

Abstract
Background:
Transforming growth factor-β (TGF-β) in addition to the C-terminal region can phosphorylate receptor-regulated Smads (R-Smads) in their linker region. The aim of the present study was to evaluate the role of signaling mediators such as NAD(P)H oxidases (reactive oxygen species [ROS] generators), ROS, and ROS-sensitive p38 mitogen-activated protein kinase (p38MAPK) in this signaling pathway in cultured human vascular smooth muscle cells (VSMCs).
Methods:
The present in vitro study was performed on human VSMCs. Proteins were detected by western blotting utilizing an anti-phospho-Smad2 (Ser245/250/255) rabbit polyclonal antibody and a horseradish peroxidase-labeled secondary antibody. Glyceraldehyde-3-phosphate dehydrogenase was used as a loading control. The phospho-Smad2 linker region (pSmad2L) was detected in all the experimental groups: a control group (untreated group), a group treated with TGF-β (2 ng/mL), and a group treated with TGF-β plus different inhibitors. The data were normalized and presented as mean±SEM. The statistical analyses were performed using SPSS, version 16.0, and the nonparametric Kruskal-Wallis test. A P value smaller than 0.05 was considered statistically significant.
Results:
The VSMCs treated with TGF-β (2 ng/mL) showed a time-dependent increase in the pSmad2L level. The highest level was observed at 15 minutes (P=0.03). The inhibitors of NAD(P)H oxidases (diphenyleneiodonium and apocynin) (P=0.04), ROS scavenger (N-acetylcysteine) (P=0.04), and p38MAPK inhibitor (SB-202190) (P=0.04) were able to reduce the increased level of the pSmad2L by TGF-β.
Conclusion:
Our results suggested that NAD(P)H oxidases played an important role in the Smad2L phosphorylation in the human VSMCs. Furthermore, our results confirmed that ROS and p38MAPK were involved in this signaling pathway. Thus, TGF-β via a ROS-dependent mechanism can transmit its signals to the pSmad2L.
Keywords: Transforming growth factor beta , Smad2 protein , Reactive oxygen species , NADPH oxidase 4 , P38 mitogen-activated protein kinases
What’s Known
TGF-β by activating several serine/threonine kinases such as MAPKs phosphorylates specific residues in the linker region of Smad2/3.
Some studies have demonstrated the role of NAD(P)H oxidases (main ROS producers in the vasculature) and also ROS in different effects mediated by TGF-β.
What’s New
NAD(P)H oxidases have an important role in the TGF-β-induced phosphorylation of the Smad2 linker region. Using ROS scavengers, we showed the involvement of ROS in this signaling pathway.
Signal transduction of TGF-β to the linker region of Smad2 depends on the ROS-related mechanism in human vascular smooth muscle cells.
Introduction
Transforming growth factor-β (TGF-β) is a pleiotropic growth factor linked to vascular diseases such as atherosclerosis. TGF-β does its biological activities through serine/threonine kinase cell surface receptors, namely TGF-β type II receptor (TβRII) and TGF-β type I receptor (TβRI), also known as activin-like kinase 5 (AlK5), and affects the regulation of target genes via Smad transcription factors.1 Smad proteins are comprised of 3 distinct regions: C-terminal, N-terminal, and central linker region.2 TGF-β signaling commences when the TGF-β dimmer binds to TβRII, which leads to TβRI phosphorylation and activation. Most studies on TGF-β signaling have concentrated on the canonical Smad pathway, whose result is the direct phosphorylation of 2 serine residues on the carboxy terminus of receptor-regulated Smads (R-Smads) (Smad2 and Smad3). Subsequently, by binding to Smad4 (co-Smad) and forming the Smad heteromeric complex, imported to the nucleus, where it regulates the expression of specific genes. Moreover, studies have shown that TGF-β indirectly via phosphorylation and activation of several serine/threonine kinases, including mitogen-activated protein kinases (MAPKs), can phosphorylate specific serine and threonine residues (in human Smad2, including Thr 220, Ser 245, Ser 250, and Ser 255) within the linker region of R-Smads.1-6 Smad linker-region signaling, also known as “non-Smad signaling”, is involved in TGF-β signaling modification and appears to play an important role in the regulation of a broad range of cellular events.1,5 It has already been specified that TGF-β has a pro-atherogenic property via its effects on the synthesis and structure of proteoglycans. TGF-β in human vascular smooth muscle cells (VSMCs) can induce the expression of proteoglycans and the elongation of their glycosaminoglycan chain via both canonical and non-Smad signaling, which enhances binding to low-density lipoproteins and traps them in the vessel wall.7,8
