroblasts were plated in a 6 well plate at 300,000 cells per well. After incubation for 24 hours, cells were loaded with 1ml of 5 μM MitoSOX Red (Invitrogen) and incubated for 10 minutes at 37°C in 5% CO2 atmosphere. Cells were washed three times with Hank’s balanced salt solution with calcium and magnesium (Gibco), trypsinized and placed in alpha MEM. Using a flow cytometer (FlowJo, LLC), MitoSOX Red was excited at 488 nm and fluorescence emission at 575 was measured. Fluorescense was quantified using fluorescence activated cell sorting (FACS).
Western Blotting
Fibroblast lysates were created using RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) with EDTA-Free protease complete tablet (Roche), PMSF and 2% 2-mercaptoethanol. A total of 20 μg of protein per sample was loaded onto 10% SDS PAGE gels. Carbonylated proteins were detected using the Oxyblot Protein Oxidation Detection Kit (EMD Millipore) as per the manufacturer’s instructions. Blots were visualized and bands quantified using the LI-COR Odyssey imaging system. Actin was used as the loading control.
Statistical analysis
All statistical analyses were performed using the IBM-SPSS (Statistical package for Social Science) software package. Overweight and lean subjects were compared with descriptive and bivariate statistics using Mann-Whitney U test for continuous variables with equal variance, Kolmogorov-Smirnov test for continuous variables with unequal variance (only maternal weight and body fat), and Fisher’s exact test for categorical variables. Continuous variables are reported as median and interquartile range (IQR), categorical variables are presented as N (%). Spearman correlations were run between the outcome variables (basal respiration, maximal respiration, ATP Production and spare capacity) and variables of interest (maternal weight, prepregnancy BMI, maternal body fat %, infant body fat %). Spearman correlations were then run between the outcome variables and all other variables in the dataset. Due to the small sample size, we were only able to include two additional variables for the regression analysis. Therefore, regression models were performed by including the outcome variable with the highest correlation and two of the variables with the highest correlation. p-values were not adjusted for the multiple comparisons.
Results
Maternal Characteristics
We initially enrolled 40 African American women carrying male fetuses (17 lean controls, 23 overweight). 1 woman voluntarily withdrew, 1 woman delivered at an outside facility and we were unable to obtain circumcision skin or collect maternal/infant metabolic data for 16 patients. We performed complete analysis on the remaining 22 subjects (8 lean, 14 overweight) Maternal pregnancy demographics are displayed in Table 1. Other than maternal weight, BMI and percent body fat, there were no significant differences between maternal characteristics. Pre-pregnancy BMI in the lean group ranged from 18.0 kg/m2 to 25 kg/m2, and the overweight group ranged from 25 kg/m2 to 39.6 kg/m2. The lean group had a median percent body fat of only 19%. Only one woman in our control group (12.5%) and one woman in our overweight cohort (7.1%) achieved their target weight based on their pre gestational BMI. While there was no statistically significant difference in GWG between groups, the overweight mothers did have a greater total weight gain during pregnancy, and a greater percent of overweight mothers were over their target weight gain (Table 1). Most women were in their 20s, and all infants delivered at term. All (100%) lean mothers delivered vaginally, whereas a majority (57.1%) of overweight mothers delivered by caesarean section. Most common reasons for caesarean section was non reassuring fetal status (50%) followed by repeat caesarean section (37.5%). There were no significant differences in maternal glucose or insulin levels measured during the third trimester clinical research unit visit (32–34 weeks).
