

Abstract
The development of various cardiovascular diseases (CVDs) are associated with chronic inflammation. Tumor necrosis factor α (TNF-α) is a pro-inflammatory cytokine that activates the nuclear factor-κB (NF-κB) signaling pathway, leading to increased inflammatory cytokine expression, such as interleukin-6 (IL-6). Interventions to reduce each of these factors have been demonstrated to reduce the development of CVD. Methylsulfonylmethane (MSM) is a naturally occurring compound that demonstrates anti-inflammatory effects in humans and various animal and cell culture models. The effects of MSM include decreased NF-κB activation, decreased expression of TNF-α, and IL-6. However, the effects of MSM within the heart have not yet been examined. Therefore, the purpose of this investigation was to determine whether MSM protects cardiac cells from inflammation that occurs in response to pro-inflammatory stimuli. A novel immortalized human ventricular cardiomyocyte cell line, designated Ac16, developed and characterized in the laboratory of Dr. Mercy Davidson, Columbia Invention Report No. 823, U.S. patent No. 7,223,599 were utilized. Cells were treated with TNF-α, alone or in combination with MSM. To confirm an appropriate dosage of MSM, the effect of various concentrations on cell viability, and IL-6 production were examined. The effect of MSM on transcript expression of pro-inflammatory markers and activation of NF-κB were examined with the established dose by real-time quantitative PCR and western blot, respectively. MSM treatment combined with TNF-α significantly decreased IL-6 production and transcript expression compared to TNF-α alone. These findings indicate that MSM may protect against inflammation in the heart, and thereby protect against inflammation-linked CVDs. Further study is warranted to determine the effect of MSM on cardiovascular health outcomes.
Keywords: Methylsulfonylmethane, MSM, cardiac, inflammation, TNF-α, IL-6, NF-κB
Introduction
Cardiovascular disease (CVD) remains the leading cause of death in the world [1]. Chronic inflammation and oxidative stress are associated with the development of various cardiovascular diseases including hypertension, heart failure, cardiac fibrosis, and diabetic cardiomyopathy [2-5].
The identification of mechanisms by which chronic inflammation and oxidative stress contribute to the development of CVDs is an active area of research for the discovery of future treatments and preventative interventions. Studies have identified pro-inflammatory signaling factors such as nuclear factor-κB (NF-κB), tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) as important mediators in the development of CVDs [6-9]. Interventions to reduce NF-κB expression, inflammatory cytokines, and reactive oxygen species (ROS) have been demonstrated to reduce the development of CVDs, including heart failure and cardiac fibrosis [10,11]. Therefore, novel strategies to decrease cardiac inflammation and ROS hold promise to inhibit the development of CVDs.
Methylsulfonylmethane (MSM) is a naturally occurring compound that demonstrates antioxidant and anti-inflammatory effects both in vivo and in vitro [12-14]. In macrophages, MSM inhibits mitochondrial ROS production, decreases protein expression of pro-IL-1β, TNF-α, and transcript expression of IL-6 [15]. Furthermore, Kim and colleagues demonstrated that MSM blocks NF-κB activation, thereby inhibiting NF-κB mediated transcription of inflammatory genes [14]. Collectively, these findings support a beneficial role of MSM in disease states associated with chronic inflammation and oxidative stress. However, the effects of MSM within cardiac cells had not yet been examined. Therefore, the purpose of this investigation was to determine whether MSM protects cardiac cells from oxidative stress and inflammation induced by pro-inflammatory stimuli.
Materials and methods
Cell culture
A novel immortalized human ventricular cardiomyocyte cell line, designated Ac16, developed and characterized in the laboratory of Dr. Mercy Davidson [16], relating to Columbia Invention Report No. 823 and covered by U.S. patent No. 7,223,599 were utilized in this study. Ac16 cells were cultured in Gibco DMEM (ThermoFisher Scientific, Carlsbad, CA.), with 5.5% glucose and 12.5% fetal bovine serum and maintained at 37°C and 5% CO2. Cells were split at 75% confluence. 24 hours prior to all treatments, medium was replaced with DMEM containing 5.5% glucose and 1% fetal bovine serum. Application of TNF-α (10 ng/ml) was utilized as a model of pro-inflammatory signaling.
