

Abstract
A new four‐step pathway for the synthesis of γ‐halo‐δ‐lactones is described from simple, commercially available substrates: aryl bromides and 3‐methyl crotonaldehyde. The halogenolactonization reaction of β,δ‐substituted, γ,δ‐unsaturated carboxylic acid 4 a–c is regio‐ and stereoselective and gives only the trans‐isomers of lactones 5 a–c, 6 a–c, and 7 a–c. The structures of all synthesized compounds were confirmed by using spectroscopic methods. For bromolactone, containing a naphthyl moiety in the structure, crystallographic analysis was also performed. The lactones were tested for their cytotoxic activity against L929 cell lines (mouse fibroblasts) and antibacterial activity against Escherichia coli strains ATCC 8739 and Staphylococcus aureus ATCC 65389. Compounds 5 a, 5 c, 7 a, and 7 b statistically significantly inhibited the metabolic activity of mouse fibroblasts L929. Compounds 5 b and 6 a were not cytotoxic towards L929 cells, but showed moderate bactericidal properties.
Keywords: antibiotics, cytotoxicity, heterocycles, lactones, synthesis
1. Introduction
Lactones represent an important class of biologically active organic compounds. The most persistent and, thus, the most frequently naturally occurring are γ‐ and δ‐lactones with five‐ and six‐membered rings. Their rich sources are plants. Compounds containing a lactone ring in the structure are the main group of secondary metabolites.1 Lactone derivatives also occur in microorganisms and animals,2 fulfilling a number of different functions. The biological activities, such as cytostatic,3, 4, 5, 6, 7, 8 antibacterial,9, 10, 11, 12 antiviral13, 14, 15, 16, 17 or antifungal18, 19, 20 activities, have applications in medicine. Sensory properties make them useful in the production of cosmetics and in the food industry, where they are responsible for the smell and taste of many products.21 An important property of lactones is the deterrent activity,22, 23, 24, 25, 26, 27 which can be utilized in the production of insect‐control agents.
Owing to the high costs of extracting cyclic esters from natural sources, methods for their preparation through chemical synthesis28, 29, 30, 31, 32, 33, 34 and by using biotechnological methods35, 36, 37, 38, 39 are still being developed. Considering the biological properties, lactones are the focus of many research groups. In this work, we present a new synthesis pathway for halogenolactones and explore their cytotoxicity and bactericidal activity.
2. Results and Discussion
Numerous methods for the preparation and modification of halogenolactones have been reported in the literature.33, 40, 41, 42 In our previous work, we presented the synthesis pathways and cytotoxic properties of iodine and hydroxylactones against the tumor cell lines HL‐60, U‐2 OS and d‐17.40, 43 Continuing this research, we present a new four‐step, regio‐ and stereoselective synthesis of halolactones 5 a–c, 6 a–c, and 7 a–c with aryl substituents. All of the obtained compounds were tested for their cytotoxic activity against cell line L929 (mouse fibroblasts) and antibacterial activity against strains of Escherichia coli ATCC 8739 and Staphylococcus aureus ATCC 65389. The synthesis pathway of new δ‐lactones is shown in Scheme 1. Aryl bromides 1 and 3‐methyl crotonaldehyde was used as substrates.
Scheme 1.
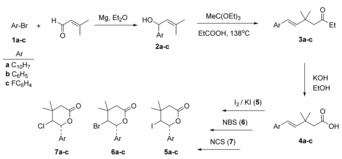
The four‐step synthesis of trans‐γ‐halo‐δ‐lactones 5 a–c, 6 a–c, and 7 a–c. The first step is the Grignard reaction of aryl magnesium bromide with 3‐methyl crotonaldehyde. The second step is a [3,3] sigmatropic rearrangement. The next stage is hydrolysis and the final stage of the synthesis is halolactonization.
The first step of the synthesis was a Grignard reaction between 3‐methyl crotonaldehyde and α‐naphthylene magnesium bromide (a), phenyl magnesium bromide (b), p‐fluorophenyl magnesium bromide (c). In this reaction, we obtained three unsaturated secondary alcohols (2 a–c) with yields of 38–65 %, already described in the literature.44, 45 The structures products 2 a–c were confirmed by using IR and NMR spectroscopy as well as mass spectrometry (HR‐MS). The presence of a hydroxyl group in the obtained alcohols was confirmed by the broad IR bands at 3600–3100 cm−1. In the 13C NMR spectrum, carbon peaks at 68.55 ppm (2 a), 70.75 ppm (2 b), and 70.11 ppm (2 c) correspond to carbon atoms bonded to the hydroxyl group.
New γ,δ‐unsaturated ethyl esters (3 a–c) were obtained with yields of 46–55 % as a result of the Johnson–Claisen rearrangement of alcohols (2 a–c). The [3,3]‐sigmatropic rearrangement reaction was carried out by using triethylorthoacetate in the presence of a catalytic amount of propionic acid. The structures of esters were confirmed by IR and NMR spectroscopies and HR‐MS. The strong IR absorption bands at 1729 cm−1 (3 a), 1730 cm−1 (3 b), and 1730 cm−1 (3 c) and signals in the 13C NMR spectrum at 172.05 ppm (3 a), 171.61 ppm (3 b), and 171.56 ppm (3 c) are characteristic for the carbonyl group. The coupling constants of the olefin protons (15.8 Hz for 3 a, 16.2 Hz for 3 b, and 16.2 Hz for 3 c) confirmed that the E configuration of double bonds remained unchanged. In the next step, esters 3 a–c were hydrolyzed in ethanolic KOH solution to the corresponding γ,δ‐unsaturated carboxylic acids 4 a–c in yields above 90 %. The products did not require further purification. Compounds 4 a and 4 c are new. Compound 4 b is known.46 The structure and configuration of the products 4 a–c were also confirmed by using IR and NMR spectroscopy as well as HR‐MS spectrometry.
The IR bands at 3300–2750 cm−1 are assigned to carboxyl groups. In the 1H NMR spectrum, the carboxylic acid proton signals appear as broad singlets at 11.02 ppm (4 a), 11.32 ppm (4 b), and 10.41 ppm (4 c). The high coupling constants of the olefin protons: 15.8 Hz (4 a), 16.2 Hz (4 b), and 16.2 Hz (4 c) indicate a trans‐configuration of double bond.
The final stage of the synthesis was halolactonization of acids 4 a–c with N‐bromosuccinimide or N‐chlorosuccinimide. The reaction proceeds via either 5‐exo or 6‐endo cyclization. In the case of bromolactonization, the mechanism involves an electrophilic attack of bromine on the double bond, which results in the formation of a bromonium ion. The reaction proceeds via 6‐endo cyclization. The reaction with chlorine takes place in the same way. In the case of iodolactonization, the key step of the reaction mechanism involves nucleophilic attack of the carboxylate ion on the iodine double‐bond complex. The kinetically controlled reaction gives a mixture of γ‐ and δ‐lactones with an excess of five‐membered‐ring lactones. The other important factor for iodolactonization of γ,δ‐unsaturated carboxylic acids is steric hindrance. Electrophilic attack on the γ‐carbon atom is hindered by substituents at the β‐position, which favors 6‐endo cyclization.47 This is also confirmed by our results. The two methyl groups at C‐3 in carboxylic acids 4 a–c prevent the formation of γ‐lactones via 5‐exo cyclization and allows formation of δ‐lactones. This reaction resulted in the selective formation of trans isomers of γ‐iodo‐δ‐lactones 5 a‐‐c, γ‐bromo‐δ‐lactones 6 a–c, and γ‐chloro‐δ‐lactones 7 a–c. All obtained products, except 5 b,46 have not been previously described in the literature. The structure of all products was confirmed by using IR and NMR spectroscopy as well as HR‐MS. The yields of the halolactonization reaction are shown in Table 1.