Table 1.
Maternal Demographic Data, Median (IQR) or number (%)
| Lean (n=8) |
Overweight (n=14) |
|
|---|---|---|
| Age, years | 24 (22–27.3) | 28.5 (25.25–35.0) |
| Prepregnancy height, cm | 167.6 (159.3–170.2) | 163.4 (162.4–165.7) |
| Prepregnancy weight, kg | 59.3 (54.2–62.9) | 81.9 (71.5–95.1)* |
| Prepregnancy BMI, kg/m2 | 21.8 (19.6–23.6) | 30.8 (26.2–35.4)* |
| Fasting Glucose. mg/dl | 80 (68.5–84.2) | 72.5 (66.5–78) |
| Fasting Insulin, uU/ml | 9.9 (7.1–13) | 11.9 (6.2–15.8) |
| Target delivery weight* | ||
| Under | 5 (62.5) | 2 (14.3) |
| Goal | 1 (12.5) | 1 (7.1) |
| Over | 2 (25) | 11 (78.5)* |
| Body fat at 34–36wks, % | 19.0 (15.9–21.3) | 33.4 (25.6–36.3) |
| Gestational age, weeks | 39.3 (38.1–40.1) | 39.1 (38.4–40) |
| Mode of delivery* | ||
| Vaginal | 8 (100) | 5 (35.8) |
| Forceps assisted | 0 | 1 (7.1) |
| C-section | 0 | 8 (57.1)* |
| Parity | ||
| 1 | 4 (50) | 4 (28.6) |
| 2 | 1(12.5) | 2 (14.6) |
| 3 | 2 (25) | 5 (35.7) |
| >3 | 1 (12.5) | 3 (21.4) |
| Number of prenatal visits | ||
| 1–5 | 1 (12.5) | 1 (7.1) |
| 5–10 | 4 (50) | 6 (42.9) |
| >10 | 3 (37.5) | 7 (50) |
Abbreviations: IQR – interquartile range, BMI – body mass index; C-section – Caesarean section
Statistical analysis done with Mann Whitney U, Kolmogorov-Smirnov or Fisher’s exact test
p < 0.05
Infant Characteristics
Infants born to overweight mothers had a greater length at birth when corrected for GA: 52nd percentile, (IQR 51st-63.5th) vs 95th percentile (IQR 80th -100th) with Z-scores of 0.05 (IQR 0.028–0.46) vs 1.8 (IQR 0.84–2.7), p <0.01). There was no significant difference in birthweight between both groups when birthweight was corrected for height and GA. However, head circumference, corrected for GA was also significantly higher in the infants of overweight mothers (Figure 1A–D). Weight and body composition were assessed at 24–72 hours post partum using the Peapod. Infants born to overweight mothers had significantly higher weights at this point compared to their lean counterparts (data not shown, 2963g (IQR 2463–3440) vs. 3446g (IQR 3131–3773), p=0.046). This difference in weight was not significant when corrected for GA (data not shown). Although total body fat and percent body fat tended to be greater in infants born to overweight mothers, this did not reach statistical significance (Figure 1E, F). We also performed indirect calorimetry to evaluate and compare the infants’ metabolic rate; there were no significant differences in oxygen consumption, resting energy expenditure (RER) and respiratory quotient between infants born to lean versus overweight mothers (Supplemental data). The majority of infants in both groups were breast fed; 62.5% in the control group and 42.9% in the overweight group. One infant in the overweight group received D10 intravenous fluids for 24 hours. The indirect calorimetry testing for this infant was performed when he was off IVF and breast and bottle feeding for >24 hours. In order to determine whether there were baseline differences in circulating metabolites or insulin levels in these infants, we measured glucose, free fatty acids (FA), triglycerides, cholesterols and insulin and c-peptide from cord blood. While there was no significant differences in free FA, triglycerides, cholesterols, (data not shown) or glucose levels (Figure 1G), insulin was significantly elevated in infants born to overweight mothers (Figure 1H, 4.6μU/ml (IQR 1.8–6.3) vs. 10.0μU/ml (IQR 5.2–12.4). The c-peptide was not significantly different between groups (data not shown, 0.785 ng/ml (IQR 0.59–1.34) vs 1.18 ng/ml (IQR 0.80 vs 1.44).