MAPKs, including Erk, p38, and Jnk, are a family of intracellular serine/threonine kinases that have roles in transmitting extracellular signals from cell-surface receptors to intracellular targets and finally affect transcription factors. MAPKs can be activated by TGF-β.5 A member of the MAPK family, p38 is strongly implicated in the progression of atherosclerosis9 and is a key signaling molecule in cellular responses to various stress signals such as oxidative stress.10,11
Reactive oxygen species (ROS) play a significant role in the initiation and development of cardiovascular diseases such as atherosclerosis.12 It has been proven that ROS, as important second messengers, can influence cellular signal-transduction pathways.13 Multiple enzyme systems are the sources of ROS generation in the vasculature; these enzymes include NAD(P)H oxidases (Noxs), xanthine oxidase, enzymes of the respiratory chain, NO synthases, lipoxygenases, and cytochrome p450 monooxygenases. Despite the importance of other vascular sources of ROS, it has been shown that Noxs are the main producers of ROS because the ROS formed by these enzymes can begin the release of ROS from other enzymes.12,14 The vascular Nox is a multiprotein complex with cytosolic and membrane-related subunits, transferring electrons from NAD(P)H, as an electron donor, to the oxygen molecule to produce superoxide radicals.15 Four isoforms of the Nox enzyme (Nox1, Nox2, Nox4, and Nox5) are typically expressed in vascular cells.16
It has been previously suggested that ROS and Nox enzymes mediate many TGF-β-induced effects.17 However the molecular mechanism of the phosphorylation of the Smad linker region (SmadL) is not very clear. Seeking to settle the long-standing question whether ROS are involved in the non-Smad signaling pathway, in the present study we evaluated the effects of ROS-sensitive p38MAPK, Noxs (main source of ROS in the vasculature), and ROS by using their chemical inhibitors in the TGF-β-induced phosphorylation of the Smad2L. The novelty of our research is that it seeks to clarify the role of Nox enzymes and ROS in the phosphorylation of the Smad2L as the main transcription factor in TGF-β signaling.
Methods
This current in vitro study was conducted in 2016 in the Department of Clinical Biochemistry of Ahvaz Jundishapur University of Medical Sciences (Ahvaz, Iran).
Cell Culture
Human aortic VSMCs were purchased from Pasteur Institute (Iran). The cells were cultured in Dulbecco’s Modified Eagle’s Medium/F-12 (DMEM/F-12; Invitrogen, Iran), containing 10% fetal bovine serum (FBS; Gibco, USA) and 1% antibiotic (Penicillin-Streptomycin; Invitrogen, Iran) at 37 °C in a 5% CO2 incubator. For experimentations, 5×105 cells were seeded in 3.5-mm culture plates and were maintained until confluent. Then, the cells were serum starved so as to synchronize cell growth via culturing in DMEM/F-12 with 0.5% FBS for 16 to 24 hours before treatment. Next, the phospho-Smad2 linker region (pSmad2L) was detected by western blotting in all the experimental groups: a control group (untreated group), a group treated with TGF-β (Cell Signaling Technology, USA), and a group treated with TGF-β plus different inhibitors. All the inhibitors, namely SB-431542, diphenyleneiodonium (DPI), apocynin, N-acetylcysteine (NAC), and SB-202190, were purchased from Sigma-Aldrich (MO-USA). First, the cells were pretreated with specific inhibitors for certain time periods and then TGF-β was added to the culture medium (with 0.5% FBS), with the exception of the control group. For the purposes of a time-course study in the TGF-β groups, the human VSMCs were treated with TGF-β (2 ng/mL) and then harvested at the time points of 5 minutes, 10 minutes, 15 minutes, 30 minutes, 1 hour, and 2 hours (figure 1). The effects of the p38 inhibitor on the pSmad2L were tested by pretreating the cells with 2 concentrations (10 and 20 µM) of SB-202190 for 30 minutes. Thereafter, TGF-β (2 ng/mL) was added to the TGF-β group and the group treated with TGF-β plus different inhibitors for 15 minutes. Another group was also selected for SB-202190 (20 µM) alone as a vehicle (figure 2). The Nox inhibitors were tested on the pSmad2L via the pre-incubation of the cells with DPI and apocynin at 10 and 20 µM for 2 hours prior to the addition of TGF-β (2 ng/mL) to the culture medium. After 15 minutes, the cells were harvested (figure 3). In the last experiment, antioxidant NAC was administered at concentrations of 1 mM, 5 mM, and 10 mM for 1 hour before the addition of TGF-β (2 ng/mL, 15 min) (figure 4). In all the experiments, 1 (SB) group was pretreated with 10 µM of the TβRl/AlK5 inhibitor (SB-431542) for 30 minutes prior to the addition of TGF-β.