Cell viability and proliferation assays
Ac16 Cells were plated in 96-well format and treated with various concentrations (0, 5, 10, 20 or 40 mg/ml) of MSM (OptiMSM®, Bergstrom Nutrition, Vancouver, WA.) for 24 hours, n=3. Assessment of cell viability and cytotoxicity was completed via the CellTiter96Aqueous assay (Promega, Madison, WI) and cell proliferation by the MTT assay (MilliporeSigma, St. Louis, MO), following manufacturer’s instructions.
Culture medium analysis
Ac16 cells were treated as Control or with TNF-α (10 ng/ml) alone and in combination with various concentrations of MSM (2.5, 5.0 or 10.0 mg/ml) for 24 hours, n=4. Culture medium was collected and stored at -80 for IL-6 analysis. IL-6 levels were assessed utilizing a commercially available kit following manufacturer’s instructions (ThermoScientific, Carlsbad, CA.).
Real time quantitative PCR
Ac16 cells were treated as Control, with MSM (10 mg/ml), with TNF-α (10 ng/ml), or MSM and TNF-α for 24 hours, n=4 per group. Cells were harvested with RNA Protect Cell Reagent (Qiagen, Valenica, CA.) and stored at -80 until analysis. Total RNA was extracted using the RNeasy Protect Cell Mini Kit (Qiagen, Valenica, CA.). cDNA was synthesized using the Verso cDNA synthesis kit (ThermoFisher Scientific, Carlsbad, CA.). Primers were purchased from Integrated DNA Technologies (IDT, Coralville, IA), sequences are provided in Table 1. Transcript expression of NF-κβ1 & 2, inhibitor of κB (IκB), IL-6 and IL-1 were evaluated as markers of inflammation and normalized to TATA binding protein. Real-time quantitative PCR was performed with SYBR green master mix (Qiagen, Valencia, CA.) and 2 ng template cDNA per reaction. Cycles were as follows: 10 min at 95°, followed by 40 cycles of 10 s at 95° and 1 min at 60°, followed by melt curve analysis beginning at 55° and increasing 0.5° for 81 cycles.
Table 1.
Primers for real time qPCR analysis
| NF-κB1 | Forward | CAGTGGTGCCTCACTGCTAA |
| Reverse | GGACAACGCAGTGGAATTTT | |
| NF-κB2 | Forward | GAAGATTGAGCGGCCTGTAA |
| Reverse | TGTCTTCCACCAGAGGGTAATA | |
| IκB | Forward | TACCAGGGCTATTCTCCCTAC |
| Reverse | TGGCAGCATCTGAAGGTTT | |
| IL-6 | Forward | ATTCTGCGCAGCTTTAAGGA |
| Reverse | GAGGTGCCCATGCTACATTT | |
| IL-1 | Forward | CAAAGGCGGCCAGGATATAA |
| Reverse | CTAGGGATTGAGTCCACATTCAG | |
| TBP | Forward | ATGTTGAGTTGCAGGGTGTG |
| Reverse | CCCAGATAGCAGCACGGTAT |
NF-κB, Nuclear factor-κB, IκB, inhibitor of κB, IL-6, interluekin-6, IL-1, interluekin-1, TBP, TATA binding Protein.
