

Abstract
Quinolinic acid (QUIN) is an endogenous neurotoxin that acts as an N-methyl-D-aspartate receptor (NMDAR) agonist generating a toxic cascade, which can lead to neurodegeneration. The action of QUIN in Caenorhabditis elegans and the neurotoxins that allow the study of glutamatergic system disorders have not been carefully addressed. The effects of QUIN on toxicological and behavioral parameters in VM487 and VC2623 transgenic, as well as wild-type (WT) animals were performed to evaluate whether QUIN could be used as a neurotoxin in C. elegans. QUIN reduced survival of WT worms in a dose-dependent manner. A sublethal dose of QUIN (20 mM) increased reactive oxygen species (ROS) levels in an nmr-1/NMDAR-dependent manner, activated the DAF-16/FOXO transcription factor, and increased expression of the antioxidant enzymes, superoxide dismutase-3, glutathione S-transferase-4, and heat shock protein-16.2. QUIN did not change motor behavioral parameters, but altered the sensory behavior in N2 and VM487 worms. Notably, the effect of QUIN on the sensory behavioral parameters might occur, at least in part, secondary to increased ROS. However, the touch response behavior indicates a mechanism of action that is independent of ROS generation. In addition, non-lethal doses of QUIN triggered neurodegeneration in glutamatergic neurons. Our findings indicate that C. elegans might be useful as a model for studies of QUIN as a glutamatergic neurotoxin in rodent models.
Keywords: C. elegans, Neurodegeneration, Quinolinic Acid, NMDA, Glutamatergic System
1. Introduction
2,3-Pyridine dicarboxylic acid, widely known as quinolinic acid (QUIN), is a metabolite of tryptophan in the kynurenine pathway, which acts as an N-methyl-D-aspartate receptor (NMDAR) agonist, preferentially on the NR2A and NR2B receptor subunits [1-3]. QUIN is produced mostly by microglia and activated macrophages in the brain [4] under pathological conditions and has been described as a potent endogenous neurotoxin. The mechanisms of action of QUIN take different forms causing excitotoxicity by elevating cytosolic calcium concentrations, depleting ATP, and forming free radicals [5]. QUIN plays a role in several neuropathological conditions, such as Alzheimer’s disease [6], Huntington’s disease [7], and neurodegenerative disorders [8].
The toxic events triggered by QUIN occur even at low concentrations [9]. Moreover, excitotoxicity generated by QUIN is associated with increased formation of reactive oxygen species (ROS) and reduced activity of antioxidant defense systems, resulting in oxidative damage [4]. Previous studies have shown that QUIN disrupts DNA and promotes lipid peroxidation. Secondary to its ability to form complexes with Fe(II), QUIN may cause oxidative events partially independent of NMDAR [10]. Furthermore, ROS can be detoxified through different mechanisms [11]. Among them, activation of transcription factors required for antioxidant enzyme synthesis and chaperones. Previous studies have suggested that the toxicity of QUIN is associated with a reduction in the transcription of genes related to antioxidant defenses [12].
The nematode Caenorhabditis elegans is an invaluable experimental model, as it is easy to culture, develops and reproduces rapidly, has a simple nervous system, and is easy to genetically manipulate. In addition, its transparent morphology allows the use of fluorescent markers for studies on various molecules. C. elegans has cellular processes and molecular mechanisms that modulate the stress response, which are highly conserved in mammals [12, 13].
The C. elegans nervous system is comprised of a set of neurotransmitters, such as acetylcholine (ACh), γ-aminobutyric acid, glutamate, nitric oxide, serotonin, and other monoamines [13-15]. Glutamatergic neurotransmission has been partially elucidated in C. elegans. Studies that have mapped neurons expressing glutamate receptors have revealed 10 ionotropic glutamate receptor subunits [16, 17]. Among them, eight are in the non-NMDA class (GLR-1–GLR-8), which are similar to AMPA or kainate subfamilies, and two are in the NMDA class (NMR-1 and NMR-2), which are homologous to the NR1 and NR2A subfamilies, respectively in mammals [17, 18]. There is no evidence of QUIN production in C. elegans astrocytes. Furthermore, there is no pharmacological model that induces glutamatergic neurotoxicity in this worm. Thus, the aim of this study was to investigate QUIN as a neurotoxin in C. elegans and establish a new model to allow the study of diseases associated with glutamatergic neurotoxicity.
There is no evidence of a neurotoxin acting on the C. elegans glutamatergic system. In our study, we evaluated the toxicity generated by QUIN in C. elegans and the mechanisms involved in this process to contribute to new models and allow a further understanding of neurodegenerative disorders.
2. Materials and methods
2.1. C. elegans strains, maintenance and treatment
The wild type strain N2 and mutant strains CL2070 (dvls70 [hsp-16.2p∷GFP + rol-6(su1006)]), TJ356 (zls356 [daf-16p∷daf-16a/b∷GFP + rol-6(su1006)]), CF1553 (muIs84 [(pAd76) sod-3p∷GFP + rol-6(su1006)]), VM487 [nmr-1(ak4)], VC2623 [nmr-2(ok3324)], CL2166 (dvIs19 [(pAF15) gst-4p∷GFP∷NLS] and OH438 (otls117 [unc-33p∷GFP + unc-4(+)] pan-neuronal GFP) were obtained from the Caenorhabditis Genetics Center (CGC, University of Minnesota, Minneapolis, MN, USA). The CL2070, TJ356, CF1553 and OH438 has GFP fused with promotor region for heat shock protein 16-2, transcription factor DAF-16/FOXO, superoxide dismutase 3 and pan-neuronal, respectively. The VM487 and VC2623 does not contain the nmr-1 and nmr-2 subunits of the NMDA ionotropic glutamate receptors gifts in C. elegans, respectively.
For all worms, age-synchronized eggs were obtained by isolating embryos from gravid hermaphrodites using bleaching solution (1% NaOCl, 0.25 M NaOH), washed three times and stored overnight in M9 to obtain all animals in stage L1. The L1 population was transferred to NGM (Nematode Growth Medium) previously seeded with Escherichia coli OP50 as food source, at 20°C, until reaching the young adult stage, about 40 hours [19].
Worms at young adult stage (after approximately 40 hours of L1 stage) were exposed acutely to QUIN at final concentrations different in plate (5, 10, 20, 50, 100 and 200 mM) or control, during periods 1 hour in plates containing NGM and OP50 culture medium (O.D.600 = 1.0) as predicted in previous assays, where it was added only in the preparation of the culture medium. The QUIN-concentration were added onto the NGM, previously seeded with OP50, 1 hour before exposing the worms. Subsequently, the worms were washed three times and the analyses were performed.
2.2. Survival
After exposure to different concentrations of QUIN, the worms were transferred to news plates containing NGM seeded with and OP50 in the absence of QUIN during 24 hours to recovery. About 100 worms were analyzed per group per experiment and the number of survivors was quantified. This assay was repeated four times in duplicate.