Figure 1. Infants born to overweight mothers are longer and have increased cord blood insulin levels.
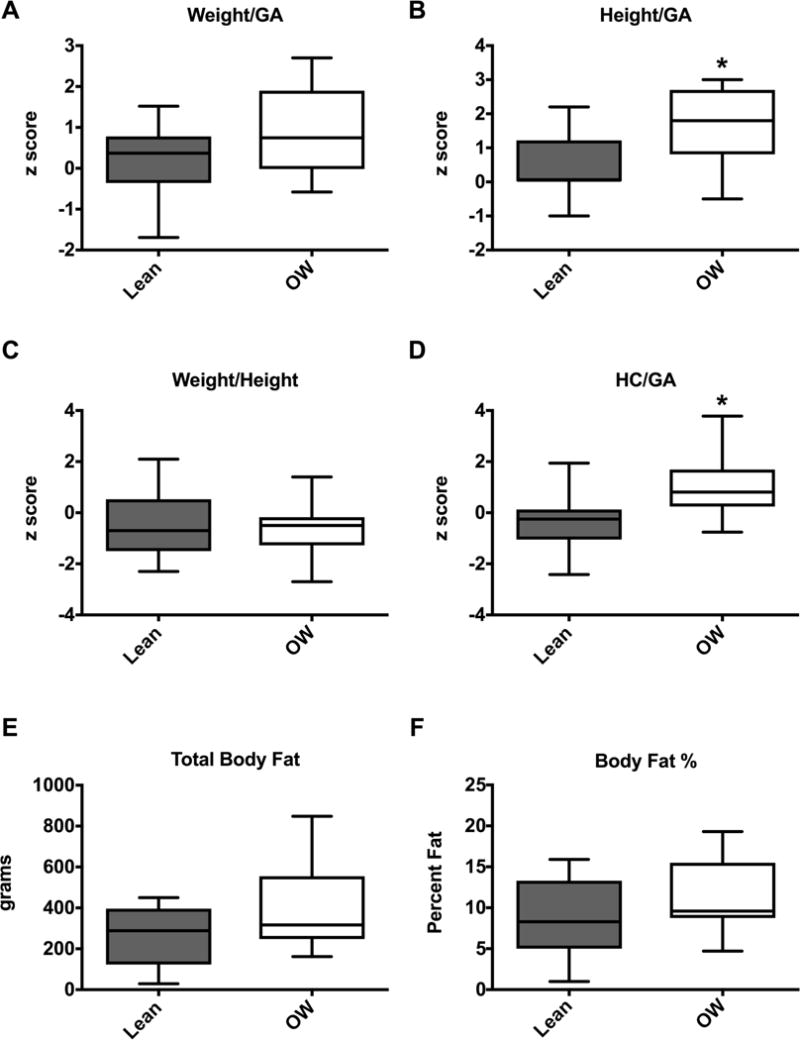
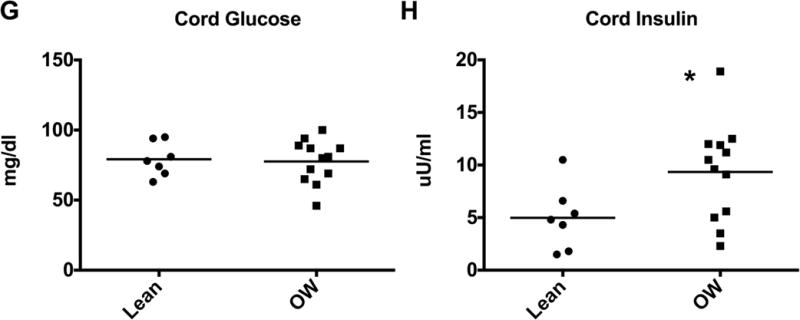
(A–D) Infant demographic data as box-and-whisker plots for lean (n=8) and overweight (OW, n=14) groups. Boxes depict 25th to 75th percentiles with line at the median. Whiskers represent the minimum and maximum data. (E-F) Cord blood glucose (E) and insulin (F) values. Symbols represent data from individual infants, line at the median. *p<0.05. GA=gestational age, HC= head circumference
Metabolic Function in Skin Fibroblasts
In order to evaluate cellular nutrient uptake, we evaluated glucose and FA uptake in a subset of fibroblasts from infants born to lean and overweight mothers. There was no significant difference in glucose or FA uptake among fibroblasts of infants born to lean mothers compared to the overweight cohort (data not shown). We next evaluated mitochondrial function in the skin fibroblasts. Figure 2 shows that skin fibroblasts of infants born to overweight mothers had significant differences when compared to skin fibroblasts from infants born to lean mothers, including higher maximal respiration (Figure 2B, 89.0 pmol/min (IQR 61.2–113.5) vs. 122.0 pmol/min (IQR 100.4–138.5), p=0.01), a higher spare respiratory capacity (Figure 2D, 44.6 pmol/min (IQR 28.0–61.3) vs. 67.8 pmol/min (IQR 58.3–77.6), p <0.01) and a trend towards significantly higher basal respiration (Figure 2A, 44.3pmol/min (IQR 34.5–53.0) vs. 54.2 pmol/min (IQR 49.8–59.5), p=0.06). Interestingly, there was no difference in ATP production (Figure 2C, 40.2 pmol/min (IQR 30.9–48.8) vs. 46.3 (IQR 44.0–53.6) in the infant skin fibroblasts from overweight mothers compared to lean mothers, suggesting that these mitochondria work inefficiently. Supporting this theory, we found a trend towards a higher proton leak in fibroblasts of infants born to overweight mothers compared to the lean cohort (lean-4.3pmol/min (IQR 3.5–5.0) vs. OW-5.5pmol/min (IQR 4.4–6.8), p= 0.1).
Figure 2. Increased fibroblast mitochondrial function in infants born to overweight mothers.

Oxygen consumption at baseline (basal), after adding oligomycin (ATP production, D) and FCCP (maximal, B). Spare capacity (C) represents the difference between maximal and basal oxygen consumption rate (OCR). Data displayed as box-and-whisker plots with line at the median. (lean, n=8, overweight (OW), n=14)) *p<0.05.
In order to further understand the association between maternal weight and body fat on infant mitochondrial function we performed correlation analyses. We noted that maternal prepregnancy weight had a significant association with maximal respiration and spare respiratory capacity (Figure 3A, B). These associations remained significant after controlling for maternal age and maternal triglyceride levels. We also noted significant correlations between maternal pre-pregnancy BMI and maternal body fat percentage and respiration (Figure 3C–F), although the correlation was less significant than pre-pregnancy weight. Gestational weight gain was not significantly correlated with respiration rates. There were also no significant correlations between maternal glucose and insulin levels and infant glucose, insulin or mitochondrial respiration rates.
Figure 3. Maternal prepregnancy weight positively correlated with infant mitochondrial function.
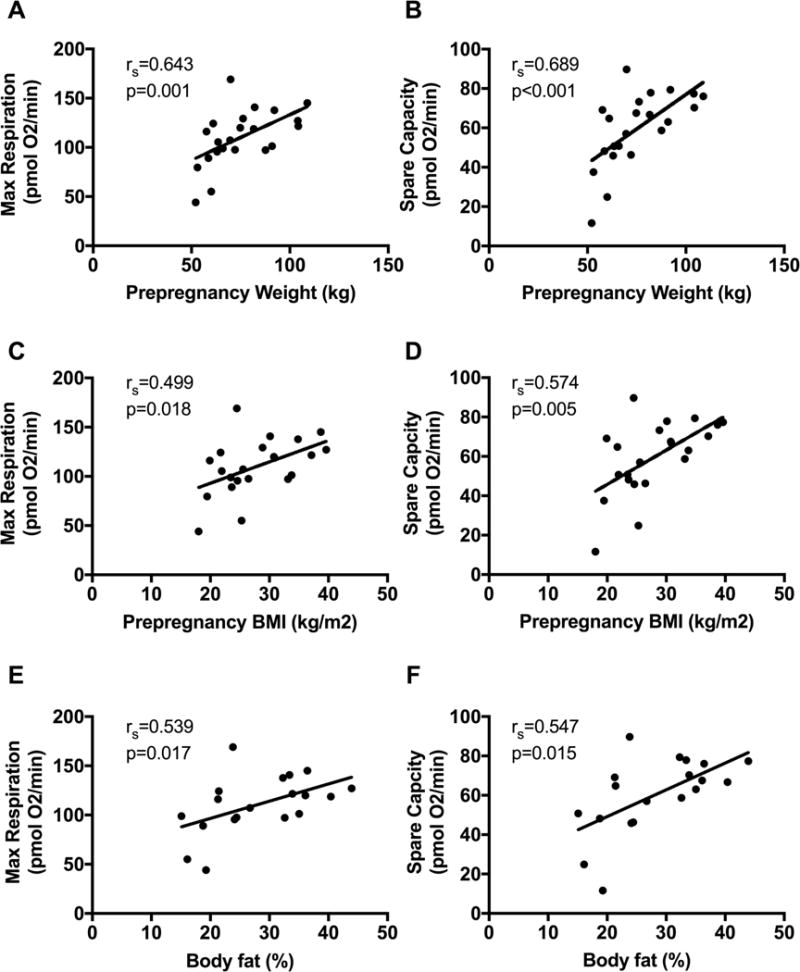
Associations of maximal respiration (left) and spare capacity (right) with maternal prepregnancy weight (A, B), prepregnancy BMI (C, D), and maternal body fat % (E, F).