Cell fractionation and western blotting
Ac16 cells were treated as Control or with TNF-α (10 ng/ml) alone or in combination with MSM (10 mg/ml), n=3. Cells were harvested at 30 minutes post treatment. Briefly, cells were trypsinized and centrifuged for 5 minutes at 37°C. The cell pellet was washed and re-suspended in ice-cold PBS, transferred to a microcentrifuge tube and centrifuged 5 minutes at 4°C. Fractionations were separated utilizing a Subcellular fractionation kit (ThermoFisher Scientific, Carlsbad, CA.), following the manufacturer’s instructions. Each fraction was stored at -80°C for future analysis. For western blot of cellular fractions, both nuclear and cytoplasmic samples were thawed on ice. Total protein content was assessed by the DC protein assay (BioRad, Irvine, CA.). Samples were diluted in phosphate buffered saline to normalize the protein concentration, mixed 1:1 with laemmli sample buffer containing 5% β-mercaptoethanol, and boiled at 90°C for 5 min. Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Equal transfer was assessed by Ponceau S Staining reagent (Sigma Aldrich, St. Louis, MO.). Blots were then blocked 30 minutes at room temperature in PBS with 5% non-fat dry milk (NFDM). All primary antibodies were applied 1:1000 in PBS 1% tween, overnight at 4°C. Secondary antibodies were applied 1:2000 in PBS 5% NFDM for 1 hour at room temperature. Blots were probed for NF-κBp65 (Cell Signaling Technologies, Danvers, MA. 8242S). Histone H3 (Cell Signaling Technologies, Danvers, MA. 4499S) and β-tubulin (Cell Signaling Technologies, Danvers, MA. 15115S) were utilized for nuclear and cytoplasmic fractions, respectively. Blots were visualized with SuperSignal Chemiluminescent substrate containing horseradish peroxidase (ThermoFisher, Carlsbad, CA.) utilizing the ChemiDocXRS imaging system (BioRad, Irvine, CA.).
Antioxidant capacity assays
Cells were cultured and treated as Control or with TNF-α (10 ng/ml) alone or in combination with MSM (10 mg/ml) for 24 hours. Total antioxidant capacity was assessed via the Oxiselect Total Antioxidant Capacity assay kit, (Cell Biolabs, San Diego, CA.). Assessment of glutathione was performed with the Oxiselect Total Glutathione assay kit, (Cell Biolabs, San Diego, CA.). Cells were harvested for each assay according to provided protocol instructions. To measure Oxidized (GSSG) versus reduced (GSH) glutathione, GSH was derivatized with 2-vinylpyridine, allowing for measurement of GSSG only. GSH was calculated as the difference between measured GSSG and total glutathione levels [17].
Statistical analysis
All statistical analysis were performed on Sigma Plot Software (San Jose, CA.). Results are presented as means ± SEM with P < 0.05 accepted as statistically significant. Equal variance was tested and normality was confirmed with the Shapiro-Wilk test. Between groups differences were evaluated with t-tests or one-way analysis of variance (ANOVA) with Bonferroni post hoc when appropriate. The Mann-Whitney rank sum test or Kruskal-Wallis ANOVA on ranks was utilized in the case of unequal variance.
Results
Cell viability and proliferation is not affected by MSM at doses of 20 mg/ml or less
Initial experiments were undertaken to determine an appropriate dosage of MSM in the Ac16 cell model. Compared to cells cultured in the absence of MSM, no differences in cell viability or proliferation were detected at doses of 5, 10, or 20 mg/ml (Figure 1, Supplementary Data). However, significant reductions in cell viability occurred at a dosage of 40 mg/ml. While a reduction in cell viability at any concentration was unexpected, it should be noted that previous studies performed in different cell types have utilized doses well below 40 mg/ml. We have confirmed that MSM does not alter cell viability at a dosage of 20 mg/ml or below in Ac16 cells. Previous studies support the rationale that doses of MSM within this range exert positive physiological effects [14,15].
Figure 1.