Figure1.
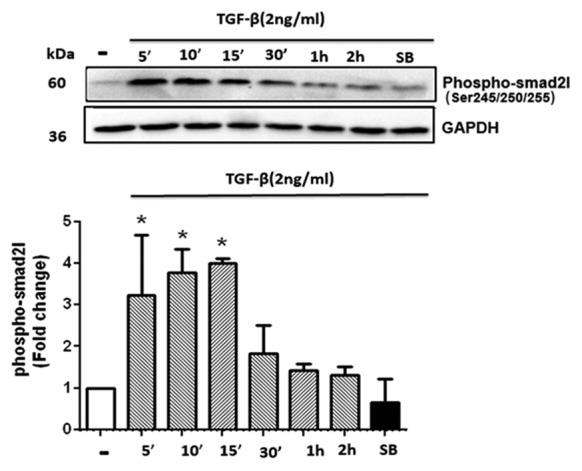
Transforming growth factor-β (TGF-β) stimulated the phosphorylation of the Smad2 linker region in cultured human vascular smooth muscle cells (VSMCs). The cells were treated with TGF-β (2 ng/mL) for a time period ranging from 5 minutes to 2 hours. For the (SB) group, the cells were pre-incubated with a TβRI inhibitor (SB-431542 [10 µM]) for 30 minutes before adding TGF-β. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. The graph represents mean ± SEM of 3 independent blots. *P<0.05 compared with the control.
Figure2.
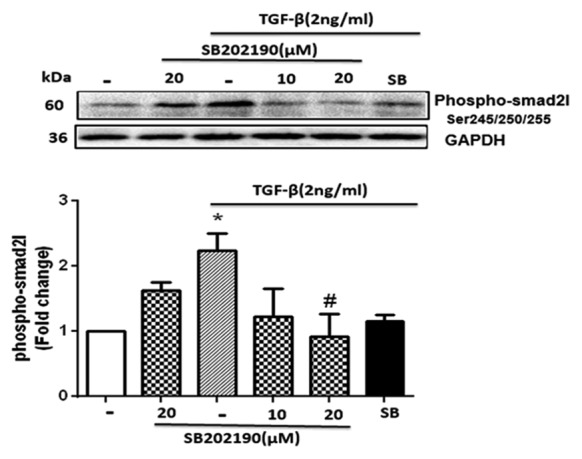
The p38 inhibitor (SB-202190) reduced the transforming growth factor-β (TGF-β)-mediated phosphorylation of the Smad2 linker region in human vascular smooth muscle cells (VSMCs). The cells were pretreated with SB-202190 at concentrations of 10 and 20 µM and an inhibitor of TβRI activity (SB-431542 [SB]) at a concentration of 10 µM (both inhibitors for 30 minutes). Thereafter, TGF-β (2 ng/mL) was added for 15 minutes. *P<0.05 compared to the control.
Figure3.