Table 1.
The yields of the halolactonization reaction.
| Compound | Yield [%] | |||
|---|---|---|---|---|
| 5 a | 93 | |||
| 5 b | 87 | |||
| 5 c | 60 | |||
| 6 a | 62 | |||
| 6 b | 59 | |||
| 6 c | 85 | |||
| 7 a | 75 | |||
| 7 b | 85 | |||
| 7 c | 65 |
Iodolactones 5 a–c were obtained through the reaction of γ,δ‐unsaturated acids 4 a–c with I2/KI and NaHCO3. The products were purified by column chromatography using mixtures of acetone and hexane in volume ratio of 1:10 or 1:15 as the eluent. The reaction yields ranged from 60 to 93 %. The lactones, obtained from the iodolactonization reaction, occur only in one isomeric form.
The stretching vibrations at 1723 cm−1 (5 a), 1723 cm−1 (5 b), and 1720 cm−1 (5 c) as well as the signals in the 13C NMR spectrum at 168.94 ppm (5 a), 168.91 ppm (5 b), and 168.65 ppm (5 c) are characteristic for the carbonyl group in δ‐lactones. The vibrations characteristic for the γ‐lactones appear around 1760–1780 cm−1 and the chemical shifts of the carbonyl carbon in the five‐membered rings are around 176 ppm.40 The 1H NMR spectrum of trans‐5‐iodo‐4,4‐dimethyl‐6‐(α‐naphthyl)tetrahydro‐2H‐pyran‐2‐one (5 a) shows signals from H‐5 and H‐6 protons as two doublets at 4.71 and 6.36 ppm. The high coupling constant (J H5/H6=11.4 Hz) confirms the axial–equatorial disposition of the protons and trans configuration of the lactone. The same configuration is observed for iodolactones 5 b and 5 c.
The reaction of carboxylic acids 4 a–c and N‐bromosuccinimide gives trans‐γ‐bromo‐δ‐lactones 6 a–c with yields in the range 59–85 %. The products were purified by column chromatography using mixtures of acetone and hexane in a volume ratio of 1:10 as the eluent. The six‐membered structure of bromolactone was confirmed by IR absorption bands at 1730–1726 cm−1 and the chemical shifts of the carbonyl carbon around 168 ppm. The high coupling constant (J H5/H6=10.8 Hz) indicates the E configuration of bromolactones. The crystallographic analysis, carried out for lactone 6 a,48 confirmed the trans configuration between the aromatic ring and the bromine atom at C6 and C5 carbon. Figure 1 shows the crystal structure of trans‐5‐bromo‐4,4‐dimethyl‐6‐(α‐naphthyl)tetrahydro‐2H‐pyran‐2‐one (6 a).
Figure 1.
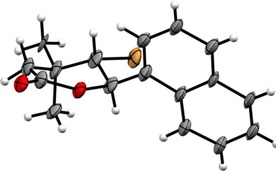
The crystal structure of trans‐5‐bromo‐4,4‐dimethyl‐6‐(α‐naphthyl)tetrahydro‐2H‐pyran‐2‐one (6 a).
In an analogous reaction with N‐chlorosuccinimide, trans‐γ‐chloro‐δ‐lactones 7 a–c were obtained with yields in the range of 65–85 %. The products were purified by column chromatography using mixtures of acetone and hexane in volume ratio of 1:10 as the eluent. The structures of all products were confirmed by using IR and NMR spectroscopy as well as HR‐MS. The structures of the δ‐lactones were confirmed by characteristic bands in the IR spectra at 1730 cm−1 (7 a), 1733 cm−1 (7 b), and 1730 cm−1 (7 c). The chemical shifts of the carbonyl carbon at 168.56 ppm (7 a), 168.35 ppm (7 b), and 168.17 ppm (7 c) also confirmed formation of the 6‐endo cyclization products. The signals of protons at C5 and C6 appear as two doublets at 4.37 and 6.06 ppm (7 a), 3.97 and 5.25 ppm (7 b), 3.92 and 5.24 ppm (7 c). The high coupling constant (J H5/H6=10.4 Hz) indicates the E configuration of lactones.
2.1. The MTT Reduction Effectiveness
The synthesized γ‐halogeno‐δ‐lactones were tested for their cytotoxic activity against the cell lines L929 (mouse fibroblasts). Statistically significant inhibition of metabolic activity of L929 fibroblasts was observed for lactones 5 a, 5 c, 7 a, and 7 b in the range of 50–0.1 μg mL−1. In the presence of these compounds, 37–93 % of the cells were not able to convert MTT [(3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide)] into formazan (Table 2). The ability to convert MTT loses 24–70 % of the cells when lactones 7 c, 5 b, and 6 c were used in a concentration range of 50–5 μg mL−1. Lactones 6 b c were not cytotoxic only at 0.5 μg mL−1, whereas 6 a did not cause a significant decrease of metabolic activity of the cells at a concentration of 10 μg mL−1 (Table 2).
Table 2.
Cytotoxic effect of investigated lactones against L929 cells.[a]
| Concentration [μg/mL] | Controls | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 50 | 20 | 10 | 5 | 0.5 | C+ | C‐ | DMSO | ||
| Viable cells [%] | IC50 [μg/mL] | ||||||||
| 5 a | 7*±0.010 | 16*±0.018 | 26*±0.011 | 32*±0.015 | 39*±0.018 | 0.39 | |||
| 5 b | 16*±0.015 | 34*±0.014 | 54*±0.011 | 60*±0.010 | 81±0.018 | 9.26 | |||
| 5 c | 15*±0.02 | 29*±0.019 | 32*±0.010 | 45*±0.010 | 54*±0.017 | 0.46 | |||
| 6 a | 45*±0.011 | 55*±0.016 | 74±0.015 | 98±0.010 | 99±0.014 | 18 | |||
| 6 b | 34*±0.011 | 39*±0.010 | 45*±0.018 | 53*±0.012 | 66±0.015 | 100±0.015 | 0 | 79±0.016 | 4.7 |
| 6 c | 14*±0.012 | 28*±0.011 | 50*±0.019 | 67*±0.017 | 70±0.012 | 10 | |||
| 7 a | 14*±0.013 | 34*±0.017 | 38*±0.014 | 45*±0.019 | 51*±0.012 | 0.49 | |||
| 7 b | 26*±0.011 | 34*±0.016 | 45*±0.017 | 53*±0.019 | 63*±0.014 | 4.7 | |||
| 7 c | 30*±0.015 | 36*±0.013 | 54*±0.015 | 66*±0.019 | 75±0.015 | 9.26 | |||
[a] The cytotoxicity was assessed by the MTT reduction assay. The cell viability was calculated for four experiments, including three repeats for each compound. Complete RMPI medium (cRPMI) was used as a positive control (C+) of cell viability (100 % viable cells) and 0.03 % H2O2 as a negative control (C−) of cell viability (100 % dead inactive cells). All values were expressed as the mean ± SD. The differences positive control and tested compounds were tested the non‐parametric Mann‐Whitney U test. Statistical significance: *p<0.05.
The cytotoxicity of new compounds against eukaryotic cells may limit their medical applications related to the potential antimicrobial activity. In our study, the lowest IC50 values were exhibited by lactones 6 a, 5 b, and 7 c. However, the high IC50 marker can help to select highly cytotoxic compounds, which can be screened toward cancer cells. Gładkowski et al.49 showed the cytotoxic effect of novel racemic iodo‐, bromo‐ and chlorolactones with β‐phenyl‐γ‐lactone or β‐phenyl‐δ‐lactone frameworks. The highest activity versus carboplatin was shown for cis‐5‐(1‐iodoethyl)‐4‐(4′‐isopropylphenyl)dihydrofuran‐2‐one towards Jurkat cell line (human leukemia). New lactones have also been estimated in terms of their antifeedant activity toward the granary weevil beetle (Sitophilus granarius L.), the khapra beetle (Trogoderma granarium Everts) and the confused flour beetle (Tribolium confusum Du Val.).50 These studies have shown the wide application possibilities of new lactones.