Metabolic Gene Expression and Mitochondrial DNA Quantification
Given the differences in mitochondrial function, we sought to determine whether there were differences in gene expression for genes involved in metabolism and mitochondrial function. We performed quantitative real time PCR for selected target genes involved in metabolic and mitochondrial function. Figure 4 demonstrates that there are several alterations in gene expression. Sterol regulatory element-binding protein 1 (SREBP1c) is a transcriptional regulator of several genes involved in glucose metabolism and FA and lipid production and its expression is regulated by insulin.(29) SREBP1c and upstream stimulatory factors 1 and 2 (USF 1 and 2) transcriptionally regulate FA synthase (FAS). Both SREBP1c and FAS were higher in fibroblasts from infants born to overweight mothers. Solute carrier family 2 member 1 (SLC2a1) is involved in glucose transport and metabolism, and it was lower in the fibroblasts of the overweight group. ATP Synthase 5a (ATPSyn5a), which converts ADP to ATP in the mitochondria, was upregulated in the fibroblasts of the overweight group compared to the lean group. Hexokinase 2 (HK2), the first enzyme in glucose metabolism, was unchanged. Interestingly, compared to the lean cohort, uncoupling protein 2 mRNA was significantly more expressed in fibroblasts of infants born to overweight mothers. There was no difference in mitochondrial DNA (mtDNA) quantity in fibroblasts from infants of lean mothers compared to overweight mothers.
Figure 4. Infants born to overweight mothers have altered mRNA expression for genes involved in metabolism and mitochondrial function.
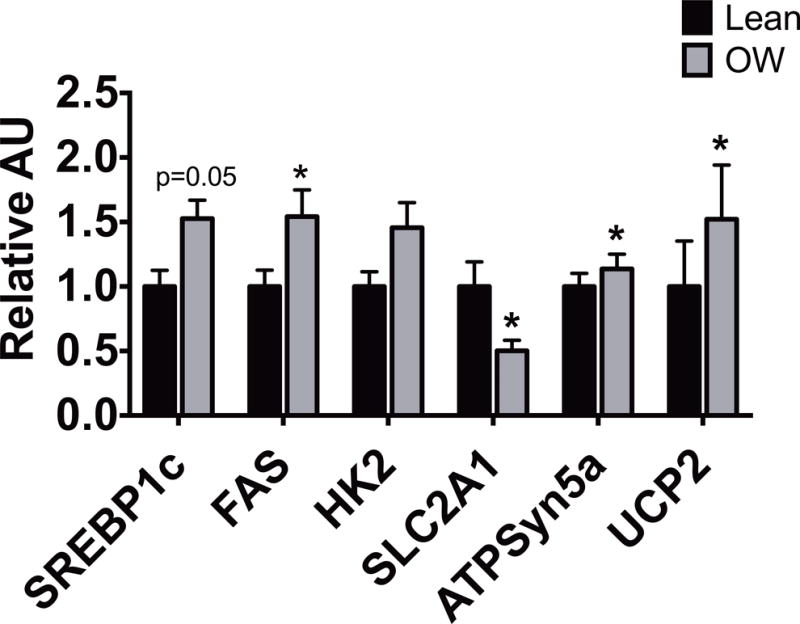
qPCR measurements of mRNA expression of selected genes (abbreviations in text) in skin fibroblasts of infants born to lean (n=8) versus overweight (n=14) mothers. Bars represent mean ± SEM arbitrary units, normalized to lean values (=1.0), *p<0.05.
Oxidative Stress
Given the increase in UCP2 and the increased oxygen consumption without increase in ATP production, we were interested in the potential for increased production of reactive oxygen species (ROS). We first performed cellular staining in a subset of our fibroblasts samples using MitoSox, which stains for mitochondrial superoxide and noted a higher total number of fluorescent cells (Figure 5A, left) and significantly increased median fluorescence intensity in fibroblasts of infants born to overweight mothers compared to infants born to lean mothers (Figure 5A, right). Given this evidence of increased superoxide in the skin fibroblasts of infants born to overweight mothers, we explored the possibility of oxidative protein damage in these cells. To that end we performed Western blotting to detect protein carbonylation using the Oxyblot kit. Protein carbonylation was higher in skin fibroblasts from infants born to overweight mothers compared to infants born to lean mothers (Figure 5B).
Figure 5. Skin fibroblasts of infants born to overweight mothers have increased superoxide and oxidative damage.