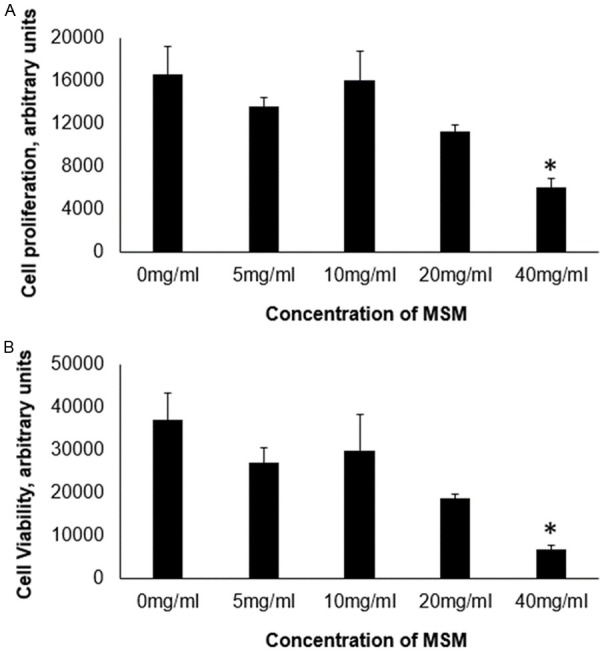
Cell proliferation and viability. Data is presented as group means, ± SEM, n=3. MSM, methylsulfonylmethane. A. Cell Titer 96 assay, B. MTT assay. MSM at concentrations of 20 mg/ml or below had no effect on cell proliferation or viability. * indicates different from 0 mg/ml, P ≤ 0.05.
MSM decreases TNF-α mediated IL-6 release in Ac16 cells
IL-6 was significantly elevated in the culture medium of cells treated with TNF-α compared to untreated control cells. Compared to cells treated with TNF-α alone, IL-6 levels were significantly decreased in culture medium from cells treated TNF-α combined with both 5.0 and 10 mg/ml MSM. However, cells treated with TNF-α in the presence of 2.5 mg/ml MSM were not different from cells treated with TNF-α alone (Figure 2, Supplementary Data). These results indicate that MSM effectively decreases activation of inflammatory pathways that occur in response to TNF-α at concentrations of 5-10 mg/ml. Based on these findings, further treatments were performed with 10 mg/ml MSM.
Figure 2.
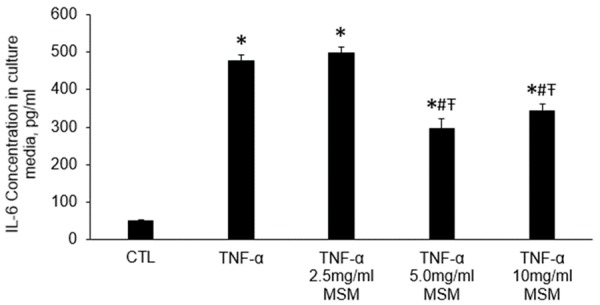
IL-6 concentration in culture media following indicated treatments for 24 hours. CTL, Control, TNF-α, tumor necrosis factor-α, MSM, methylsulfonylmethane. Data represents group means ± SEM. Cell treatment with TNF-α resulted in elevated IL-6 production, which was attenuated by either 5.0 mg/ml or 10 mg/ml MSM. * indicates different from CTL, # indicates different from TNF-α, Ŧ indicates different from TNF-α 2.5 mg/ml MSM P ≤ 0.05.
MSM decreases TNF-α induced IL-6 mRNA transcript expression
MSM alone resulted in lower expression of NFκβ1 and IL-1 compared to control cells. No other transcript levels were different between MSM and control cells (Figure 3, Supplementary Data). Treatment with TNFα resulted in elevated transcript expression of IL-6, IL-1, NF-κβ1 & 2, and Iκβ compared to controls. Co-treatment of cells with TNFα and MSM significantly decreased IL-6 transcript expression compared to cells treated with TNFα alone. However, co-treatment with MSM did not significantly impact transcript expression of IL-1, NF-κβ1 and 2, or Iκβ (Figure 4, Supplementary Data). It is important to note that this data reflects only a single time point, where differences may exist at different time points following treatment. Overall, the results support the notion that MSM reduces inflammatory signaling under basal conditions and in response to pathological stimuli.