2.3. Measurement of reactive oxygen species (ROS)
Intra-worm ROS generation was measured in C. elegans wild-type and transgenic strains using CM-H2DCFDA, as previously described [20] with minor modifications [21]. Briefly, the worms were washed with M9 buffer three times and transferred to micro tubes. After, 10 μL of 2 mM CM-H2DCFDA were added to the Eppendorf tubes containing 1000 worms in 990 μL M9 (20 μM CM-H2DCFDA - final concentration) and incubated for 2 h. The worms were washed and transferred to 96-well plates (100 worms per well). The fluorescence intensity was measured with a plate reader in SpectraMax i3 (Excitation: 488 nm; Emission: 510 nm).
2.4. Subcellular Daf-16 localization
About 50 worms per group were transferred to a microscopic slide in M9 buffer. Fluorescence images was acquired with an OLYMPUS® FLUOVIEW FV10i Confocal Microscope housed in air-conditioned rooms (20°C). Localization DAF-16∷GFP in animals was classified in three category: as total localization in the cytoplasm, partially in the nucleus (intermediate) or located totally in the nucleus. Four assays was performed and 10 worms for group were randomly picked for evaluation. The worms exposed to heat stress at 35 ° C for 1 hour on NGM plates with OP50 was used as a positive control.
2.5. Quantitation of SOD-3, GST-4, HSP-16.2 and pan-neuronal expression
Approximately 50 worms in M9 buffer were transferred to microscopic slides and 10 uL of 10 mM sodium azide was added as a paralyzing agent were used to better visualize the whole animal. The positive control utilized in strains are worms exposed to heat stress at 35 ° C for 1 hour on NGM plates with OP50. The expression of HSP-16.2, SOD-3 and GST-4 [22, 23] was measured by quantifying the fluorescence of the reporter protein GFP, according to fluorescence specificity location for each transgenic strain. In the pan-neuronal strain neuronal integrity was evaluated through the quantification and evaluation of the neurons located in the region of the head of the animals, where there is presence of the majority of the neurons that express glutamate receptors of type NMDA. The integrity of the neurons present in the ventral nerve cord of the animals was also analyzed by observing the presence or absence of neuron fluorescence.
The data are expressing in % AFU of Ctrl refers to the amount of arbitrary fluorescence units (AFU) present as a percentage of the control values (non-QUIN-treated animal), considering the mean of the control values as 100%. The intensity of fluorescence of each worm in each image was analyzed using ImageJ2X (ImageJ2X software; Rawak Software, Inc., Stuttgart, Germany). From each group, 20 worms were randomly picked to measure the mean pixel density.
2.6. Behavior analysis
After 1h-QUIN exposure, the worm behavior worms were evaluated in the same exposure plates. Behaviors of locomotion and pharyngeal beating of approximately 20 animals, in each plate, were evaluated using optical microscopy and later classified in: animals as having standard behavior (pharyngeal pumps and locomotion unchanged) and non-standard behavior (paralysis or absence of pharyngeal pumps). The locomotion unchanged were considered ability to traverse the plaque in search of food by the presence of movement/locomotion of the animal, and non-standard behavior were animals no present movement of the body musculature even after stimulated with brush bristles [24]. Animals that had omega turns and cork screws were not considered abnormal. The pharyngeal pumps were classified according to the presence or absence of contraction of the pharyngeal muscle.
The assay was repeated four days independently, by duplicate and approximately 10 worms of each group were evaluated in each assay. In addition, worms that presented standard behavior were submitted to an additional behavioral task, nose touch response, in order to test potential effects related with on the glutamatergic system.
2.6.2. Pharyngeal pumping
The number of pharyngeal contractions during a 10-sec interval in triplicate was measured [25]. The assay was performed at three independent times with 10 worms per group and results are shown as pumping/minute.
2.6.3. Defecation cycle
The defecation cycle length was assessed by observing the duration between the pBoc steps (posterior body muscle contraction) of two consecutive defecations. Ten animals per group were scored for three consecutive cycles (three successive pBoc steps) per assay. Experiments were performed at three independent times.
2.6.4. Body Bends
Well-fed worms were transferred to food-free NGM plate and allowed to freely move. After 30 s adaptation, they were scored for the number of body-bends generated in 1-min interval. A body bend was defined as change in the direction of propagation along the y axis, assuming the worm was traveling along the x axis [26]. Twenty worms of each group were evaluated were repeated three times by duplicate.
2.6.5. Touch Response
Nematode touch response was performed by touching gently the head-region of the animal with a bristle brush. The backward was considered as a positive response a touch-sensitive in the opposite direction to the stimulated site in total ten touches and as non-response animals were considered that did not present any adverse reaction when stimulated, as previously described [23]. Four assays were realized at different times, they mean different synchronization and days and 10 worms analyzed for each experiment. 10-second resting period between trials.
2.7. Statistical analysis
Statistical analysis was performed using GraphPad (Version 5.0, San Diego, CA). Significance was assessed using t-tests, or one-way analysis of variance (ANOVA), followed by Newman-Keuls Test, or two-way ANOVA, followed by Bonferroni or Tukey Test for post hoc comparison. Values of p<0.05 were considered statistically significant.
3. Results
3.1. QUIN induces toxicity in wild-type C. elegans
We first investigated whether QUIN was toxic to wild-type (N2) worms. Thus, the percentage of worms that survived exposure to QUIN was determined. Wild-type worms treated with QUIN exhibited a significant decrease in survival (11.3%, 17.7%, and 20.3% decrease for 50, 100, and 200 mM QUIN, respectively) when compared to control worms (p < 0.01; Fig. 1).
Figure 1.
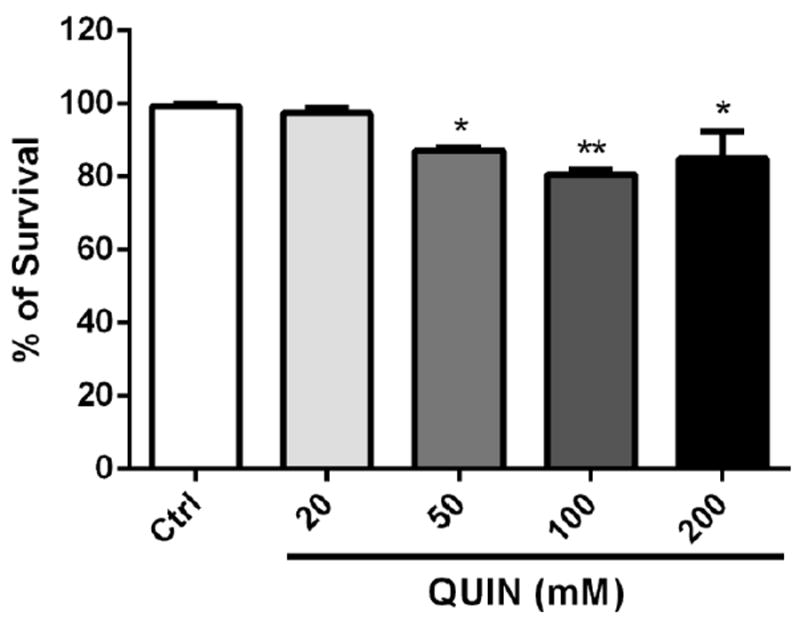
Effects of QUIN on survival assay of wild-type (N2) worms. Data are expressed as percentage of living worms derived from four independent assays of 100 worms per group in each experiment (n=4 experiments per group). Error bars represent as means ± S.E.M. *p<0.01 and **p<0.001 compared to Ctrl (One-Way ANOVA followed by post-hoc Bonferroni).