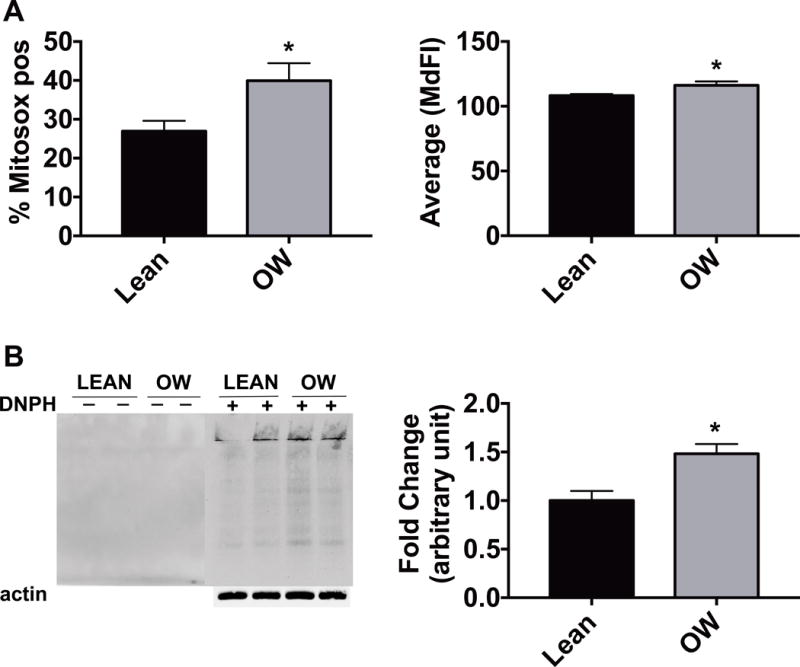
(A) Results of mitochondrial superoxide staining quantified by flow cytometery. Total number of fluorescent cells (left) and median fluorescence intensity (right), n=6–8/group, *p<0.05). (B) Protein carbonylation of skin fibroblast lysates treated with (+) or without (−) DNPH and visualized by Western blot. Representative blot is shown (left) as well as quantification (right), normalized to the lean values. n=4/group *p<0.05. All bars represent mean ± SEM.
Discussion
Evidence indicates that maternal obesity alters oocyte and offspring mitochondrial function,(20, 22, 23) but the impact of maternal obesity on mitochondrial function in human neonates is unknown. As an initial step to answer this question, we evaluated mitochondrial function in skin fibroblasts of healthy neonates born to either overweight or lean mothers. To our knowledge, this is the first study to show that mitochondrial function is altered in human infants born to overweight mothers. We demonstrate that skin fibroblasts in infants born to overweight mothers have increased mitochondrial respiration without a significant increase in ATP production, indicating inefficient respiration. Given the correlation between maternal weight and pre-pregnancy BMI and maximal oxygen consumption rates, this increase in mitochondrial respiration appears related to maternal pre-pregnancy nutrition status. Although, the higher frequency of women that were over their target weight in the overweight group, does make it more difficult to be certain that only pre-pregnancy nutrition status impacts the mitochondria. Previous animal models have shown damaged mitochondria and decreased mitochondrial function in oocytes of obese mothers.(20, 23) Additionally, Saben et al. found decreased mitochondrial function in the skeletal muscle of F1 offspring of mothers fed a high fat/high sugar diet.(22) It is possible that we found increased mitochondrial function as a result of a compensatory response to inheritance of damaged maternal mitochondria. Previous studies have also demonstrated that oxidative stress can drive mitochondrial biogenesis (Igosheva, Lee HC 2000). Thus, one might consider that an increase in mitochondrial mass might drive increased respiration. While we did not note a difference in mtDNA quantity or transcriptional regulators of mitochondrial biogenesis (data not shown), it is still reasonable to consider that maternal oxidative stress might impact offspring respiration rates.
We were able to detect these differences in mitochondrial metabolism in tissue with relatively low metabolic rate. One could speculate that differences maybe even more evident in tissues with higher metabolic rates, such as myocardial or brain tissue. However, we cannot be certain that these specific mitochondrial function differences will exist in all tissues. The lack of difference in RER or oxygen consumption by indirect calorimetry, despite finding differences in fibroblast oxygen consumption, is likely related to variability in measurements in neonates, particularly related to technical aspects of the study and standardization of feeding. A future study to evaluate these parameters in older infants would be ideal. The lack of increase in ATP production suggests more uncoupled respiration, which was also noted by Saben et al., as they noted reduced respiratory control ratio in offspring of mice with metabolic syndrome.(22) Previous work in murine models of obesity and diabetes noted a progression from normal to decreased mitochondrial function in insulin resistant to frank diabetic heart muscle.(30) This study also noted inefficient respiration in the earlier insulin resistant animals. Given that we studied early neonates, and the previous offspring data was in 8 week-old animals, we speculate that there may be an early compensatory response that later leads to decreased mitochondrial function.