Figure 3.
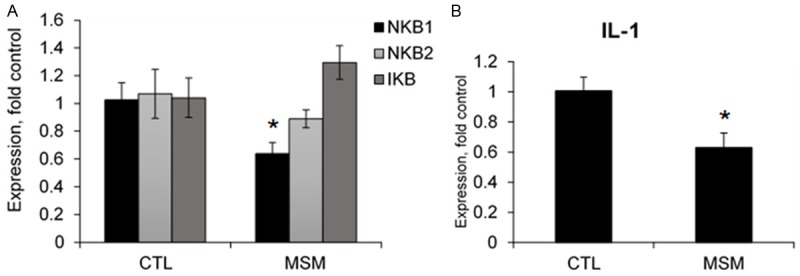
Transcript expression in MSM treated cells compared to control. CTL, control, MSM, methylsulfonylmethane, NFκB, nuclear factor-κB, IκB, inhibitor of κB, IL-1, interleukin-1. Data represents group means ± SEM, n=4. A. MSM treatment resulted in lower expression of NFκβ1. B. MSM treatment resulted in lower expression of IL-1 compared to control (CTL). * indicates significantly different from CTL, P ≤ 0.05.
Figure 4.
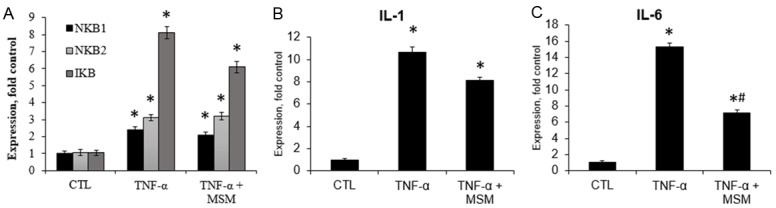
Transcript expression following TNF-α treatment alone, or in combination with MSM. CTL, control, TNF-α, tumor necrosis factor-α, MSM, methylsulfonylmethane, NF-κB, nuclear factor-κB, IκB, inhibitor of κB, IL-1, interleukin-1. Data represents group means ± SEM, n=4. A. Co-treatment of cells with TNF-α and MSM did not affect transcript expression of NF-κB1 and 2, or IκB compared to TNF-α alone. B. Co-treatment of cells with TNF-α and MSM did not affect IL-1 compared to TNF-α alone. C. Co-treatment of cells with TNF-α and MSM significantly decreased IL-6 expression compared to TNF-α alone. * denotes significantly different from control, # denotes significantly different from TNF-α, P ≤ 0.0.5.
NF-κB activation
TNF-α treatment increased NF-κB within the nuclear fraction, indicating that NF-κB activation is increased by TNF-α. Co-treatment of TNF-α with MSM did not significantly decrease the amount of NF-κB present in the nuclear fraction compared to TNF-α alone. Also, MSM treatment alone had no effect on NF-κB compartmentalization compared to control treated cells.
Antioxidant capacity assays
Treatment of cells with MSM did not significantly impact the GSSG/GSH ratio, or the total antioxidant capacity. Furthermore, no differences were found for these variables following treatment with TNF-α alone, or in combination with MSM.
Discussion
The key findings of this study indicate that MSM decreases both protein and transcript expression of IL-6 in response to pathological stimuli. Given the involvement of IL-6 in cardiac inflammation and cardiovascular disease, this finding supports the hypothesis that MSM may provide protection against inflammatory-mediated cardiovascular pathologies. Continued investigation of the potential benefits of MSM on the development and progression of cardiovascular diseases is highly warranted.