3.2. QUIN increases ROS generation in an NMR-1R-dependent manner
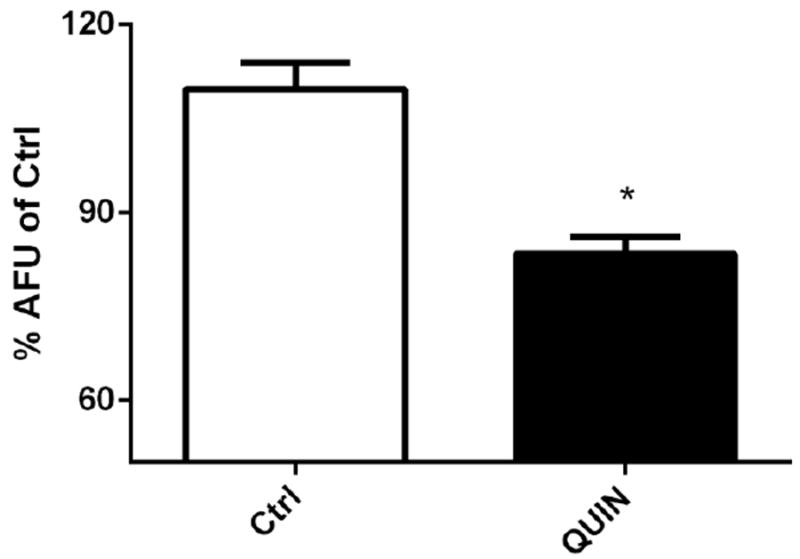
As shown in Fig. 2, wild-type (N2) worms exposed to 20 mM QUIN exhibited a prominent increase in DCF fluorescence (78%), whereas untreated worms exhibited basal fluorescence levels that corresponded to the physiological production of ROS. Upon exposure to 20 mM QUIN, the VM487 transgenic worms had indistinguishable levels of ROS compared to those in the untreated control. Analogous to the wild-type, ROS levels increased in VC2623 knockout worms after exposure to 20 mM QUIN (p < 0.05).
Figure 2.
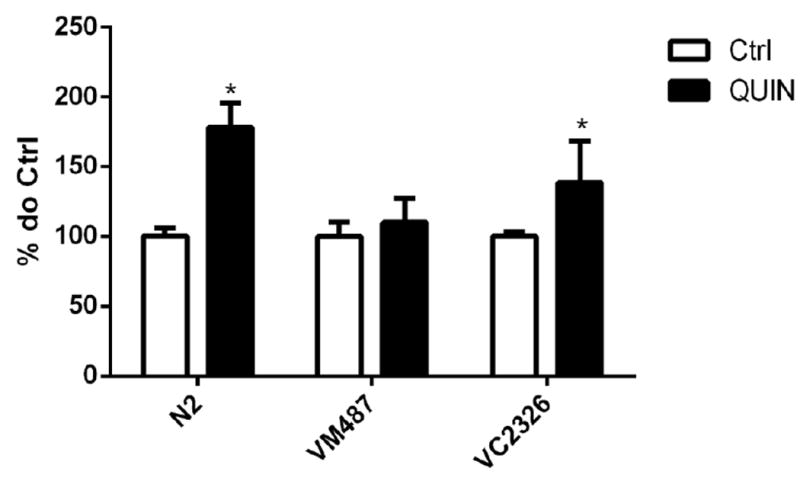
Effect of QUIN on reactive oxygen species (ROS) production. Data are expressed as percentage of the arbitrary fluorescence units (AFU) of Ctrl of each strain derived from 5 independent assays (n=5). Levels of basal ROS production in wild-type (N2), VM487 (nmr-1) and VC2623 (nmr-2) mutants treated or not with the QUIN 20 mM. Error bars represent as means ± S.E.M. *p<0.05 compared to untreated group (Test-t).
3.3. QUIN partially induces activation of DAF-16/FOXO
Figure 3A shows localization of the DAF-16∷GFP protein and the percentage of worms in each category. The DAF-16∷GFP protein was located in the cytoplasm under normal control culture conditions (96.4%). However, the DAF-16∷GFP protein was located entirely in the nucleus during exposure to high temperature (35°C; nuclear localization in 100% of the worms). After the QUIN-treatment, the DAF-16 protein partially migrated to the nucleus (54.2% cytoplasmic localization), and significant DAF-16 activation and migration to the nucleus was observed (p < 0.01, Fig. 3). The DAF-16∷GFP protein was localized in the cytoplasmic, intermediate, and nuclear categories (Fig. 3B).
Figure 3.
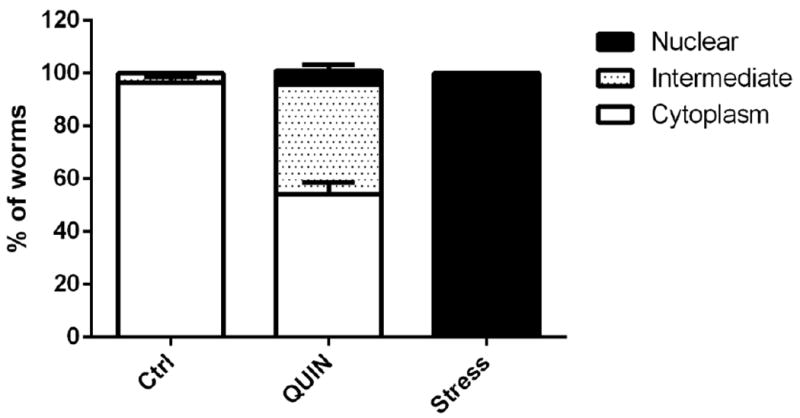
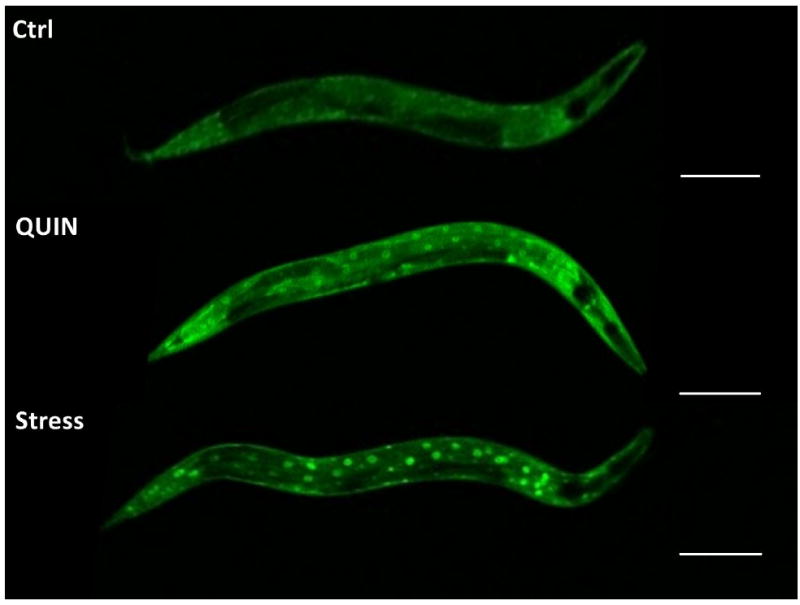
Effect of QUIN in migration of daf-16 protein. A) Effect of QUIN (20 mM) and thermal stress (35° C) on DAF-16 subcellular localization in in the transgenic C. elegans TJ356 (daf-16;;GFP) strain. B) Cytosolic, intermediate and nuclear localization. Data are expressed as percent of worms exhibiting cytosolic, intermediate or nuclear pattern of location, derived from five independent assays with 10 worms per group in each experiment (n=5). Error bars represent as means ± S.E.M (Two-way ANOVA followed by post-hoc Bonferroni).