2.2. Antibacterial Properties
The effect of synthesized lactones on growth of Escherichia coli ATCC 8739 and Staphylococcus aureusin ATCC 65389 was tested. Antibacterial properties of the compounds were tested for the concentration 50–0.1 μg mL−1 of lactones. After 18 h incubation, an increase of turbidity and bacterial sediment were observed in all examined cases, with the exception of sterility control. In the next stage of the study, the number of living bacterial cells in the mixtures was calculated. The tests were carried out for the highest concentration of investigated lactones (50 μg mL−1). The highest bactericidal activity against both E. coli ATCC 8739 and S. aureus ATCC 65389 was found in the case of lactone 6 a. The average number of colony‐forming units (CFUs) decreased by about 50 % compared to the number of CFUs in the DMSO control mixture. Compounds 5 b, 6 b, and 7 a exhibited moderate bactericidal properties. The bactericidal effect of the investigated lactones against E. coli and S. aureus is shown in Table 3.
Table 3.
The bactericidal effect of the investigated compounds towards Escherichia coli ATCC 8739 and Staphylococcus aureus ATCC 65389 in concentration 50 μg/mL.
| Compounds | E. coli ATCC 8739 | S. aureus ATCC 65389 |
|---|---|---|
| [number of CFU/mL (×107)] | ||
| 5 a | 6.30 | 7.24 |
| 5 b | 4.74 | 5.44 |
| 5 c | 6.30 | 9.02 |
| 6 a | 3.38 | 3.11 |
| 6 b | 5.56 | 6.10 |
| 6 c | 6.10 | 8.83 |
| 7 a | 5.84 | 7.26 |
| 7 b | 5.83 | 9.04 |
| 7 c | 6.14 | 9,07 |
| Positive control | 6.92 | 9.19 |
| Negative control | 0.00 | 0.00 |
| DMSO | 6.61 | 9.08 |
The antibacterial activity of other synthetic lactones has also been described.50 Lactones without a halogen atom were effective towards S. auresus and Listeria momocytogenes. Antimicrobial activity has also been reported for halolactones and hydroxylactone obtained through biotransformation.51 These compounds inhibit growth of different bacterial species (e.g. E. coli, S. aureus, B. subtilis), yeast (e.g. Candida albicans and Saccharomyces cerevisiae), as well as fungi (e.g. Fusarium linii and Aspergillus niger). Recently, promising antibacterial properties was also shown for bicyclic lactones with three or four methyl groups.39 Halolactones were also considered as potential inhibitors of HIV‐1 non‐nucleoside reverse transcriptase.52
3. Conclusions
This paper describes the regio‐ and stereoselective synthesis of γ‐halogeno‐δ‐lactones 5 a–c, 6 a–c, and 7 a–c from commercially available reagents. The structure of all products was confirmed by using IR and NMR spectroscopy as well as HR‐MS. Crystallographic analysis was carried out for lactone 5 a. All synthesized γ‐halogeno‐δ‐lactones were tested for cytotoxic activity against the cell lines L929. Statistically significant inhibition of metabolic activity of L929 fibroblasts was observed for lactones 5 a, 5 c, 7 a, and 7 b. The antimicrobial activity of obtained lactones against the strains Escherichia coli ATCC 8739 and Staphylococcus aureus ATCC 65389 was also checked. Lactones 5 b and 6 a showed moderate antibacterial activity. Investigated lactones have antibacterial potential. However, it is necessary to carefully control and exclude their cytotoxic activity against eukaryotic cells in the case of future application perspective.
Experimental Section
General Methods
The reagents used for the synthesis were purchased from Fluka and Aldrich. The 1H and 13C NMR spectra were measured by an NMR Bruker Avance DRX 500 MHz using chloroform‐d as the solvent. IR spectra were recorded on a PerkinElmer Spectrum 400 spectrometer. The ESI mass spectra were recorded on a Bruker micrOTOF‐Q II equipped with syringe pump. Gas chromatography was performed on a Thermo Scientific‐Trace 1310 chromatograph equipped with a TG‐5HT column (30 m × 0.25 mm). The gas chromatograph was temperature programmed from 150 to 300 °C at a rate of 10 °C min−1. Melting points were determined with a Boetius micro melting point apparatus and were uncorrected. The refractive index was determined with Atago RX‐7000CX refractometer. TLC analysis was performed on silica gel 60 F254 plates (Merck). Chromatographic separations were carried out by using silica gel 60 (particle size 0.040–0.063 mm, Merck). Column chromatography was performed on silica gel (Kieselgel 60, 230–400 mesh, Merck) using mixtures of acetone, hexane, and ethyl acetate as eluents. The crystal structure was determined by single‐crystal X‐ray diffraction (Xcalibur, Mo‐Kα, Sapphire2 detector) and deposited with the Cambridge Crystallographic Data Centers CCDC No. 1841378.48
Grignard Reaction
The synthesis of unsaturated secondary alcohols 2 a‐c was carried out following the procedure described by Gaylord and Becker.53 A solution of aryl bromide (0.08 mol) in diethyl ether was added dropwise to magnesium turnings (0.08 mol) and heated under reflux for 4 hours. The mixture was cooled on ice and a solution of 3‐methyl crotonaldehyde in diethyl ether was added dropwise and stirred for 24 h. The mixture was dissolved in diethyl ether and added to ice and water. The separated organic layer was dried over anhydrous magnesium sulfate and evaporated under vacuum. The crude products was purified by column chromatography on silica gel [ethyl acetate/hexane, 1:8 (v/v)]. The spectral data are given below.
Compound 2 a: 3‐Methyl‐1‐(α‐naphthyl)but‐2‐en‐1‐ol
Yield 38 %, yellow oil, n20 D=1.5572; R f 0.24 (EtOAc : hexane,1:8), 1H NMR (CDCl3, 500 MHz) δ [ppm]: 1.73 (d, J=1.3 Hz, CH 3, 3 H),1.92 (d, J=1.3 Hz, CH3, 3 H), 2.87 (s, OH,1 H), 5.53 (m, CH=C(CH3)2, 1 H), 6.10 (dd, J=8.6 Hz, J=3.9 Hz, CH(OH), 1 H), 7.46–7.52 (m, HAr, 3 H), 7.75 (d, J=7.1, HAr, 1 H), 7.80 (d, J=8.2, HAr, 1 H), 7.88 (d, J=7.8, HAr, 1 H), 8.23 (d, J=8.4, HAr, 1 H). 13C NMR (125 MHz, CDCl3) δ [ppm]: 17.48, 25.02, 67.81, 123.19, 124.33, 125.27, 125.41, 125.45, 127.29, 128.52, 128.98, 130.86, 133.43, 134.05, 141.25. IR 3590‐3125, 1660, 1598, 1506, 1450, 1370, 1160, 1135, 1045, 970, 792, 770 cm−1. HR‐MS (ESI‐TOF) calculated for C15H16O, m/z [M+Na]+: 235.1098786; experimental value: 235.111019.