This was the first investigation to examine the effects of MSM in cardiac cells, specifically the AC16 cell model. In order to identify a proper treatment concentration of MSM, we first performed cell viability assays with varying concentrations of MSM. The concentrations tested were based upon work in different cell models [14,15]. Our results indicated that MSM may decrease cell viability at very high concentrations, but had no effect on cell viability at most concentrations tested. The concentrations in which cell viability was unaffected are well within the concentrations found most frequently reported in studies utilizing different cell models [14,15]. Next, the effect of various concentrations of MSM on IL-6 production following TNF-α treatment were examined to further identify an appropriate dose of MSM for the remainder of the study. The findings from these two experiments indicated that 5-10 mg/ml MSM is effective at diminishing TNF-α induced IL-6 production, while remaining well below the threshold that could impact cell viability. Therefore, a concentration of 10 mg/ml MSM was utilized for the remainder of the experiments.
The next objective of the study was to determine whether MSM had an effect on transcript expression of inflammatory markers, either alone or in combination with TNF-α treatment. MSM treatment resulted in decreased transcript expression of NF-κB1 and IL-1. These results indicate that MSM may have a role in decreasing inflammatory markers during basal conditions. TNF-α increased transcript expression of inflammatory pathway components (NF-κB1, NF-κB2, IκB, IL-1 and IL-6). Relative to cells treated with TNF-α, co-treatment with TNF-α and MSM decreased the transcript expression of IL-6, while the expression of NF-κB1, NF-κB2, IκB, and IL-1 was unaffected. Interestingly, while MSM decreased transcript expression of NF-κB and IL-1 under basal conditions, there was no effect when combined with TNF-α. Therefore, it appears the effect of TNF-α overrides the effect of MSM to decrease transcription of NF-κB and IL-1. It is important to note that these studies were performed at only one time point. It may be that transcriptional changes occur at times other than that which was observed in this study, and therefore MSM may have effects that were not observed.
We hypothesized that MSM diminishes IL-6 transcript and protein expression following TNF-α treatment by decreasing the level of NF-κB activation that occurs in response to TNF-α. NF-κB is a well-studied super family of transcription factors involved in transcriptional regulation of various inflammatory cytokines, such as TNFα, IL-6, and IL-1 [18], as well as genes regulating oxidative status [19,20]. Under basal conditions, inactive NF-κB is bound to inhibitor of κB (IκB) within the cytoplasm [21]. Activation of NF-κB occurs due to phosphorylation and degradation of IκB by IκB kinase (IKK), followed by translocation of NF-κB to the nucleus [21]. Therefore, the level of NF-κB activation was measured by examining the cellular location of NF-κB. TNF-α treatment resulted in significantly elevated NF-κB within the nuclear compartment compared to control treated cells. Although group means are decreased with TNF-α and MSM co-treatment compared to TNF-α alone, we did not find significant differences in NF-κB within the nuclear fraction. Samples were measured at 30 min post treatment application. This timeframe is based upon previous findings that indicate NF-κB is maximally activated at 30 min, and follows am oscillating pattern of activation [22]. Despite this, it is possible that the timing of sample collection may have been slightly different from when the peak activation occurred. Therefore, although the data does not support an effect of MSM on decreased NF-κB activation, further study is needed to confirm this finding and sample various timeframes following treatment. Additionally, future investigations should aim to determine the mechanisms by which MSM decreases expression of inflammatory markers.
The results of our antioxidant capacity assay did not reveal any effect associated with MSM treatment. Likewise, MSM treatment had no effect on the GSSG/GSH ratio, which is commonly used as a marker of oxidative stress. These findings were somewhat unexpected, as many have documented improved antioxidant capacity and decreased oxidative stress following MSM treatment. However, differences in our study and others could be due to both the difference in study model and timing. We have utilized an immortalized model of human heart cells, compared to other cell types or consumption in humans, as well as difference in treatment times, we examined 24 hours, while other studies have examined more prolonged supplementation in humans [12,15]. Additional studies are needed to further clarify effects of MSM on oxidative status and antioxidant capacity in heart tissues.