3.4. QUIN induces SOD-3, GST-4, and HSP-16.2 expression
Animals treated with QUIN showed increased GFP-labeled SOD-3, HSP-16.2/GFP, and GST-4/GFP fluorescence (30.6%, 12.2%, and 46.5% respectively) compared to that in control animals (p < 0.01, Fig. 4A–C).
Figure 4.
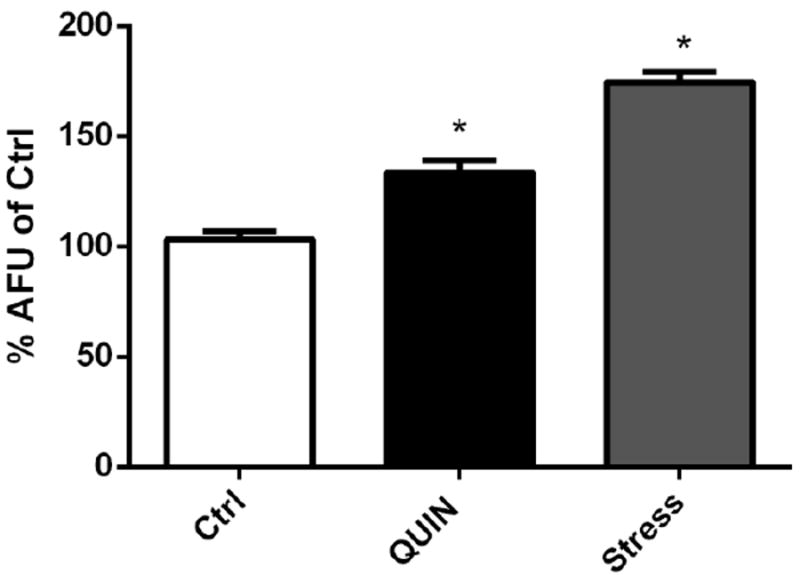
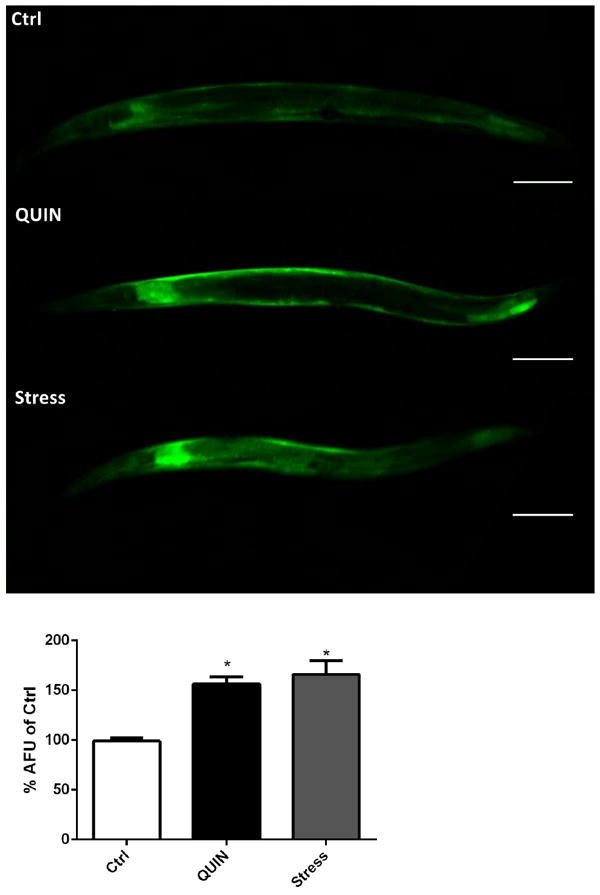
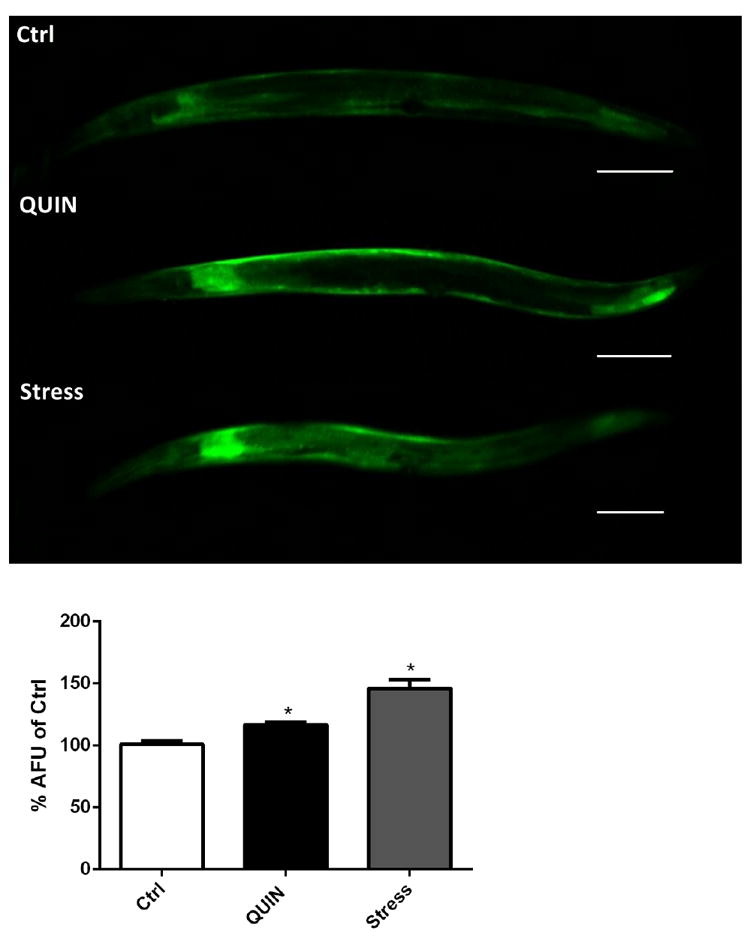
Effect of QUIN (20mM) and thermal stress (35° C) on expression of antioxidant enzymes. (A) Quantification of SOD-3 expression and (B) representative images of the transgenic strain CF1553. (C) Quantification of GST-4 expression and (D) representative images of the transgenic strain CL2166. (E) Quantification of HSP-16.2 expression and (F) representative images of the transgenic strain CL2070. Data are expressed as percentage of the arbitrary fluorescence units (AFU) of control derived from three independent assays with 5 worms per group in each experiment (n=3). Error bars represent as means ± S.E.M. *p<0.01 compared to ctrl untreated group (One-way ANOVA followed by post-hoc Bonferroni).