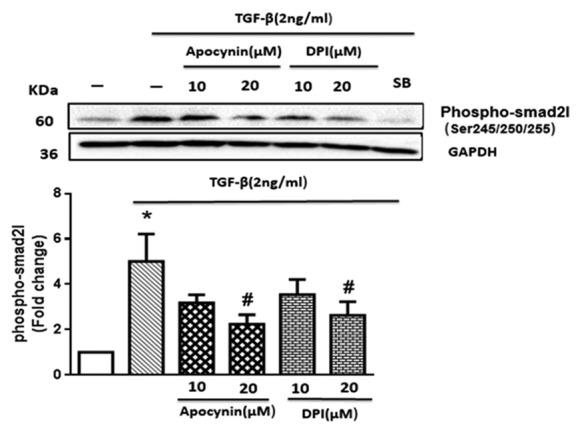
NAD(P)H oxidase (Nox) inhibitors (diphenyleneiodonium [DPI] and apocynin) reduced the transforming growth factor-β (TGF-β)-induced phosphorylation of the Smad2 linker region in human vascular smooth muscle cells (VSMCs). The VSMCs were pretreated with DPI and apocynin at 2 concentrations of 10 and 20 µM for 2 hours and SB-431542 (SB, 10 µM) for 30 minutes prior to being incubated with TGF-β (2 ng/mL) for 15 minutes. The graph represents mean ± SEM of 3 independent blots. *P<0.05 compared to the control; #P<0.05 inhibitors vs. TGF-β.
Figure4.
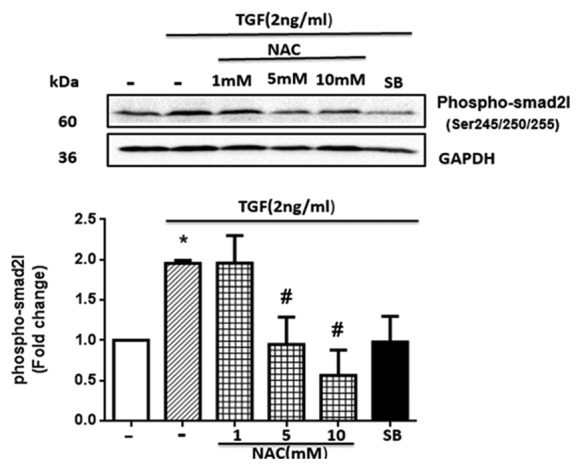
Antioxidant N-acetylcysteine (NAC) blocked the transforming growth factor-β (TGF-β)- induced phosphorylation of the Smad2 linker region in human vascular smooth muscle cells (VSMCs). The cultured VSMCs were pre-incubated with NAC at concentrations of 1, 5, and 10 mM for 1 hour and a TβRI antagonist (SB-431542 [SB]) at a concentration of 10 µM for 30 minutes before treatment with TGF-β (2 ng/mL) for 15 minutes. *P<0.05 compared to the control; #P<0.05 inhibitors vs. TGF-β.
Western Blotting
After treatment, the whole cell lysate was harvested and total concentration protein was measured using a BCA Protein Quantification kit (Parstous Biotechnology, Iran). Samples with equal concentrations of protein (60 µg) were separated on 10% SDS-PAGE. After electrophoresis, the proteins on gel were transferred to a polyvinylidene difluoride membrane (PVDF membrane; Roche Diagnostics, Germany). Next, the membrane was placed in a blocking solution (containing 3% skim milk powder in TBST; Tris-buffered saline+0.1% tween20) for 1 hour at room temperature. The membrane was washed with TBST and incubated with a primary antibody, anti-pSmad2 (Ser245/250/255) rabbit polyclonal antibody (Cell Signaling Technology, USA) (1:1000 dilution with 3% BSA in TBST) at 4°C overnight. After being washed once again, the membrane was incubated with an anti-rabbit IgG horseradish peroxidase (HRP)-labeled secondary antibody (Sigma-Aldrich, MO-USA) (1:10000 dilution with 3% skim milk in TBST) for 1 hour at room temperature, followed by enhanced chemiluminescence (ECL; Bio-Rad, USA) detection. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), as internal control, was used to normalize the results. The stripped membrane was incubated with an anti-GAPDH polyclonal antibody (Abcam, USA) (1:5000 dilution with 3% skimmed milk in TBST), followed by an HRP-conjugated secondary antibody and the ECL detection system. Protein bands were imaged by ChemiDoc (Bio-Rad, USA) and were quantified via densitometric analysis software, ImageJ.