Compound 2 b: 3‐Methyl‐1‐phenylbut‐2‐en‐1‐ol45
Yield 42 %, yellow oil, n20 D=1.5543; R f 0.27 (EtOAc : hexane, 1:8), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.75 (d, J=1.2 Hz, CH 3, 3 H),1.80 (d, J=1.2 Hz, CH 3, 3 H), 5.43–5.39 (m, CH=C(CH3)2, 1 H), 5.84–5.93 (m, CH(OH), 1 H), 7.27–7.36 (m, HAr, 5 H). 13C NMR (125 MHz, CDCl3), δ [ppm]: 18.30, 25.83, 70.75, 126.39, 127.26, 128.45, 128.54, 137.51, 144.24. IR: 3600‐3100, 1667, 1495, 1450, 1376, 1200, 1030, 1003, 863, 756, 747, 699 cm−1. HR‐MS (ESI‐TOF) calculated for C11H14O, m/z [M+Na]+: 185.0942294; experimental value: 185.094086.
Compound 2 c: 1‐(p‐Fluorophenyl)‐3‐methylbut‐2‐en‐1‐ol44
Yield 65 %, yellow oil, n20 D=1.5266; R f 0.25 (EtOAc : hexane, 1:8), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.76 (d, J=1.2 Hz, CH 3, 3 H), 1.80 (d, J=1.2 Hz, CH 3, 3 H), 5.35–5.40 (m, CH=C(CH3)2, 1 H), 5.45 (d, J=8.7 Hz, CH(OH), 1 H), 6.94–7.07 (m, HAr, 2 H), 7.28–7.43 (m, HAr, 2 H). 13C NMR (125 MHz, CDCl3) δ [ppm]: 18.26, 25.81, 70.20, 115.22, 127.74, 127.80, 133.53, 139.04, 162.15. IR: 3623‐3087, 1605, 1501, 1459, 1370, 1221, 1150, 970, 855, 816 cm−1. HR‐MS (ESI‐TOF) calculated for C11H13FO, m/z [M+Na]+: 203.084808; experimental value: 203.085423.
Claisen Rearrangement
γ,δ‐Unsaturated ethyl esters 3 a–c were obtained from a Claisen rearrangement.54 A mixture of the alcohol (0.02 mol) and triethyl orthoacetate (0.15 mol) with a drop of propionic acid was heated at 138 °C for 5 h while distilling ethanol. When the reaction was completed (as monitored by TLC), the excess triethyl orthoacetate was distilled off and the crude product was purified by column chromatography on silica gel [ethyl acetate/hexane 1:80 (v/v)]. Spectral data are given below.
Compound 3 a: Ethyl 3,3‐Dimethyl‐5‐(α‐naphthyl)pen‐4‐enoate
Yield 54 %, yellow oil, n20 D=1.5753; R f 0.46 (EtOAc : hexane, 1:20), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.17 (t, J=7.2 Hz, OCH2CH 3, 3 H), 1.38 (s, C(CH 3)2, 6 H), 2.50 (s, CH 2COOCH2CH3, 2 H) 4.16 (q, J=7.2 Hz, OCH 2CH3, 2 H), 6.33 (d, J=15.8 Hz, CH=CH, 1 H), 7.11 (d, J=15.8 Hz, Ar‐CH=CH, 1 H), 7.43–7.46 (m, HAr, 1 H), 7.47–7.53 (m, HAr, 2 H), 7.56 (d, J=8.1 Hz, HAr, 1 H), 7.76 (d, J=8.1 Hz, HAr, 1 H), 7.85 (d, J=8.1 Hz, HAr, 1 H), 8.11 (d, J=8.1 Hz, HAr, 1 H). 13C NMR (125 Hz, CDCl3) δ [ppm]: 14.34, 27.60, 36.37, 47.40, 60.15, 123.57, 123.80, 123.95, 125.68, 125.84, 127.43, 128.48, 128.98, 131.32, 133.62, 135.67, 142.27, 171.72. IR: 1729, 1507, 1394, 1367, 1235, 1120, 1035, 970, 789, 777 cm−1. HR‐MS (ESI‐TOF) calculated for C18H20O2, m/z [M+Na]+: 291.136092; experimental value: 291.136897.
Compound 3 b: Ethyl 3,3‐Dimethyl‐5‐phenylpent‐4‐enoate
Yield 46 %, yellow oil, n20 D=1.5201; R f 0.51 (EtOAc : hexane, 1:20), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.21 (t, J=7.1 Hz, OCH2CH 3, 3 H) 1.25 (s, C(CH 3)2, 6 H), 2.38(s, CH 2COOCH2CH3, 6 H), 4.09 (q, J=7.1 Hz, OCH 2CH3, 2 H), 6.30 (d, J=16.2 Hz, CH=CHC(CH3)2, 1 H), 6.34 (d, J=16.2 Hz, CH=CHC(CH3)2, 1 H), 7.19 (t, J=7.3 Hz, HAr, 1 H), 7.29 (t, J=7.7 Hz, HAr, 2 H), 7.35 (d, J=7.2 Hz, HAr, 2 H). 13C NMR (125 Hz, CDCl3) δ [ppm]: 14.29, 27.39, 29.26, 47.29, 60.01, 126.10, 126.14, 126.99, 128.45, 137.62, 138.80, 171.61. IR: 1730, 1463, 1447, 1360, 1230, 1200, 1150, 1120, 1030, 960, 750, 960 cm−1. HR‐MS (ESI‐TOF) calculated for C15H20O2, m/z [M+Na]+: 255.136092; experimental value: 255.137098.
Compound 3 c: Ethyl 5‐(p‐Fluorophenyl)‐3,3‐dimethylpent‐4‐enoate
Yield 55 %, yellow oil, n20 D=1.5103; R f 0.49 (EtOAc : hexane, 1:20), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.20 (t, J=7.1 Hz, OCH2CH 3, 3 H), 1.24 (s, C(CH 3)2, 6 H), 2.37 (s, CH 2CO2CH2CH3, 2 H), 4.09 (q, J=7.1 Hz, OCH 2CH3, 2 H), 6.22 (d, J=16.2 Hz, CH=CHC(CH3)2, 1 H), 6.30 (d, J=16.2 Hz, CH=CHC(CH3)2, 1 H), 6.98 (t, J=8.7 Hz, HAr, 2 H), 7.28–7.34 (m, HAr, 2 H). 13C NMR (125 Hz, CDC3), δ [ppm]: 14.28, 27.39, 35.81, 47.29, 60.02, 115.29, 124.98, 127.54, 138.54, 138.55, 161.99, 171.56. IR: 1730, 1505, 1365, 1220, 1160, 1030, 973, 850, 815 cm−1. HR‐MS (ESI‐TOF) calculated for C15H19FO2, m/z [M+Na]+: 273.1266706; experimental value: 273.127710.
Basic Hydrolysis
γ,δ‐Unsaturated carboxylic acids were obtained by carrying out alkaline hydrolysis of esters 3 a–c. The ester was dissolved in ethanolic KOH and heated for 2 h under reflux. When the reaction was complete, the ethanol was evaporated and the mixture was acidified with 0.1 m hydrochloric acid. The product was extracted with diethyl ether and dried over anhydrous magnesium sulfate. The product did not require purification. Spectral data are given below.
Compound 4 a: 3,3‐Dimethyl‐5‐(α‐naphthyl)pent‐4‐enoic Acid
Yield 97 %, yellow oil, n20 D=1.5830; R f 0.24 (EtOAc : hexane, 1:5), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.37 (s, C(CH 3)2, 6 H),2.52 (s, CH 2COOH, 2 H), 6.29 (d, J=15.8 Hz, ArCH=CH, 1 H), 7.10 (d, J=15.8 Hz, ArCH=CH, 1 H), 7.38–7.53 (m, HAr, 4 H), 7.73 (d, J=8.1 Hz, HAr, 1 H), 7.82 (dd, J=7.42 Hz, J=2.0 Hz, HAr, 1 H), 8.07 (d, J=7.9 Hz, HAr, 1 H), 11.02 (s, COOH, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 27.54, 36.16, 46.90, 123.84, 123.92, 124.00, 125.64, 125.82, 127.45, 127.76, 128.42, 131.28, 133.53, 135.55, 141.76, 177.51. IR: 3300‐2745, 1701, 1507, 1394, 1288, 1168, 1115, 968, 794, 778 cm−1. HR‐MS (ESI‐TOF) calculated for C17H18O2, m/z [M+Na]+: 277.1204428; experimental value: 277.127429.