3.5. QUIN induces behavioral deficits
Significant behavioral deficits in locomotion and pharyngeal contraction were observed in wild-type worms treated with 20 mM QUIN (p < 0.05, 46.4%) compared to that in the control group (Fig. 5A). Knockout animals of the nmr-1 subunit of the ionotropic glutamate receptor NMDA type (VM487) exposed to various concentrations of QUIN demonstrated no significant difference in behavioral phenotype (Fig. 5B). The worms that changed the number of pharyngeal beats and locomotion were grouped for later evaluation because they presented with low survival.
Figure 5.
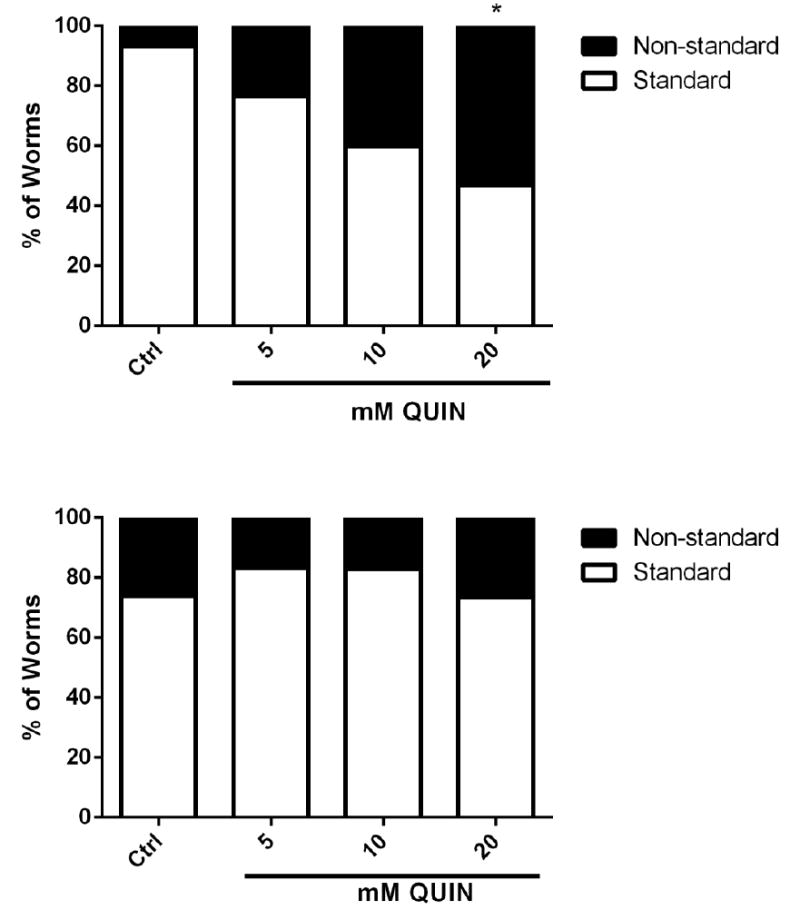
Effect QUIN on behavioral analysis in wild-type (N2) and transgenic VM487(nmr-1) strain. Data are expressed as percent of worms with standard or altered behavior in N2 (A) and VM487 (B) derived from three independent assays with 20 worms per group in each experiment (n=5). *p<0.05 compared to non-standard behavior control (One-way ANOVA, followed by post-hoc Tukey).
The number of pharyngeal beats, duration of defecation cycles, and body bend frequency in the worms that presented standard behavior were not different between the QUIN-treated N2 and VM487 knockout worms (data not shown), compared to those in untreated worms. However, a reduction in the touch response was noted in the N2 and VM487 strains treated with QUIN compared with their respective controls (p < 0.05, Fig. 6A, B).
Figure 6.
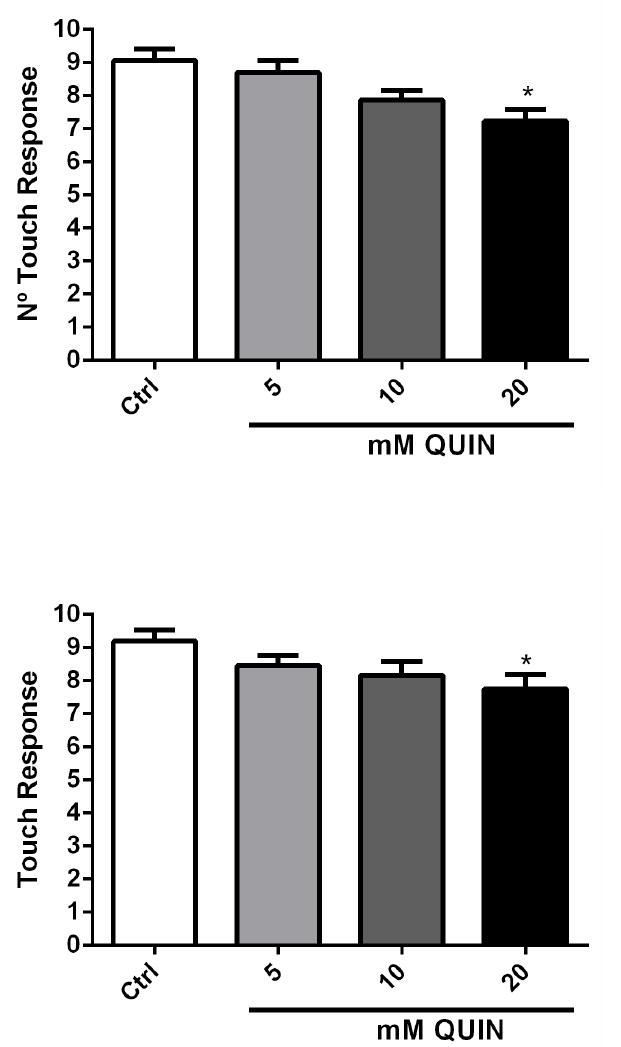
Effect of QUIN on touch response behavior assays in the N2 and VM487 strains. Touch response in N2 (A) and in VM487 (B) worms. Data are derived from three independent assays with 5 worms per group in each experiment (n=15). Error bars represent as means ± S.E.M. *p<0.05 compared to control for each behavior evaluated. (One-way ANOVA followed by post-hoc Bonferroni).