The potential for increased uncoupled respiration is supported by the robust increase in UCP2 mRNA expression and the trend towards increased proton leak in skin fibroblasts of neonates born to overweight mothers compared to lean mothers. The family of uncoupling proteins extrude protons from the intermembrane space into the mitochondrial matrix, thus dissipating the proton gradient and decreasing ATP and superoxide production.(31, 32) Thus, increased UCP2 expression is considered an adaptive response to attenuate mitochondrial ROS production and protect against oxidative damage.(33, 34) Evidence suggests that metabolic diseases, such as obesity or diabetes, are associated with elevated ROS levels and therefore elevated oxidative stress.(35, 36) Data also show that increased ROS production, specifically superoxide radicals, activate UCP2 expression.(37) Consistent with these findings, we detected an increase in superoxide molecules in the skin fibroblasts of infants born to overweight mothers. This increase in superoxide may be driving the increase in UCP2 expression as an adaptive mechanism to protect the mitochondria against oxidative damage. Previous studies have shown that overproduction of ROS is associated with oxidative damage to proteins, lipids and DNA,(38) which we detected as increased protein carbonylation in the skin fibroblasts. Moreover, accumulation of ROS can lead to mitochondrial DNA damage, which contributes to abnormal electron transport chain function and compromises cell viability. This can eventually result in apoptotic cell death.(39–41) We postulate, that this progression in mitochondrial dysfunction in the setting of oxidative stress might be the reason why our study found increased mitochondrial function in these early neonates, whereas Saben et al. detected decreased mitochondrial function in older animals.
In addition to altered mitochondrial oxygen consumption, we noted significant changes in metabolic gene expression as well as cord blood insulin in infants born to overweight mothers. Many of the gene changes we noted are consistent with previous findings. Upregulation of genes involved in lipid and mitochondrial metabolism was previously demonstrated in human umbilical vein endothelial cells of infants born to obese mothers.(42) Long et. al demonstrated greater mRNA expression for FAS in adipose depots of offspring from obese ewes.(43) Additionally, SREBP1c, a transcriptional regular involved in FA metabolism, is known to be activated in the setting of overnutrition, resulting in accumulation of FAs and secondarily insulin resistance.(44, 45) While we did not see a difference in FA uptake in our cells, we did see upregulation of both SREBP1c and FAS. Evidence suggests that diabetic rats have a decreased expression of GLUT1, also known as SLC2a1.(46) The increased expression of enzymes mediating FA biosynthesis along with a decrease in glucose transporters may represent an adaptation to altered nutrient availability associated with obese pregnancy. ATP synthase is a central enzyme which converts ADP to ATP. Data suggests that obesity and diabetes is associated with an upregulation of enzymes involved in oxidative phosphorylation.(47) We suspect that this increase in ATP synthase might be a compensatory response to the inefficient mitochondria. The mechanism leading to the altered gene expression is not clear. There is strong evidence demonstrating that short-term exposure to hyperinsulinemia in healthy subjects results in increased mRNA expression for several inflammatory genes and transcription factors in human muscle biopsies.(48, 49) It is interesting that maternal insulin levels were not different between the groups, yet we detected a significant increase in insulin in the cord blood of the offspring from overweight mothers. This suggests evidence of early insulin resistance, which may be related to the early mitochondrial dysfunction. It is unclear at this point whether elevated insulin levels and altered nutrient availability during development cause modified gene expression or whether this could be due to a programming effect, such as epigenetic changes. Future studies should focus on small RNA expression, histone modification or DNA methylation as a mechanism for driving gene expression and metabolic changes in infants born to overweight mothers.
Our study has several limitations. The small sample size may limit the power to detect differences, and potentially limits the generalizability of our findings. We only enrolled male neonates; a future aim will be to investigate whether umbilical cells can be used in a similar manner to evaluate mitochondrial function in all neonates. We also only enrolled African American mothers. African Americans have a higher incidence of obesity,(50, 51) and are at a higher risk of developing oxidative stress related diseases, such as type 2 diabetes,(52) thus our results are very relevant for this population. However, it is possible that our results cannot be generalized to other races. To our knowledge there is no current data regarding ethnic differences in mitochondrial function. Additionally, the overweight group had a higher rate of caesarean delivery, which may have impacted the metabolic response in the offspring. Enrolling a heterogenous group of both overweight and obese females may limit our ability to detect smaller differences between our lean and overweight cohort. Using skin fibroblasts to assess global mitochondrial activity has not been validated, but skin biopsies are one method for diagnosing mitochondrial diseases.(53) It is unclear whether our findings could be applied to other cells in these infants. Since mitochondria are maternally inherited and our findings are in skin fibroblasts at a very young age, it is suggestive of a global process.
Conclusion
This prospective study in healthy human neonates demonstrates altered mitochondrial function and oxidative stress in skin fibroblasts of infants born to overweight mothers. Future studies are needed to determine the long-term impact of this finding on the metabolic health of these children. In particular, it will be important to determine if early mitochondrial function can predict later body composition and whether mitochondrial function changes over time.
Supplementary Material
Acknowledgments
This project was supported by the NIH/National Center for Advancing Translational Sciences (NCATS) grant UL1 TR000448. We thank the Women’s and Infant’s Health Consortium for assistance with cord blood collection and Lana Mehanovic for help with statistical analyses. LIFE-Moms is supported by the NIH through the NIDDK (U01 DK094418, U01 DK094463, U01 DK094416, 5U01 DK094466 (RCU)), the NHLBI (U01 HL114344, U01 HL114377), the NICHD (U01 HD072834), the NCCIH, the NIH Office of Research in Women’s Health (ORWH), the Office of Behavioral and Social Science Research (OBSSR), the Indian Health Service, and the Intramural Research Program of the NIDDK. We thank the LIFE-Moms consortium members for their contributions to the development and oversight of the common measures and procedures shared across trials.