In conclusion, the findings from this investigation indicate that MSM is effective in decreasing IL-6 transcript and protein expression following TNF-α treatment. This is the first investigation to examine the effects of MSM in a cardiac model. Given the role of chronic inflammation in cardiovascular disease, these findings support the importance of future studies to robustly identify the beneficial effects of MSM as a novel option for prevention and treatment of cardiovascular disease.
Acknowledgements
This work was supported by the College of Nursing at Washington State University and Bergstrom Nutrition, Vancouver, WA.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bahrami H, Bluemke DA, Kronmal R, Bertoni AG, Lloyd-Jones DM, Shahar E, Szklo M, Lima JA. Novel metabolic risk factors for incident heart failure and their relationship with obesity: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;51:1775–1783. doi: 10.1016/j.jacc.2007.12.048. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki T, Katz R, Jenny NS, Zakai NA, LeWinter MM, Barzilay JI, Cushman M. Metabolic syndrome, inflammation, and incident heart failure in the elderly: the cardiovascular health study. Circ Heart Fail. 2008;1:242–248. doi: 10.1161/CIRCHEARTFAILURE.108.785485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frati G, Schirone L, Chimenti I, Yee D, Biondi-Zoccai G, Volpe M, Sciarretta S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. 2017;113:378–388. doi: 10.1093/cvr/cvx011. [DOI] [PubMed] [Google Scholar]
- 5.Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review) Vascul Pharmacol. 2015;71:40–56. doi: 10.1016/j.vph.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 9.McTiernan CF, Feldman AM. The role of tumor necrosis factor alpha in the pathophysiology of congestive heart failure. Curr Cardiol Rep. 2000;2:189–97. doi: 10.1007/s11886-000-0068-4. [DOI] [PubMed] [Google Scholar]
- 10.Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol. 2008;375:637–649. doi: 10.1016/j.jmb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong P, Wu L, Qian Y, Fang Q, Liang D, Wang J, Zeng C, Wang Y, Liang G. Blockage of ROS and NF-kappaB-mediated inflammation by a new chalcone L6H9 protects cardiomyocytes from hyperglycemia-induced injuries. Biochim Biophys Acta. 2015;1852:1230–1241. doi: 10.1016/j.bbadis.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Nakhostin-Roohi B, Barmaki S, Khoshkhahesh F, Bohlooli S. Effect of chronic supplementation with methylsulfonylmethane on oxidative stress following acute exercise in untrained healthy men. J Pharm Pharmacol. 2011;63:1290–1294. doi: 10.1111/j.2042-7158.2011.01314.x. [DOI] [PubMed] [Google Scholar]
- 13.Nakhostin-Roohi B, Niknam Z, Vaezi N, Mohammadi S, Bohlooli S. Effect of single dose administration of methylsulfonylmethane on oxidative stress following acute exhaustive exercise. Iran J Pharm Res. 2013;12:845–853. [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YH, Kim DH, Lim H, Baek DY, Shin HK, Kim JK. The anti-inflammatory effects of methylsulfonylmethane on lipopolysaccharide-induced inflammatory responses in murine macrophages. Biol Pharm Bull. 2009;32:651–656. doi: 10.1248/bpb.32.651. [DOI] [PubMed] [Google Scholar]
- 15.Ahn H, Kim J, Lee MJ, Kim YJ, Cho YW, Lee GS. Methylsulfonylmethane inhibits NLRP3 inflammasome activation. Cytokine. 2015;71:223–231. doi: 10.1016/j.cyto.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol. 2010;648:269–277. doi: 10.1007/978-1-60761-756-3_18. [DOI] [PubMed] [Google Scholar]
- 18.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 19.Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, Yamaguchi O, Mano T, Matsumura Y, Ueno H, Tada M, Hori M. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105:509–515. doi: 10.1161/hc0402.102863. [DOI] [PubMed] [Google Scholar]
- 20.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.