3.6. QUIN induces neurodegeneration
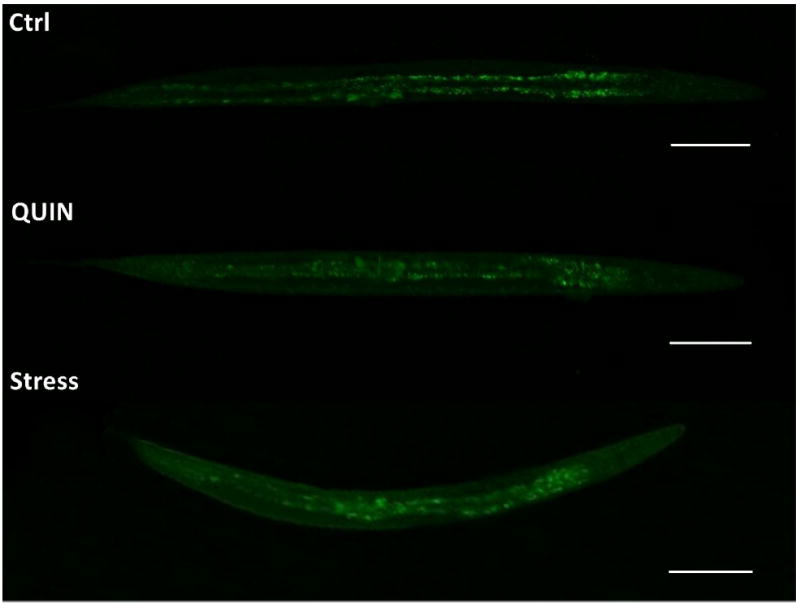
Reduced neuronal fluorescence at the anterior region of the head was observed in 25% of pan-neuronal/GFP animals treated with 20 mM QUIN compared to the mean of all arbitrary fluorescence unit values obtained in the QUIN-untreated control animals. (*p < 0.05, Fig. 7A, B). In contrast, no significant change in fluorescence was observed in the ventral cord neurons (Fig. 7C).
Figure 7.
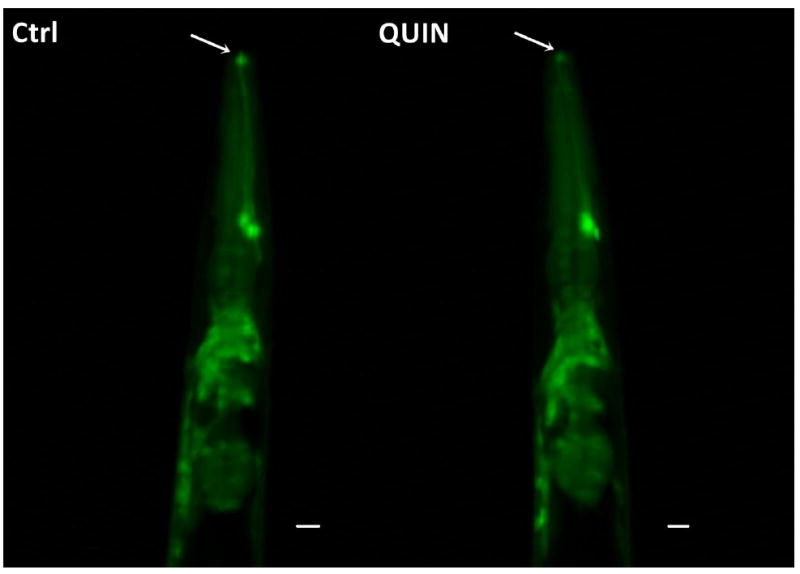
Effect QUIN on neurodegeneration in the OH438 (pan-neuronal∷GFP) strain. The OH438 strain shows all GFP-labeled neurons. A) Quantification of the neurons head fluorescence, indicated neurodegeneration by reducing fluorescence, in the dates expressed as percentage of the arbitrary fluorescence units (AFU) of Ctrl. B) Images the previous region of the worm head. The arrows indicate reduction GFP-fluorescence in neurons. C) GFP-labeled ventral nerve cord without morphological alteration in QUIN-treated worms. Data are derived from three independent assays with 10 worms per group at each experiment (n=30, *p<0.020).
4. Discussion
In this study, we evaluated the adverse effects of the glutamatergic neurotoxin QUIN in the nematode C. elegans. Our data indicate that QUIN adversely affected survival of C. elegans and increased ROS generation in an nmr-1-dependent manner. QUIN caused behavioral disturbances involving the glutamatergic system. In addition, QUIN caused neuron-specific neurodegeneration in C. elegans.
The QUIN treatment resulted in 12.28%, 18.62%, and 21.28% decreased survival of worms exposed to the 50, 100 and 200 mM QUIN doses, respectively (Fig. 1). QUIN is known to exert neurotoxic effects at nanomolar concentrations in rats and humans [3]. However, the response to neurotoxicity-triggering molecules in C. elegans is only visible when the worm is exposed to high concentrations [27, 28]. These worms can absorb compounds through the cuticle, in addition to ingesting them orally. The cuticle acts as a highly impermeable barrier, making it difficult to pass external agents into the animal [29]. It is likely that the lethality associated with QUIN exposure in the worms reflects the nature of their cuticle, as only the highest QUIN concentrations tested decreased survival.
Pathologies related to excitotoxicity often develop by hyperstimulation of glutamatergic membrane receptors, which increase the glutamate concentration in the synaptic cleft [30]. Excitotoxicity, ATP depletion, and oxidative stress are toxic events that commonly lead to neuronal death in several neurodegenerative diseases [31]. The toxic cascades triggered in the glutamatergic system by QUIN involve formation of ROS and nitrogen reactive species, which cause oxidative damage as part of the neurodegenerative process. It is well known that ROS generated by QUIN is due to activation of NMDAR, and that QUIN causes excitotoxicity characteristic of excess activation of the glutamatergic system [3, 9]. In this regard, C. elegans expresses the nmr-1 and nmr-2 subunits of the NMDA-like receptors, which belong to the subfamilies NR1 and NR2A, respectively [17, 18, 32, 33].
Our data demonstrate that wild-type worms treated with 20 mM QUIN presented with increased ROS levels (Fig. 2). This effect has already been demonstrated in different brain regions of rats exposed to QUIN [34]. However, when the QUIN evaluation was performed in nmr-1 transgenic worms, no increase in ROS levels was observed. In contrast, an increase in ROS was also observed in the transgenic nmr-2 treated worms after exposure to 20 mM QUIN, which resembled the levels observed in wild-type animals.
Previous studies in hippocampal cell cultures exposed to Aβ oligomers (inducer of Alzheimer’s disease) also show that increased ROS occurs in a manner dependent on activation of NMDA-type receptors [35], specifically the NR1 receptor homologous to nmr-1 in C. elegans. Thus, our data suggest that QUIN increased ROS levels in C. elegans by stimulating nmr-1/NMDA receptors, which were toxic to the worms. Therefore, C. elegans is an invaluable model to evaluate oxidative stress generated by QUIN, analogous to other mammals. We established that acute treatment with 20 mM QUIN increased ROS generation in wild-type C. elegans dependent on the nmr-1 receptor.