Footnotes
Conflict of Interest: None of the authors declare any conflict of interest
References
- 1.Seidell JC. Obesity, insulin resistance and diabetes–a worldwide epidemic. Br J Nutr. 2000;83(Suppl 1):S5–8. doi: 10.1017/s000711450000088x. [DOI] [PubMed] [Google Scholar]
- 2.Vahratian A. Prevalence of overweight and obesity among women of childbearing age: results from the 2002 National Survey of Family Growth. Matern Child Health J. 2009;13(2):268–73. doi: 10.1007/s10995-008-0340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branum AM, Kirmeyer SE, Gregory EC. Prepregnancy Body Mass Index by Maternal Characteristics and State: Data From the Birth Certificate, 2014. Natl Vital Stat Rep. 2016;65(6):1–11. [PubMed] [Google Scholar]
- 4.Chen A, Feresu SA, Fernandez C, Rogan WJ. Maternal obesity and the risk of infant death in the United States. Epidemiology. 2009;20(1):74–81. doi: 10.1097/EDE.0b013e3181878645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metwally M, Ong KJ, Ledger WL, Li TC. Does high body mass index increase the risk of miscarriage after spontaneous and assisted conception? A meta-analysis of the evidence. Fertil Steril. 2008;90(3):714–26. doi: 10.1016/j.fertnstert.2007.07.1290. [DOI] [PubMed] [Google Scholar]
- 6.Stothard KJ, Tennant PW, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301(6):636–50. doi: 10.1001/jama.2009.113. [DOI] [PubMed] [Google Scholar]
- 7.Oken E. Maternal and child obesity: the causal link. Obstet Gynecol Clin North Am. 2009;36(2):361–77. ix–x. doi: 10.1016/j.ogc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Perng W, Gillman MW, Mantzoros CS, Oken E. A prospective study of maternal prenatal weight and offspring cardiometabolic health in midchildhood. Ann Epidemiol. 2014;24(11):793–800 e1. doi: 10.1016/j.annepidem.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32(6):1076–80. doi: 10.2337/dc08-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modi N, Murgasova D, Ruager-Martin R, Thomas EL, Hyde MJ, Gale C, et al. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res. 2011;70(3):287–91. doi: 10.1203/PDR.0b013e318225f9b1. [DOI] [PubMed] [Google Scholar]
- 11.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261(5):412–7. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 12.Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311(6998):171–4. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51(2):383–92. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 14.Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2008;294(2):R528–38. doi: 10.1152/ajpregu.00316.2007. [DOI] [PubMed] [Google Scholar]
- 15.Zambrano E, Nathanielsz PW. Mechanisms by which maternal obesity programs offspring for obesity: evidence from animal studies. Nutrition reviews. 2013;71(Suppl 1):S42–54. doi: 10.1111/nure.12068. [DOI] [PubMed] [Google Scholar]
- 16.Muhlhausler BS. Programming of the appetite-regulating neural network: a link between maternal overnutrition and the programming of obesity? J Neuroendocrinol. 2007;19(1):67–72. doi: 10.1111/j.1365-2826.2006.01505.x. [DOI] [PubMed] [Google Scholar]
- 17.Guan H, Arany E, van Beek JP, Chamson-Reig A, Thyssen S, Hill DJ, et al. Adipose tissue gene expression profiling reveals distinct molecular pathways that define visceral adiposity in offspring of maternal protein-restricted rats. Am J Physiol Endocrinol Metab. 2005;288(4):E663–73. doi: 10.1152/ajpendo.00461.2004. [DOI] [PubMed] [Google Scholar]
- 18.Desai M, Gayle D, Babu J, Ross MG. Programmed obesity in intrauterine growth-restricted newborns: modulation by newborn nutrition. Am J Physiol Regul Integr Comp Physiol. 2005;288(1):R91–6. doi: 10.1152/ajpregu.00340.2004. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Frolova AI, Purcell S, Adastra K, Schoeller E, Chi MM, et al. Mitochondrial dysfunction and apoptosis in cumulus cells of type I diabetic mice. PLoS One. 2010;5(12):e15901. doi: 10.1371/journal.pone.0015901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, et al. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7(11):e49217. doi: 10.1371/journal.pone.0049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu LL, Dunning KR, Yang X, Russell DL, Lane M, Norman RJ, et al. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology. 2010;151(11):5438–45. doi: 10.1210/en.2010-0551. [DOI] [PubMed] [Google Scholar]
- 22.Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, et al. Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations. Cell Rep. 2016;16(1):1–8. doi: 10.1016/j.celrep.2016.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Igosheva N, Abramov AY, Poston L, Eckert JJ, Fleming TP, Duchen MR, et al. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One. 2010;5(4):e10074. doi: 10.1371/journal.pone.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gemma C, Sookoian S, Alvarinas J, Garcia SI, Quintana L, Kanevsky D, et al. Mitochondrial DNA depletion in small- and large-for-gestational-age newborns. Obesity (Silver Spring) 2006;14(12):2193–9. doi: 10.1038/oby.2006.257. [DOI] [PubMed] [Google Scholar]
- 25.Lopez LB, Calvo EB, Poy MS, del Valle Balmaceda Y, Camera K. Changes in skinfolds and mid-upper arm circumference during pregnancy in Argentine women. Matern Child Nutr. 2011;7(3):253–62. doi: 10.1111/j.1740-8709.2009.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thame M, Trotman H, Osmond C, Fletcher H, Antoine M. Body composition in pregnancies of adolescents and mature women and the relationship to birth anthropometry. Eur J Clin Nutr. 2007;61(1):47–53. doi: 10.1038/sj.ejcn.1602484. [DOI] [PubMed] [Google Scholar]