Compound 4 b: 3,3‐Dimethyl‐5‐phenylpent‐4‐enoic Acid46
Yield 95 %, yellow oil, n20 D=1.5830; R f 0.33 (EtOAc : hexane, 1:5), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.26 (s, C(CH 3)2, 6 H), 2.43 (s, CH 2COOH, 2 H), 6.30 (d, J=16.2 Hz, ArCH=CH, 1 H), 6.32 (d, J=16.2 Hz, ArCH=CH, 1 H), 7.18–7.22 (m, HAr, 1 H), 7.18–7.22 (m, HAr, 1 H), 7.34–7.36 (m, HAr, 2 H), 11.32 (s, COOH, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 27.36, 35.67, 46.88, 126.22, 126.44, 127.12, 128.50, 137.52, 138.42, 177.77. IR: 3280‐2760, 1697, 1488, 1445, 1409, 1318, 1270, 965, 750, 670 cm−1. HR‐MS (ESI‐TOF) calculated for C13H16O2, m/z [M+Na]+: 227.1047936; experimental value: 227.103997.
Compound 4 c: 5‐(p‐Fluorophenyl)‐3,3‐dimethylpent‐4‐enoic Acid
Yield 97 %, yellow oil, n20 D=1.5205; R f 0.30 (EtOAc : hexane, 1:5), 1H NMR (CDCl3, 500 MHz), δ [ppm]:1.26 (s, C(CH 3)2, 6 H), 2.42 (s, CH 2COOH, 2 H), 6.22 (d, J=16.2 Hz, ArCH=CH, 1 H), 6.32 (d, J=16.2 Hz, ArCH=CH, 1 H), 6.93–7.02 (m, HAr, 1 H), 7.28–7.34 (m, HAr, 2 H), 10.41 (s, COOH, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 27.36, 35.63, 46.78, 115.32, 125.34, 127.63, 133.61, 138.10, 177.09, 162.05. IR: 3280‐2780, 1697, 1506, 1315, 1275, 1230, 1160, 967, 811 cm−1. HR‐MS (ESI‐TOF) calculated for C13H15FO2, m/z [M+Na]+: 245.0953722; experimental value: 245.096365.
Procedure of Iodolactonization
Iodolactones 5 a–c were prepared according to the procedure described in Ref. 55. The mixed solution of the above‐mentioned acid in diethyl ether and saturated aqueous NaHCO3 was stirred for 30 min. A solution of iodine in potassium iodide was added dropwise and the reaction mixture was heated under reflux for 24 h. When the reaction was complete, the mixture was dissolved in diethyl ether and washed with saturated Na2S2O3 solution. The organic layer was washed with saturated NaHCO3 solution and dried over anhydrous magnesium sulfate. The crude products were purified by column chromatography on silica gel [acetone/hexane, 1:15 (v/v)]. Spectral data are given below.
Compound 5 a: trans‐5‐Iodo‐4,4‐dimethyl‐6‐(α‐naphthly)tetrahydro‐2H‐pyran‐2‐one
Yield 93 %, yellow crystals, m.p.=118–119 °C, R f 0.16 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.26 (s, C(CH 3)2, 3 H), 1.40 (s, C(CH 3)2, 3 H), 2.78 (d, J=17.3 Hz, CH 2, 1 H), 2.96 (d, J=17.3 Hz, CH 2, 1 H), 4.71 (d, J=11.4 Hz, CHI, 1 H), 6.36 (d, J=11.4 Hz, CHAr, 1 H), 7.48–7.55 (m, HAr, 2 H), 7.56–7.61 (m, HAr, 2 H), 7.90 (d, J=8.2 Hz, HAr, 2 H), 8.10 (d, J=8.6 Hz, HAr, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 23.60, 32.58, 35.75, 42.59, 43.57, 77.23, 122.95, 124.98, 125.90, 125.95, 126.60, 129.16, 130.24, 131.24, 133.13, 133.92, 168.94. IR: 1723 (carbonyl band), 1352, 1235, 1155, 1020, 1002, 800, 775, 735, 650, 620, 610 cm−1. HR‐MS (ESI‐TOF) calculated for C17H17IO2, m/z [M+Na]+: 403.0170952; experimental value: 403.017215.
Compound 5 b: trans‐5‐Iodo‐4,4‐dimethyl‐6‐phenylo‐tetra‐ hydro‐2H‐pyran‐2‐one46
Yield 87 %, yellow crystals, m.p.=72–73 °C, R f 0.19 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.21 (s, C(CH 3)2, 3 H), 1.29 (s, C(CH 3)2, 3 H), 2.65 (d, J=17.3 Hz, CH 2, 1 H), 2.87 (d, J=17.3 Hz, CH 2, 1 H), 4.28 (d, J=11.3 Hz, CHBr, 1 H), 5.56 (d, J=11.3 Hz, CHAr, 1 H), 7.34–7.48 (m, HAr, 5 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 23.28, 32.57, 35.44, 42.38, 44.97, 84.98, 127.78, 128.53, 129.40, 137.75, 168.91. IR: 1723 (carbonyl band), 1460, 1350, 1240, 1160, 1070, 1005, 925, 851, 750, 700, 620, 609 cm−1. HR‐MS (ESI‐TOF) calculated for C13H15IO2, m/z [M+Na]+: 353.003446; experimental value: 353.004945.
Compound 5 c: trans‐6‐(p‐Fluorophenyl)‐5‐iodo‐4,4‐dimethyltetrahydro‐2H‐pyran‐2‐one
Yield 60 %, yellow crystals, m.p.=81–82 °C, R f 0.18 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz) δ [ppm]: 1.20 (s, C(CH 3)2, 3 H), 1.28 (s, C(CH 3)2, 3 H), 2.64 (d, J=17.3 Hz, CH 2, 1 H), 2.76 (d, J=17.3 Hz, CH 2, 1 H), 4.22 (d, J=11.4 Hz, CHI, 1 H), 5.54 (d, J=11.4 Hz, CHAr, 1 H), 7.04–7.14 (m, HAr, 2 H), 7.32–7.35 (m, HAr, 2 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 23.26, 32.56, 35.43, 42.31, 45.01, 84.16, 115.54, 129.59, 133.77, 163.09, 168.65. IR: 1720 (carbonyl band), 1605, 1515, 1368, 1342, 1300, 1220, 1154, 1078, 1009, 904, 850, 805, 615 cm−1. HR‐MS (ESI‐TOF) calculated for C13H14FIO2, m/z [M+Na]+: 370.9920246; experimental value: 371.001004.
Procedure of Bromolactonization
Bromolactones 6 a–c were obtained according to the procedure described in Ref. 50. The mixture of the above‐mentioned acid (0.005 mol) and N‐bromosuccinimide (0.01 mol) was dissolved in 60 mL of tetrahydrofuran (THF). Acetic acid was added dropwise and mixture was stirred at room temperature for 48 h. When the reaction was complete, the mixture was dissolved in diethyl ether, washed with saturated NaHCO3 solution, and dried over anhydrous magnesium sulfate. The crude products were purified by column chromatography on silica gel [acetone/hexane, 1:10 (v/v)]. Spectral data are given below.