In turn, DAF-16 is responsible for regulating metabolism, resistance to stress, and longevity in C. elegans [36]. The DAF-16/FOXO transcription factor is known to play a key role in oxidative and heat stress resistance, ageing, and other biological functions. DAF-16/FOXO is a target of insulin/insulin-like growth factor signaling in C. elegans, which when reduced or absent, results in dephosphorylated DAF-16/FOXO translocating to the nucleus [37, 38]. We determined whether QUIN was able to affect the location of DAF-16 in the TJ356 transgenic worm strain. We observed partial migration of DAF-16 to the nucleus in C. elegans (Fig. 3). Our findings corroborate the hypothesis that QUIN acts as a pro-oxidant compound. We also observed increased expression of SOD-3, GST-4, and HSP-16.2 (Fig. 4), which are directly related to stress resistance by reducing insulin-like signaling and increasing lifespan [39-42]. These enzymes, which are also transcriptional targets of DAF-16 activation, reflect the stress response when elevated. Thus, our data demonstrate that the generation of ROS triggered by QUIN induced activation of daf-16 and its subsequent translocation to the nucleus and increased transcription of these antioxidant enzymes.
We observed paralysis and the absence of pharyngeal contractions in a large number of the treated worms when evaluating the general behavior of the N2 worms in plates containing QUIN. This effect of QUIN was not observed in the VM487 strain. We tested the standard behavior of worms (N2 and VM487), such as pharyngeal pumping, duration of defecation cycle, body bends, and the nose touch response to evaluate if QUIN altered a more specific glutamatergic behavior. As expected, we showed that QUIN did not change behaviors related with muscular contraction (pharyngeal pumping, duration of defecation cycle, or body bends). This result is not surprising, as the main neurotransmitter that drives these behaviors is acetylcholine ACh [43-45]. In addition, we speculate that the absence of a toxic effect on motor behavior may reflect additional factors, such as the low concentrations of QUIN used and the low density of NMDA receptors in the C. elegans nervous system [46]. In contrast, QUIN was able to reduce the touch response in worms independent of the nmr-1 NMDA receptor. The touch response is mediated by sensory neurons, such as ASH. ASH neurons are located in the lateral ganglia, one in each side of the head where they express glutamate, the neurotransmitter responsible for sensory responses in C. elegans. Although glutamate NMDA receptors are not directly related to the mechanical sensory response, non-NMDA glutamate type receptors, such as glr-1, are mainly responsible for regulating this behavior [18, 47-49]. Thus, changes in the glr-1 receptor result in worms that are defective in the touch response task [46, 50]. Considering these observations, our data suggest that the mechanism of action of the QUIN-induced reduction in the touch response may represent an nmr-1 independent pathway that is not likely caused by an increase in ROS levels.
A classic toxic effect induced by QUIN is glutamate-mediated neurotoxicity, with ensuing neurodegeneration [51]. This occurs once QUIN triggers activation of the NMDA receptor, leading to intracellular increases in Ca2+ levels. The increase in Ca2+ is dependent on NMR-1 hyperactivation and may be caused by QUIN secondary to direct phosphorylation of NMDA receptor subunits via a cAMP/PKA-dependent pathway [51]. Here, we observed reduced GFP fluorescence in neurons of the anterior region of C. elegans treated with QUIN, with no changes detected in the ventral nerve cord (Fig. 7C). As pan-neuronal∷GFP labels a variety of neurons, we evaluated the main region associated with loss of GFP fluorescence. The mechanosensory ASH neurons, as well as interneurons responsible for synodal movement and locomotion against mechanical stimuli (AVA, AVB, AVD, AVE, and PVC) are located in the anterior region of the worm’s head [26]. These neurons predominantly express glutamate [17]. Therefore, we suggest that the neurodegeneration observed upon QUIN exposure was likely associated with the demise of glutamatergic neurons, and may account for the reduced touch response behavior associated with QUIN exposure. Taken together, our findings indicate that QUIN acts on glutamatergic neurons in the anterior region, affecting the touch response in C. elegans by interfering with glutamatergic neurotransmission.
In conclusion, the present study demonstrated the toxicity of a known neurotoxin in worms and corroborated that analogous mechanisms are involved in the neurotoxic effects of QUIN to those inherent in cell cultures and rodents models. Specifically, we found that QUIN caused mortality, changed behavioral patterns, and increased the generation of ROS in a manner consistent with glutamatergic dysfunction in C. elegans. These results suggest that QUIN could be a useful tool for understanding NMDA-associated pathologies or involvement of the glutamatergic system in disorders of the nervous system [52].
Highlights.
Quinolinic Acid induces an increase in oxidative stress in C. elegans mediate by glutamate receptor NMDA
Quinolinic Acid alters behavioral parameters in C. elegan
Quinolinic Acid exposition reduces neuronal viability in the worms head
C. elegans glutamatergic system is affected similarly to that of mammals when exposed to Quinolinic Acid
Acknowledgments
The authors are thankful to Instituto Nacional de Ciência e Tecnologia (INCT) for Excitotoxicity and Neuroprotection—MCT/CNPq, Programa de Apoio a Núcleos Emergentes (PRONEM/FAPERGS) 16/2551-0000248-7 for providing financial assistance. F.A.A.S., and T.L.S., receive a fellowship from CNPq, D.C.Z., M.L.M. and L.P.A. receive a fellowship from CAPES and T.C.S. receive a fellowship from FAPERGS. M.A. was supported in part by grants from NIEHS, R01 ES10563, 1R03 ES024849, ES07331 and R21 ES025415.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans: disease and healthy States. Int J Tryptophan Res. 2009;2:1–19. doi: 10.4137/ijtr.s2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarcz R, et al. Of mice, rats and men: Revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog Neurobiol. 2010;90(2):230–45. doi: 10.1016/j.pneurobio.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, Meininger V, Guillemin GJ. Recent advances in the treatment of amyotrophic lateral sclerosis. Emphasis on kynurenine pathway inhibitors. Cent Nerv Syst Agents Med Chem. 2009;9(1):32–9. doi: 10.2174/187152409787601941. [DOI] [PubMed] [Google Scholar]
- 4.Braidy N, et al. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res. 2009;16(1):77–86. doi: 10.1007/s12640-009-9051-z. [DOI] [PubMed] [Google Scholar]
- 5.Perez-De La Cruz V, Konigsberg M, Santamaria A. Kynurenine pathway and disease: an overview. CNS Neurol Disord Drug Targets. 2007;6(6):398–410. doi: 10.2174/187152707783399229. [DOI] [PubMed] [Google Scholar]
- 6.Guillemin GJ, Brew BJ. Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep. 2002;7(4):199–206. doi: 10.1179/135100002125000550. [DOI] [PubMed] [Google Scholar]
- 7.Finkbeiner S, et al. Disease-modifying pathways in neurodegeneration. J Neurosci. 2006;26(41):10349–57. doi: 10.1523/JNEUROSCI.3829-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarcz R, et al. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13(7):465–77. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-De La Cruz V, Carrillo-Mora P, Santamaria A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res. 2012;5:1–8. doi: 10.4137/IJTR.S8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–13. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 2016;15(1):71. doi: 10.1186/s12937-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tasset I, et al. Protective effect of tert-butylhydroquinone on the quinolinic-acid-induced toxicity in rat striatal slices: role of the Nrf2-antioxidant response element pathway. Neurosignals. 2010;18(1):24–31. doi: 10.1159/000243650. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Kim K, Nelson LS. FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. Brain Res. 1999;848(1-2):26–34. doi: 10.1016/s0006-8993(99)01972-1. [DOI] [PubMed] [Google Scholar]