Statistical Analysis
The data were normalized and reported as mean±standard error of the mean (SEM) of 3 independent experiments. The statistical evaluation of the results to determine significant differences between the groups was performed using the nonparametric Kruskal-Wallis test in SPSS, version 16.0. A P value smaller than 0.05 was considered statistically significant.
Results
TGF-β Stimulated the Phosphorylation of the Smad2L in the Cultured Human VSMCs
The pSmad2L level in the TGF-β treated cells significantly increased at 5 minutes (P=0.037), 10 minutes, and 15 minutes (P=0.034) compared with the baseline value and reached a peak (approximately fourfold) at 15 minutes. In the presence of the TβRl/AlK5 inhibitor (SB-431542), the phosphorylation of the Smad2L had completely abolished. Accordingly, we used an incubation time of 15 minutes with TGF-β in the subsequent experiments.
The p38 Inhibitor (SB-202190) Reduced the TGF-β-Mediated Phosphorylation of the Smad2L in the Human VSMCs
The VSMCs treated with TGF-β showed an increase in the pSmad2L level to almost 2.3-fold (P=0.034) by comparison with the control cells. SB-202190 at 10 µM reduced the increased level of the pSmad2L by TGF-β, but the difference was not significant. At the dose of 20 µM, SB-202190 significantly inhibited the increased level of the pSmad2L by TGF-β (P=0.043). The increased level of the pSmad2L, which was stimulated by TGF-β, was inhibited in the cells pretreated with SB-431542. These data indicated the role of ROS-sensitive p38MAPK in the TGF-β-mediated linker region of Smad2 signaling.
The Nox Inhibitors (DPI and apocynin) Reduced the TGF-β-Induced Phosphorylation of the Smad2L in the Human VSMCs
The cells treated with TGF-β exhibited a significant increase (nearly fivefold) in comparison with the control group (P=0.037) in the phosphorylation residues of the Smad2L. DPI and apocynin inhibitors at 10 µM could not significantly reduce the increased level of the pSmad2L following TGF-β treatment. Nonetheless, in the presence of a 20-µM concentration of DPI and apocynin, the level of the pSmad2L significantly decreased (P=0.043). In this experiment, SB-431542 attenuated the intensity of the pSmad2L band. These data demonstrated the potential role of Nox enzymes in the TGF-β-mediated phosphorylation of the Smad2L.
Antioxidant NAC Blocked the TGF-β-Stimulated Phosphorylation of the Smad2L in the Human VSMCs
As is shown in figure 4, TGF-β caused a nearly twofold increase (P=0.034) in the pSmad2L level. It was observed that NAC caused the inhibition in a concentration-dependent manner. Two concentrations (5 mM and 10 mM) of NAC effectively suppressed the TGF-β-stimulated pSmad2L levels (P=0.043), while the 1-mM dose of NAC had no inhibitory effect on the pSmad2L. SB-431542, as a positive control, reduced the Smad2L phosphorylation to the baseline level. Given the inhibition of the Smad2L phosphorylation in the presence of NAC, it can be concluded that ROS are involved in this TGF-β-mediated signaling pathway. The overall schema of the current study is depicted in figure 5.
Figure5.
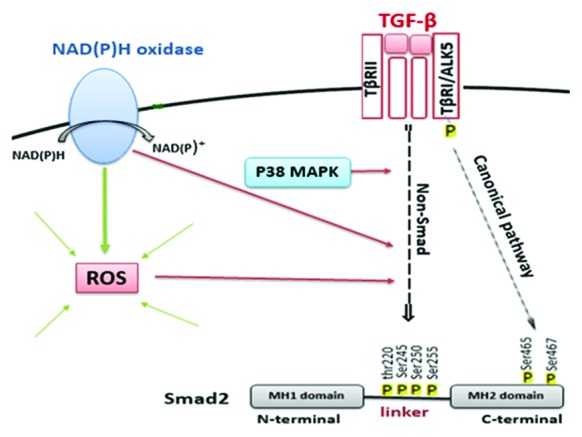
A schema of the effects of NAD(P)H oxidases, ROS, and p38 mitogen-activated protein kinase (MAPK) on transforming growth factor-β (TGF-β)-induced Smad2 linker region phosphorylation in human vascular smooth muscle cells (VSMCs) is shown.