Compound 6 a: trans‐5‐Bromo‐4,4‐dimethyl‐6‐(α‐naphthyl)‐ tetrahydro‐2H‐pyran‐2‐one
Yield 62 %, colorless crystals, m.p.=197–198 °C, R f 0.20 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.29 (s, C(CH 3)2, 3 H), 1.42 (s, C(CH 3)2, 3 H), 2.76 (d, J=17.4 Hz, CH 2, 1 H), 2.94 (d, J=17.4 Hz, CH 2, 1 H), 4.56 (d, J=10.7 Hz, CHBr, 1 H), 6.25 (d, J=10.7 Hz, CHAr, 1 H), 7.47–7.53 (m, HAr, 2 H), 7.53–7.59 (m, HAr, 2 H), 7.89 (d, J=8.2 Hz, HAr, 2 H), 8.08 (d, J=8.6 Hz, HAr, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 21.74, 29.68, 35.65, 44.18, 60.51, 79.69, 122.93, 125.00, 125.85, 125.95, 126.60, 129.12, 130.12, 131.22, 132.53, 133.88, 168.59. IR: 1730 (carbonyl band), 1460, 1355, 1240, 1160, 1100, 1020, 1007, 908, 802, 780, 740, 660, 630 cm−1. HR‐MS (ESI‐TOF) calculated for C17H17BrO2, m/z [M+K]+: 371.0048922; experimental value: 371.004088.
Crystal Data for 6 a
C17H17BrO2, M=333.21, colorless block, orthorhombic, space group P212121, a=6.2726(10) Å, b=7.6137(16) Å, c=29.901(4) Å, β=90°,V=1428.0(4) Å3, Z=4, ρcald=1.5497, Mgm−3, T=298(2) K, R=0.1065, wR=0.2092 [2893 reflections with I>2 sigma(I)] for 181 variables).48
Compound 6 b: trans‐5‐Bromo‐4,4‐dimethyl‐6‐phenyl‐tetra‐ hydro‐2H‐pyran‐2‐one
Yield 59 %, colorless crystals, m.p.=107–108 °C, R f 0.25 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.20 (s, C(CH 3)2, 3 H), 1.28 (s, C(CH 3)2, 3 H), 2.61 (d, J=17.4 Hz, CH 2, 1 H), 2.81 (d, J=17.4 Hz, CH 2, 1 H), 4.11 (d, J=10.8 Hz, CHBr, 1 H), 5.40 (d, J=10.8 Hz, CHAr, 1 H), 7.35–7.42 (m, HAr, 5 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 21.40, 29.64, 35.53, 44.02, 61.33, 83.39, 168.40, 127.59, 128.50, 129.25, 137.19. IR: 1730 (carbonyl band), 1462, 1340, 1235, 1180, 1017, 923, 860, 806, 755, 670, 640 cm−1. HR‐MS (ESI‐TOF) calculated for C13H15BrO2, m/z [M+Na]+: 305.015305; experimental value: 305.013815.
Compound 6 c: trans‐5‐Bromo‐6‐(p‐fluorophenyl)‐4,4‐di‐ methyltetrahydro‐2H‐pyran‐2‐one
Yield 85 %, colorless crystals, m.p.=102–103 °C, R f 0.23 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.21 (s, C(CH 3)2, 3 H), 1.28 (s, C(CH 3)2, 3 H), 2.60 (d, J=17.4 Hz, CH 2, 1 H), 2.82 (d, J=17.4 Hz, CH 2, 1 H), 4.05 (d, J=10.8 Hz, CHBr, 1 H), 5.39 (d, J=10.8 Hz, CHAr, 1 H), 7.03–7.12 (m, HAr, 2 H), 7.32–7.39 (m, HAr, 2 H). 13C NMR (125 Hz, CDCl3), δ[ppm]: 21.38, 29.55, 35.34, 44.01, 61.35, 82.61, 115.52, 129.42, 133.12, 163.05, 168.25. IR: 1726 (carbonyl band), 1510, 1360, 1230, 1160, 1020, 905, 855, 805, 650, 620 cm−1. HR‐MS (ESI‐TOF) calculated for C13H14BrFO2, m/z [M+Na]+: 323.0058836; experimental value: 323.008034.
Procedure of Chlorolactonization
Chlorolactones 7 a–c were obtained according to the procedure described in Ref. 50. The mixture of the above‐mentioned acid (0.005 mol) and N‐chlorosuccinimide (0.009 mol) was dissolved in 60 mL of THF. Acetic acid was added dropwise and mixture was stirred at room temperature for 48 h. When the reaction was complete, the mixture was dissolved in diethyl ether, washed with saturated NaHCO3 solution, and dried over anhydrous magnesium sulfate. The crude products was purified by column chromatography on silica gel [acetone/hexane, 1:10 (v/v)]. Spectral data are given below.
Compound 7 a: trans‐5‐Chloro‐4,4‐dimethyl‐6‐(α‐naphthyl)‐ tetrahydro‐2H‐pyran‐2‐one
Yield 75 %, colorless crystals, m.p.=192–193 °C, R f 0.20 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.23 (s, C(CH 3)2, 3 H), 1.35 (s, C(CH 3)2, 3 H), 2.68 (d, J=17.4 Hz, CH 2, 1 H), 2.84 (d, J=17.4 Hz, CH 2, 1 H), 4.37 (d, J=10.4 Hz, CHCl, 1 H), 6.06 (d, J=10.4 Hz, CHAr, 1 H), 7.47–7.60 (m, HAr, 4 H), 7.88 (d, J=7.8 Hz, HAr, 2 H), 8.05 (d, J=8.5 Hz, HAr, 1 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 20.83, 29.54, 35.72, 44.35, 66.68, 79.59, 122.92, 125.01, 125.84, 125.91 126.61, 129.10, 130.06, 131.24, 132.24, 133.87, 168.56. IR: 1730 (carbonyl band), 1350, 1240, 1216, 1170, 1140, 1035, 930, 910, 850, 790, 775, 730, 675, 640 cm−1. HR‐MS (ESI‐TOF) calculated for C17H17ClO2, m/z [M+Na]+: 311.0814712; experimental value: 311.080645.
Compound 7 b: trans‐5‐Chloro‐4,4‐dimethyl‐6‐phenylotetra‐ hydro‐2H‐pyran‐2‐one
Yield 85 %, colorless crystals, m.p.=96–97 °C, R f 0.24 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.19 (s, C(CH 3)2, 3 H), 1.26 (s, C(CH 3)2, 3 H), 2.57 (d, J=17.4 Hz, CH 2, 1 H), 2.76 (d, J=17.4 Hz, CH 2, 1 H), 3.97 (d, J=10.4 Hz, CHCl, 1 H), 5.25 (d, J=10.4 Hz, CHAr, 1 H), 7.33–7.44 (m, HAr, 5 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 20.45, 28.22, 35.39, 44.26, 67.26, 83.01, 127.48, 128.53, 129.22, 136.92, 168.35. IR: 1733 (carbonyl band), 1460, 1350, 1250, 1215, 1174, 1143, 1020, 930, 910, 863, 830, 760, 700, 660 cm−1. HR‐MS (ESI‐TOF) calculated for C13H15ClO2, m/z [M+Na]+: 261.065822; experimental value: 261.065139.