- 14.Brownlee DJ, Fairweather I. Exploring the neurotransmitter labyrinth in nematodes. Trends Neurosci. 1999;22(1):16–24. doi: 10.1016/s0166-2236(98)01281-8. [DOI] [PubMed] [Google Scholar]
- 15.Nathoo AN, et al. Identification of neuropeptide-like protein gene families in Caenorhabditiselegans and other species. Proc Natl Acad Sci U S A. 2001;98(24):14000–5. doi: 10.1073/pnas.241231298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockie PJ, et al. Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J Neurosci. 2001;21(5):1510–22. doi: 10.1523/JNEUROSCI.21-05-01510.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brockie PJ, Maricq AV. Ionotropic glutamate receptors in Caenorhabditis elegans. Neurosignals. 2003;12(3):108–25. doi: 10.1159/000072159. [DOI] [PubMed] [Google Scholar]
- 18.Brockie PJ, et al. The C. elegans glutamate receptor subunit NMR-1 is required for slow NMDA-activated currents that regulate reversal frequency during locomotion. Neuron. 2001;31(4):617–30. doi: 10.1016/s0896-6273(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 19.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakaue Y, Kim J, Miyamoto Y. Effects of TAT-conjugated platinum nanoparticles on lifespan of mitochondrial electron transport complex I-deficient Caenorhabditis elegans, nuo-1. Int J Nanomedicine. 2010;5:687–95. [PMC free article] [PubMed] [Google Scholar]
- 21.Zamberlan DC, et al. Diphenyl-diselenide suppresses amyloid-beta peptide in Caenorhabditis elegans model of Alzheimer’s disease. Neuroscience. 2014;278:40–50. doi: 10.1016/j.neuroscience.2014.07.068. [DOI] [PubMed] [Google Scholar]
- 22.Chaudhuri J, et al. A Caenorhabditis elegans Model Elucidates a Conserved Role for TRPA1-Nrf Signaling in Reactive alpha-Dicarbonyl Detoxification. Curr Biol. 2016;26(22):3014–3025. doi: 10.1016/j.cub.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang E, Wink M. Chlorophyll enhances oxidative stress tolerance in Caenorhabditis elegans and extends its lifespan. PeerJ. 2016;4:e1879. doi: 10.7717/peerj.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Link CD, et al. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging. 2003;24(3):397–413. doi: 10.1016/s0197-4580(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 25.Huang C, Xiong C, Kornfeld K. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2004;101(21):8084–9. doi: 10.1073/pnas.0400848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chalfie M, et al. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5(4):956–64. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Finley EJ, et al. The Role of skn-1 in methylmercury-induced latent dopaminergic neurodegeneration. Neurochem Res. 2013;38(12):2650–60. doi: 10.1007/s11064-013-1183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Severiano F, et al. S-Allylcysteine, a garlic-derived antioxidant, ameliorates quinolinic acid-induced neurotoxicity and oxidative damage in rats. Neurochem Int. 2004;45(8):1175–83. doi: 10.1016/j.neuint.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Page AP, Johnstone IL. The cuticle. WormBook. 2007:1–15. doi: 10.1895/wormbook.1.138.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin CL, et al. Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med Chem. 2012;4(13):1689–700. doi: 10.4155/fmc.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uttara B, et al. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baidya M, et al. Dopamine modulation of avoidance behavior in Caenorhabditis elegans requires the NMDA receptor NMR-1. PLoS One. 2014;9(8):e102958. doi: 10.1371/journal.pone.0102958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kano T, et al. Memory in Caenorhabditis elegans is mediated by NMDA-type ionotropic glutamate receptors. Curr Biol. 2008;18(13):1010–5. doi: 10.1016/j.cub.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrachinski F, et al. Cooperation of non-effective concentration of glutamatergic system modulators and antioxidant against oxidative stress induced by quinolinic acid. Neurochem Res. 2012;37(9):1993–2003. doi: 10.1007/s11064-012-0820-3. [DOI] [PubMed] [Google Scholar]
- 35.De Felice FG, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282(15):11590–601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 36.DiLoreto R, Murphy CT. The cell biology of aging. Mol Biol Cell. 2015;26(25):4524–31. doi: 10.1091/mbc.E14-06-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120(4):449–60. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Kenyon CJ. The genetics of ageing. Nature. 2010;464(7288):504–12. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 39.Oh SW, et al. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102(12):4494–9. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Blanco A, Kim SK. Variable pathogenicity determines individual lifespan in Caenorhabditis elegans. PLoS Genet. 2011;7(4):e1002047. doi: 10.1371/journal.pgen.1002047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leiers B, et al. A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans. Free Radic Biol Med. 2003;34(11):1405–15. doi: 10.1016/s0891-5849(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 42.Rea SL, et al. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005;37(8):894–8. doi: 10.1038/ng1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winnier AR, et al. UNC-4/UNC-37-dependent repression of motor neuron-specific genes controls synaptic choice in Caenorhabditis elegans. Genes Dev. 1999;13(21):2774–86. doi: 10.1101/gad.13.21.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hallam S, et al. The C. elegans NeuroD homolog cnd-1 functions in multiple aspects of motor neuron fate specification. Development. 2000;127(19):4239–52. doi: 10.1242/dev.127.19.4239. [DOI] [PubMed] [Google Scholar]
- 45.McKay JP, et al. eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics. 2004;166(1):161–9. doi: 10.1534/genetics.166.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maricq AV, et al. Mechanosensory signalling in C. elegans mediated by the GLR-1 glutamate receptor. Nature. 1995;378(6552):78–81. doi: 10.1038/378078a0. [DOI] [PubMed] [Google Scholar]
- 47.Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22(6):2165–73. doi: 10.1523/JNEUROSCI.22-06-02165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kass J, et al. The EGL-3 proprotein convertase regulates mechanosensory responses of Caenorhabditis elegans. J Neurosci. 2001;21(23):9265–72. doi: 10.1523/JNEUROSCI.21-23-09265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mellem JE, et al. Decoding of polymodal sensory stimuli by postsynaptic glutamate receptors in C. elegans. Neuron. 2002;36(5):933–44. doi: 10.1016/s0896-6273(02)01088-7. [DOI] [PubMed] [Google Scholar]
- 50.Hart AC, Sims S, Kaplan JM. Synaptic code for sensory modalities revealed by C. elegans GLR-1 glutamate receptor. Nature. 1995;378(6552):82–5. doi: 10.1038/378082a0. [DOI] [PubMed] [Google Scholar]
- 51.Pierozan P, et al. Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim Biophys Acta. 2016;1863(12):3001–3014. doi: 10.1016/j.bbamcr.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Kotlar I, et al. Comparison of the Toxic Effects of Quinolinic Acid and 3-Nitropropionic Acid in C. elegans: Involvement of the SKN-1 Pathway. Neurotox Res. 2017 doi: 10.1007/s12640-017-9794-x. [DOI] [PubMed] [Google Scholar]