Discussion
The present study was designed to find out what main components are involved in the TGF-β-mediated Smad2L signaling pathway. Our results showed that the TGF-β-induced phosphorylation of the Smad2L was inhibited by using Nox inhibitors, ROS scavengers, and p38MAPK inhibitors in human VSMCs.
TGF-β superfamily members transfer signals via the serine/threonine kinase receptor complex. SB-431542, as a potent competitive inhibitor, by attaching to the ATP-binding domain of TβRI/AlK5 specifically inhibits Smad2/3 activation and blocks TGF-β signal transduction.18,19 We utilized the SB-431542 inhibitor as a positive control in all our experiments: it was able to completely prevent the phosphorylation of the Smad2L. This shows that TGF-β via its receptor affects the Smad2L.
Previous research shows that p38 is a member of the MAPK family, and ROS and TGF-β play a role in the activation of p38MAPK.5,20 We examined the effects of p38MAPK, as a downstream of TβRI activation, on the phosphorylation of the Smad2L. We demonstrated in cultured human VSMCs that SB-202190 (a potent and selective p38 inhibitor) significantly decreased the pSmad2L, which was stimulated by TGF-β. This finding chimes in with the results obtained by Little et al.,21 who observed that in human VSMCs, whereas SB-202190 could not alter the increased band intensity by TGF-β in the C-terminal of Smad2, it was able to reduce it in the Smad2L. The same authors recently demonstrated that by using a specific antibody against each phosphorylated residue (Ser 245/250/255 and Thr 220) in the Smad2L, SB-202190 inhibited only the serine 245 phosphorylation level.4
Evidence has shown that ROS can influence TGF-β signaling via various pathways, including the Smad pathway, MAPK (such as p38) pathway, and Rho-GTPase pathway. On the other hand, studies have demonstrated that TGF-β contributes to oxidative stress by increasing ROS production through Nox induction and also suppresses antioxidant systems such as GSH (glutathione).22 Noxs constitute a membrane-related enzymatic complex, which is a major source of vascular ROS generation.12 Some studies have indicated the role of Noxs and ROS in different effects mediated by TGF-β. Groth et al.23 in a study on human pancreatic carcinoma cells observed that DPI (a pharmacological inhibitor of Noxs) suppressed TGF-β-induced biglycan expression and p38 activation. They suggested that the effects of TGF-β on biglycan expression presumably depended on Rac-1(as a subunit of Noxs required for its enzymatic activity).
In the current investigation, to test whether vascular Noxs and generally ROS are involved in TGF-β-induced Smad2L signaling, we assessed the effects of Nox inhibitors (DPI and apocynin) and also antioxidant NAC on the pSmad2L in cultured human VSMCs. Frequently used as a general inhibitor of flavoenzymes, including Noxs, DPI is not specific only for the inhibition of ROS production by Noxs, whereas apocynin is more specific insofar as it can prevent the association between the p47phox subunit and the membrane-bound heterometric complex of Nox enzymes.14,24 NAC is a nontoxic aminothiol with strong reductive capacity that is widely used as potent antioxidant, because it is a precursor of GSH and also direct ROS scavenging.25
Cheuk et al.26 reported that TGF-β-induced p38 activation was mediated by the Rac1-regulated generation of ROS in cultured human keratinocytes; nonetheless, the authors observed that DPI and pyrrolidine dithiocarbamate (known as an antioxidant) had no inhibitory effects on the TGF-β induced phosphorylation of Smad2. Yuanquan et al.27 observed the inhibitory effects of ROS inhibitors (DPI and NAC) on TGF-β-induced Smad2 phosphorylation in corneal cells. In an investigation conducted by Rhyu et al.20 on renal tubular epithelial cells, it was shown that TGF-β increased cellular ROS generation and the use of antioxidants (NAC and catalase) significantly reduced the TGF-β-induced p38 and Smad2 phosphorylation levels. These studies have not mentioned the exact phosphorylation region (carboxy or linker) in the Smad2 transcription factor. We have already observed in our laboratory that Nox inhibitors (DPI and apocynin) have no inhibitory effect on the phosphorylation of the Smad2 carboxy terminus (phospho-Ser465/467) for dissimilar concentrations in TGF-β-stimulated VSMCs (results not published yet). However, in the present study, DPI and apocynin significantly reduced the increased level of the pSmad2L by TGF-β. In this work, we also showed that NAC at 2 concentrations abolished the TGF-β-stimulated pSmad2L in human VSMCs. Thus, based on these results, we suggest that vascular Noxs and ROS may be signaling mediators in the TGF-β-induced Smad2L pathway but not at canonical TGF-β signaling, which leads to Smad2 carboxy phosphorylation.