Compound 7 c: trans‐5‐Chloro‐6‐(p‐fluorophenyl)‐4,4‐di‐ methyltetrahydro‐2H‐pyran‐2‐one
Yield 65 %, colorless crystals, m.p.=89–90 °C, R f 0.22 (EtOAc : hexane, 1:7), 1H NMR (CDCl3, 500 MHz), δ [ppm]: 1.19 (s, C(CH 3)2, 3 H), 1.26 (s, C(CH 3)2, 3 H), 2.56 (d, J=17.4 Hz, CH 2, 1 H), 2.76 (d, J=17.4 Hz, CH 2, 1 H), 3.92 (d, J=10.5 Hz, CHCl, 1 H), 5.24 (d, J=10.5 Hz, CHAr, 1 H), 6.98–7.14 (m, HAr, 2 H), 7.31–7.42 (m, HAr, 2 H). 13C NMR (125 Hz, CDCl3), δ [ppm]: 20.43, 28.22, 35.38, 44.24, 67.28, 82.23, 115.53, 129.30, 132.80, 163.05, 168.17. IR: 1730 (carbonyl band), 1605, 1515, 1370, 1340, 1250, 1230, 1160, 1025, 913, 850, 805, 722, 660, 625 cm−1. HR‐MS (ESI‐TOF) calculated for C13H14ClFO2, m/z [M+Na]+: 279.0564006; experimental value: 279.057500.
In Vitro Cell Study
The L929 mouse fibroblasts (LGC Standards, Middlesex, UK) were used for in vitro cytotoxicity testing. The cells were maintained under standard conditions (37 °C, 5 % CO2) in 25 mL tissue culture flasks in RPM 1640 medium supplemented with 10 % fetal bovine serum (FBS) and antibiotics: 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cell suspension for cytotoxicity assay or to start a new culture was obtained by treatment of confluent monolayers with 0.25 % trypsin solution washed and subcultured at the cell density 108 cells mL−1. Cell cultures were supplemented with fresh medium two or three times per week to maintain them in log phase. The viability of the cells was assessed by exclusion of trypan blue dye and was in the range 93–95 %.
Measurements of Cellular Metabolic Activity and Global Growth Inhibition
Lactones 5 a–c, 6 a–c, and 7 a–c were used for the assessment of L929 cells metabolic activity. Cells in culture medium were seeded in the 96‐well plates (2×105 cells/well) for 24 h, at 37 °C, 5 % CO2. All tested compounds were diluted in RPM‐1640 medium in concentrations of 50, 20, 10, 5, 0.5, and 0.1 μg mL−1, added to the cells (100 μL/well), and incubated under standard conditions for 24 h. Following incubation, the cell monolayers were carefully screened using light microscopy, as recommended by ISO norm 10993‐5 to evaluate the morphology of cells.56 They were estimated by measuring the ability of the cells to reduce MTT [(3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide)], which is one of the tests recommended by the Food and Drug Administration (FDA) and the International Organization for Standardization (IOS). Fresh MTT solution (5 mg mL−1 in sterile PBS) was added to each well and incubated for 4 h at 37 °C. Formazan crystals were dissolved with acidic isopropanol (0.1 m HCl in absolute isopropanol). Optical density values were measured at 570 nm with a plate reader Victor2. The results are presented as the average number of cells able to convert MTT obtained from four independent experiments, including three repeats for each compound.
Assessment of Antibacterial Activity
The study of the antibacterial properties of chemical compounds was carried out by using spectrophotometric methods and counting of bacterial colonies.57 The cultures of E. coli ATCC 8739 and S.aureus ATCC 65389 were grown on LB Agar (Biocorp) for 18 h at 37 °C. A single colony was used to prepare a cell suspension in saline (0.85 %), which was adjusted to 0.5 on the McFarland scale. Added to the wells of the microtiter plate were: 180 μL of sterile enriched broth (BTL), 10 μL of the tested compound dissolved in DMSO, and 10 μL of the bacterial suspension. Control solutions were also prepared: a) 180 μL of BTL and 20 μL saline (sterility control); b) 80 μL of BTL, 10 μL of DMSO and 10 μL bacterial suspension (control of antibacterial activity of DMSO); c) 180 μlBTL 10 μL saline and 10 μL bacterial suspension (bacterial growth controls). The microtiter plate was incubated for 18 h at 37 °C. The experiment was repeated three times. The bacteriostatic effect of lactones was determined by measuring of bacterial suspension turbidity with Infinite PRO Tecanmicroplate reader at 600 nm. The CFU method was used to indicate the number of living bacterial cells after the exposition at the highest amine concentration.
Microbial media: LB Agar composition: tryptone −10 g l−, sodium chloride −5 g l−, yeast extract −5 g l−, agar −15 g l−; enriched broth (BTL), composition: beef extract −0.4 g l−, peptone −4 g l−, sodium chloride −3.5 g l−, peptone K−5.4 g l−, yeast extract −1.7 g l−. Media were prepared in accordance with the manufacturer's instructions.
Statistical Analysis
Statistical significance was accepted at a p value <0.05 on the non‐parametric Mann‐Whitney U test. Data are presented as mean values ± standard deviation (SD). For statistical analysis, the STATISTICA 10 PL software was used (Stat Soft, Poland).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
A. Kamizela, B. Gawdzik, M. Urbaniak, Ł. Lechowicz, A. Białońska, W. Gonciarz, M. Chmiela, ChemistryOpen 2018, 7, 543.
References
- 1. Yadav P. A., Suresh G., Rao M. S. A., Shankaraiah G., Usha R. P., Babu K. S., Bioorg. Med. Chem. Lett. 2014, 24, 888–892. [DOI] [PubMed] [Google Scholar]
- 2. Wirth R., Muscholl A., Wanner G., Trends Microbiol. 1996, 4, 96–103. [DOI] [PubMed] [Google Scholar]
- 3. Dirsch V. M., Stuppner H., Vollmar A. M., Planta Med. 2001, 67, 557–558. [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez A. D., Pina I. C., Barnes C. L., J. Org. Chem. 1995, 60, 8096–8100. [Google Scholar]
- 5. Chung H. S., Food Sci. Biotechnol. 2001, 10, 433–436. [Google Scholar]
- 6. Matsuo K., Ohtsuki K., Yoshikawa T., Shisho K., Yokotani-Tomita K., Shinto M., Tetrahedron 2010, 66, 8407–8419. [Google Scholar]
- 7. Mayer A. M. S., Gustafson K. R., Eur. J. Cancer 2004, 40, 2676–2704. [DOI] [PubMed] [Google Scholar]
- 8. Kitson R. A. R., Millemaggi A., Taylor R. J. K., Angew. Chem. Int. Ed. 2009, 48, 9426–9451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9590–9615. [Google Scholar]
- 9. Basanagouda M., Kulkarni M., Sharma D., Gupta V., Sandhyarani P., Rasal V., J. Chem. Sci. 2009, 121, 485–495. [Google Scholar]
- 10. Xiao Z.-P., Yi l.-C., Yi T.-F., Xiang K.-S., Zhu H.-L., J. Chem. Crystallogr. 2012, 42, 323–329. [Google Scholar]