- 27.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brookheart RT, Swearingen AR, Collins CA, Cline LM, Duncan JG. High-sucrose-induced maternal obesity disrupts ovarian function and decreases fertility in Drosophila melanogaster. Biochim Biophys Acta. 2017;1863(6):1255–63. doi: 10.1016/j.bbadis.2017.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou Y, et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem. 2014;289(9):5510–7. doi: 10.1074/jbc.M113.541110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115(7):909–17. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37(6):755–67. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 32.Ricquier D, Bouillaud F. Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. J Physiol. 2000;529(Pt 1):3–10. doi: 10.1111/j.1469-7793.2000.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26(4):435–9. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 34.Pheiffer C, Jacobs C, Patel O, Ghoor S, Muller C, Louw J. Expression of UCP2 in Wistar rats varies according to age and the severity of obesity. J Physiol Biochem. 2016;72(1):25–32. doi: 10.1007/s13105-015-0454-4. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract. 2013;7(5):e330–41. doi: 10.1016/j.orcp.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Le Lay S, Simard G, Martinez MC, Andriantsitohaina R. Oxidative stress and metabolic pathologies: from an adipocentric point of view. Oxid Med Cell Longev. 2014;2014:908539. doi: 10.1155/2014/908539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415(6867):96–9. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 38.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 39.Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta. 1998;1366(1–2):53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- 40.Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7(1–2):106–18. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 41.Majima HJ, Oberley TD, Furukawa K, Mattson MP, Yen HC, Szweda LI, et al. Prevention of mitochondrial injury by manganese superoxide dismutase reveals a primary mechanism for alkaline-induced cell death. J Biol Chem. 1998;273(14):8217–24. doi: 10.1074/jbc.273.14.8217. [DOI] [PubMed] [Google Scholar]
- 42.Costa SM, Isganaitis E, Matthews TJ, Hughes K, Daher G, Dreyfuss JM, et al. Maternal obesity programs mitochondrial and lipid metabolism gene expression in infant umbilical vein endothelial cells. Int J Obes (Lond) 2016;40(11):1627–34. doi: 10.1038/ijo.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Long NM, Rule DC, Zhu MJ, Nathanielsz PW, Ford SP. Maternal obesity upregulates fatty acid and glucose transporters and increases expression of enzymes mediating fatty acid biosynthesis in fetal adipose tissue depots. J Anim Sci. 2012;90(7):2201–10. doi: 10.2527/jas.2011-4343. [DOI] [PubMed] [Google Scholar]
- 44.Khvorostov ED, Morozov SA. A rare complication of metallo-osteosynthesis of the clavicle. Klin Khir. 1989;(12):47–8. [PubMed] [Google Scholar]
- 45.Prodanovic R, Koricanac G, Vujanac I, Djordjevic A, Pantelic M, Romic S, et al. Obesity-driven prepartal hepatic lipid accumulation in dairy cows is associated with increased CD36 and SREBP-1 expression. Res Vet Sci. 2016;107:16–9. doi: 10.1016/j.rvsc.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Pedersen O, Kahn CR, Kahn BB. Divergent regulation of the Glut 1 and Glut 4 glucose transporters in isolated adipocytes from Zucker rats. J Clin Invest. 1992;89(6):1964–73. doi: 10.1172/JCI115804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takamura T, Misu H, Matsuzawa-Nagata N, Sakurai M, Ota T, Shimizu A, et al. Obesity upregulates genes involved in oxidative phosphorylation in livers of diabetic patients. Obesity (Silver Spring) 2008;16(12):2601–9. doi: 10.1038/oby.2008.419. [DOI] [PubMed] [Google Scholar]
- 48.Tsintzas K, Norton L, Chokkalingam K, Nizamani N, Cooper S, Stephens F, et al. Independent and combined effects of acute physiological hyperglycaemia and hyperinsulinaemia on metabolic gene expression in human skeletal muscle. Clin Sci (Lond) 2013;124(11):675–84. doi: 10.1042/CS20120481. [DOI] [PubMed] [Google Scholar]
- 49.Coletta DK, Balas B, Chavez AO, Baig M, Abdul-Ghani M, Kashyap SR, et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. Am J Physiol Endocrinol Metab. 2008;294(5):E910–7. doi: 10.1152/ajpendo.00607.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samson R, Qi A, Jaiswal A, Le Jemtel TH, Oparil S. Obesity-Associated Hypertension: the Upcoming Phenotype in African-American Women. Curr Hypertens Rep. 2017;19(5):41. doi: 10.1007/s11906-017-0738-x. [DOI] [PubMed] [Google Scholar]
- 51.Fowler BA. Obesity in African-American Women–The Time Bomb is Ticking: An Urgent Call for Change. J Natl Black Nurses Assoc. 2015;26(2):42–50. [PubMed] [Google Scholar]
- 52.Nwobu CO, Johnson CC. Targeting obesity to reduce the risk for type 2 diabetes and other co-morbidities in African American youth: a review of the literature and recommendations for prevention. Diab Vasc Dis Res. 2007;4(4):311–9. doi: 10.3132/dvdr.2007.058. [DOI] [PubMed] [Google Scholar]
- 53.Rodenburg RJ. Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2011;34(2):283–92. doi: 10.1007/s10545-010-9081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.