Despite the significance of the mechanism which we found in our current study, the final outcome of ROS and Nox inhibition should also be determined on TGF-β target genes such as proteoglycans in VSMCs. Accordingly, in our other project, which is currently underway, our aim is to examine target genes such as ChSy-1 and C4ST-1, enzymes responsible for the synthesis and elongation of the glycosaminoglycan chain of biglycan proteoglycan. In the present study, we were only able to show that Noxs, ROS, and p38MAPK are signaling molecules in the phosphorylation of the TGF-β-mediated Smad2L. Consequently, more research is required to further illustrate this signaling pathway; for example by investigating the effects of TGF-β on the expression and activity of Nox enzymes (especially Nox1 isoform, which is highly expressed in VSMCs) via measuring ROS produced by these enzymes and also by probing into the effects of Noxs and ROS on p38MAPK activation in the presence of TGF-β by utilizing the phospho-p38 antibody, which was not possible for us due to financial and time constraints.
Conclusion
Our results suggest that Noxs have an important role in the phosphorylation the Smad2L in human VSMCs. In addition, by using NAC (ROS scavenger), we confirmed that ROS also play a role in this signaling pathway. Based on the role of the SmadL in the regulation of TGF-β target genes, including proteoglycans in VSMCs, and the involvement of proteoglycans in the subendothelial retention of atherogenic lipoproteins in the atherosclerosis process, identification of the factors affecting the TGF-β-induced SmadL-dependent signaling is important. Our results indicated that the effects of TGF-β on the linker region of the Smad2 transcription factor might depend on ROS (derived from Noxs and other sources) and ROS-related signaling such as p38MAPK in human VSMCs.
Acknowledgement
This study was financially supported by Ahvaz Jundishapur University of Medical Sciences (Grant no. CVRC-9418).
Conflict of Interest:None declared.
References
- 1.Rostam M. TGF-β signalling pathways controlling glycosaminoglycan chain synthesizing enzyme gene expression as a therapeutic target for atherosclerosis [dissertation] Melbourne: RMIT University; 2016. [Google Scholar]
- 2.Rezaei HB, Kamato D, Ansari G, Osman N, Little PJ. Cell biology of Smad2/3 linker region phosphorylation in vascular smooth muscle. Clin Exp Pharmacol Physiol. 2012;39:661–7. doi: 10.1111/j.1440-1681.2011.05592.x. [DOI] [PubMed] [Google Scholar]
- 3.Osman N, Getachew R, Burch M, Lancaster G, Wang R, Wang H, et al. TGF-beta stimulates biglycan core protein synthesis but not glycosaminoglycan chain elongation via Akt phosphorylation in vascular smooth muscle. Growth Factors. 2011;29:230–10. doi: 10.3109/08977194.2011.615747. [DOI] [PubMed] [Google Scholar]
- 4.Rostam MA, Kamato D, Piva TJ, Zheng W, Little PJ, Osman N. The role of specific Smad linker region phosphorylation in TGF-beta mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell Signal. 2016;28:956–66. doi: 10.1016/j.cellsig.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, et al. Transforming growth factor-beta signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017–24. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Krstic J, Trivanovic D, Mojsilovic S, Santibanez JF. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid Med Cell Longev. 2015;2015:654594. doi: 10.1155/2015/654594. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Little PJ, Tannock L, Olin KL, Chait A, Wight TN. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol. 2002;22:55–60. doi: 10.1161/hq0102.101100. [DOI] [PubMed] [Google Scholar]
- 8.Kamato D, Babaahmadi Rezaei H, Getachew R, Thach L, Guidone D, Osman N, et al. (S)-[6]-Gingerol inhibits TGF-beta-stimulated biglycan synthesis but not glycosaminoglycan hyperelongation in human vascular smooth muscle cells. J Pharm Pharmacol 2013;65:1026–36. doi: 10.1111/jphp.12060. [DOI] [PubMed] [Google Scholar]
- 9.Mei S, Gu H, Ward A, Yang X, Guo H, He K, et al. p38 mitogen-activated protein kinase (MAPK) promotes cholesterol ester accumulation in macrophages through inhibition of macroautophagy. J Biol Chem. 2012;287:11761–8. doi: 10.1074/jbc.M111.333575. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–26. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 11.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–25. doi: 10.1016/j.cardiores.2006.02.033. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. 2014;20:2794–814. doi: 10.1089/ars.2013.5607. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gimenez M, Schickling BM, Lopes LR, Miller FJ Jr. Nox1 in cardiovascular diseases: regulation and pathophysiology. Clin Sci (Lond) 2016;130:151–65. doi: 10.1042/CS20150404. [DOI] [PubMed] [Google Scholar]
- 16.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–61. doi: 10.1161/ATVBAHA.108.181610. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baltanas A, Miguel-Carrasco JL, San Jose G, Cebrian C, Moreno MU, Dotor J, et al. A synthetic peptide from transforming growth factor-beta(1) type III receptor inhibits NADPH oxidase and prevents oxidative stress in the kidney of spontaneously hypertensive rats. Antioxid Redox Signal. 2013;19:1607–18. doi: 10.1089/ars.2012.4653. [DOI] [PubMed] [Google Scholar]
- 18.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 19.Du J, Wu Y, Ai Z, Shi X, Chen L, Guo Z. Mechanism of SB431542 in inhibiting mouse embryonic stem cell differentiation. Cell Signal. 2014;26:2107–16. doi: 10.1016/j.cellsig.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–75. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 21.Burch ML, Yang SN, Ballinger ML, Getachew R, Osman N, Little PJ. TGF-beta stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell Mol Life Sci. 2010;67:2077–90. doi: 10.1007/s00018-010-0315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565–77. doi: 10.1016/j.redox.2015.09.009. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groth S, Schulze M, Kalthoff H, Fandrich F, Ungefroren H. Adhesion and Rac1-dependent regulation of biglycan gene expression by transforming growth factor-beta. Evidence for oxidative signaling through NADPH oxidase. J Biol Chem 2005;280:33190–9. doi: 10.1074/jbc.M504249200. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–9. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 25.Sugiura H, Ichikawa T, Liu X, Kobayashi T, Wang XQ, Kawasaki S, et al. N-acetyl-L-cysteine inhibits TGF-beta1-induced profibrotic responses in fibroblasts. Pulm Pharmacol Ther. 2009;22:487–91. doi: 10.1016/j.pupt.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Chiu C, Maddock DA, Zhang Q, Souza KP, Townsend AR, Wan Y. TGF-beta-induced p38 activation is mediated by Rac1-regulated generation of reactive oxygen species in cultured human keratinocytes. Int J Mol Med. 2001;8:251–5. [PubMed] [Google Scholar]
- 27.Yang Y, Wang Z, Yang H, Wang L, Gillespie SR, Wolosin JM, et al. TRPV1 potentiates TGFbeta-induction of corneal myofibroblast development through an oxidative stress-mediated p38-SMAD2 signaling loop. PLoS One. 2013;8:e77300. doi: 10.1371/journal.pone.0077300. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]