- 11. Djeddi S., Karioti A., Sokovic M., Koukoulitsa C., Skaltsa H., Bioorg. Med. Chem. Lett. 2008, 16, 3725–3731. [DOI] [PubMed] [Google Scholar]
- 12. Rabe T., Mullholland D., Staden J., J. Ethnopharmacol. 2002, 80, 91–94. [DOI] [PubMed] [Google Scholar]
- 13. Özçelik B., Gürbüz I., Karaoglu T., Yeşiloda E., Microbiol. Res. 2009, 164, 545–552. [DOI] [PubMed] [Google Scholar]
- 14. Tseng Y. P., Kuo Y. H., Hu C. P, Jeng K. S., Janmanchi D., Lin C. H., Chou C. K., Yeh S. F., Antiviral Res. 2008, 77, 206–214. [DOI] [PubMed] [Google Scholar]
- 15. Li Y., Fu L., Yeo H., Zhu J. L., Chou C. K., Kou Y. H., Yeh S. F., Gullen E., Austin D., Cheng Y. C., Antiviral Chem. Chemother. 2005, 16, 193–201. [DOI] [PubMed] [Google Scholar]
- 16. Liu J. F., Wang Y. F., Bi Y. P., Li H. J., Jia L. A., Bi Y. F., Zhang Y. B., Tetrahedron Lett. 2013, 54, 4834–4836. [Google Scholar]
- 17. González-González C. A., Fuentes-Benítez A., Cuevas-Yáńez E., Corona-Becerril D., González-Romero C., González-Calderón D., Tetrahedron Lett. 2013, 54, 2726–2728. [Google Scholar]
- 18. Barrero A. F., Oltra J. E., Alvarez M., Raglan D. S., Akssira M., Fitoterapia 2000, 71, 60–64. [DOI] [PubMed] [Google Scholar]
- 19. Vajs V., Todorovi N., Risti M., Tesevi V., Todorovi B., Janakovi P., Marin P., Milosavlijevi S., Phytochemistry, 1999, 52, 383–386. [DOI] [PubMed] [Google Scholar]
- 20. Feng J.-T., Wang D.-L., Wu Y.-L., Yan H., Zhang X., Bioorg. Med. Chem. Lett. 2013, 23, 4393–4397. [DOI] [PubMed] [Google Scholar]
- 21. Gawdzik B., Kamizela A., Szyszkowska A., Chemik 2015, 69, 342–349. [Google Scholar]
- 22. Cis J., Nowak G., Kisiel W., Biochem. Syst. Ecol. 2006, 34, 862–867. [Google Scholar]
- 23. Datta S., Saxena D., Pest Manage. Sci. 2001, 57, 95–101. [DOI] [PubMed] [Google Scholar]
- 24. Grudniewska A., Dancewicz K., Białońska A., Ciuniuk Z., Gabryś B., Wawrzeńczyk C., RSC Adv. 2011, 1, 498–510. [Google Scholar]
- 25. Skrobiszewski A., Gładkowski W., Walczak P., Gliszczyńska A., Maciejewska G., Klejdysz T., Nawrot J., Wawrzeńczyk C., J. Chem. Sci. 2015, 127, 687–699. [Google Scholar]
- 26. Paruch E., Ciuniuk Z., Nawrot J., Wawrzeńczyk C., J. Agric. Food Chem. 2000, 48, 4973–4977. [DOI] [PubMed] [Google Scholar]
- 27. Paruch E., Nawrot J., Wawrzeńczyk C., Pest Manage. Sci. 2001, 57, 776–780. [DOI] [PubMed] [Google Scholar]
- 28. Yang C. G., Reich N. W., Shi Z., He C., Org. Lett. 2005, 7, 4553–4556. [DOI] [PubMed] [Google Scholar]
- 29. Tiseni S. P., Peters R., Chem. Eur. J. 2010, 16, 2503–2517. [DOI] [PubMed] [Google Scholar]
- 30. Hopkins C. D., Guan L., Malinakova H., J. Org. Chem. 2005, 70, 6848–6862. [DOI] [PubMed] [Google Scholar]
- 31. Liu H., Pan Y., Tan C., Tetrahedron Lett. 2008, 49, 4424–4426. [Google Scholar]
- 32. Huang L., Jiang H., Qi C., Liu X., J. Am. Chem. Soc. 2010, 132, 17652–17654. [DOI] [PubMed] [Google Scholar]
- 33. Reddy M. S., Kumar Y. K., Thirupathi N., Org. Lett. 2012, 14, 824–827. [DOI] [PubMed] [Google Scholar]
- 34. Chen L., Zhou F., Shi T.-D., Zhou J., J. Org. Chem. 2012, 77, 4354–4362. [DOI] [PubMed] [Google Scholar]
- 35. Arakawa N. S., Gobello-Neto L., Ambrosio S. R., Antonucci G. A., Sampaio S. V., Pupo M. T., Said S., Schmidt T. J., F. B. Da Costa Phytochemistry 2013, 96, 92–100. [DOI] [PubMed] [Google Scholar]
- 36. Gladkowski W., Grabarczyk M., Wińska K., Ratuś B., Białońska A., Ciunik Z., Wawrzeńczyk C., J. Mol. Catal. B-Enzym. 2007, 49, 79–87. [Google Scholar]
- 37. Gładkowski W., Grabarczyk M., Konopka M., Wawrzeńczyk C., J. Mol. Catal. B-Enzym. 2004, 29, 13–17. [Google Scholar]
- 38. Grabarczyk M., Wińska K., Mączka W., Żarowska B., Maciejewska G., Dancewicz K., Gabryś B., Anioł M., Tetrahedron 2016, 72, 637–644. [Google Scholar]
- 39. Wińska K., Grabarczyk M., Mączka W., Żarowska B., Maciejewska G., Anioł M., J. Saudi Chem. Soc. 2018, 22, 363–371. [Google Scholar]
- 40. Wzorek A., Gawdzik B., Gładkowski W., Urbaniak M., Barańska A., Malińska M., Woźniak K., Kempińska K., Wietrzyk J., J. Mol. Struct. 2013, 1047, 160–168. [Google Scholar]
- 41. Gładkowski W., Skrobiszewski A., Mazur M., Siepka M., Białońska A., Eur. J. Org. Chem. 2015, 605–615. [Google Scholar]
- 42. Dohi T., Takenaga N., Goto A., Maruyama A., Kita Y., Org. Lett. 2007, 9, 3129–3132. [DOI] [PubMed] [Google Scholar]
- 43. Gawdzik B., Wzorek A., Kamizela A., Urbaniak M., Gładkowski W., Lis M., Obmińska-Mrukowicz B., Białońska A., Curr. Org. Synth. 2016, 13, 901–906. [Google Scholar]
- 44. Watson A. J. A., Atkinson B. N., Maxwell A. C., Williams J. M., J. Adv. Synth. Catal. 2013, 355, 734–740. [Google Scholar]
- 45. Maeno M., Shibata N., Cahard D., Org. Lett. 2015, 17, 1990–1993. [DOI] [PubMed] [Google Scholar]
- 46. Julia M., Guy-Rousault A., Compt. Rend. 1964, 258, 3728–3730. [Google Scholar]
- 47. Snider B. B., Johnston M. I., Tetrahedron Lett. 1985, 45, 5497–5500. [Google Scholar]
- 48.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/open.201800110 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
- 49. Gładkowski W., Skrobiszewski A., Mazur M., Siepka M., Pawlak A., Obmińska-Mrukowicz B., Białońska A., Poradowski D., Drynda A., Urbaniak M., Tetrahedron 2013, 69, 10414–10423. [Google Scholar]
- 50. Mazur M., Gładkowski W., Podkowik M., Bania J., Nawrot J., Białońska A., Wawrzeńczyk C., Pest Manage. Sci. 2014, 70, 286–294. [DOI] [PubMed] [Google Scholar]
- 51. Grabarczy M., Wińska K., Mączka W., Żołnierczyk A. K., Żarowska B., Anioł M., Molecules 2015, 20, 3335–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han X., Wu H., Dong C., Tien P., Xie W., Wu S., Zhou H., RSC Adv. 2015, 5, 10005–10013. [Google Scholar]
- 53. Gaylord N. G., Becker E. I., J. Org. Chem. 1950, 15, 305–316. [Google Scholar]
- 54. Johnson W. S., Werthemann L., Bartlett W. R., Brocksom T. J., Li T.-T., Faulkner D. J., Petersen M. R., J. Am. Chem. Soc. 1970, 92, 741–743. [Google Scholar]
- 55. Mori K., Nakazono Y., Tetrahedron 1986, 42, 283–290. [Google Scholar]
- 56.ISO 10993–10995:2009. Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity. Available at: http://www.iso.org.
- 57. Jablonska-Wawrzycka A., Barszcz B., Zienkiewicz M., Hodorowicz M., Jezierska J., Stadnicka K., Lechowicz L., Kaca W., Spectrochim. Acta Part A 2014, 129, 632–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary