

Abstract
Telomeres are small repetitive DNA sequences at the ends of chromosomes which act as a buffer in age-dependent DNA shortening. Insufficient telomere repeats will be recognized as double-strand breaks. Presently, it is becoming more evident that telomere attrition, whether or not caused by mutations in telomere maintenance genes, plays an important role in many inflammatory and age-associated diseases. In this report, a method to (semi)quantitatively assess telomere length and DNA double-strand breaks in formalin-fixed paraffin-embedded (FFPE) tissue is described. Therefore, a novel combination of quantitative fluorescence in situ hybridization, tissue elution, and immunofluorescence staining techniques was developed. Caveolin-1 (type 1 pneumocytes), pro-surfactant protein C (type 2 pneumocytes), club cell-10 (club cells), and alpha smooth muscle actin (smooth muscle cells) markers were used to identify cell types. To visualize all the different probes, restaining the tissue by heat-mediated slide elution is essential. Fluorescent signals of telomeres and DNA double-strand breaks were quantified using the Telometer plugin of ImageJ. As example, we analyzed lung tissue from a familial pulmonary fibrosis patient with a mutation in the telomere-associated gene poly(A)-specific ribonuclease (PARN). The protocol displays a novel opportunity to directly quantitatively link DNA double-strand breaks to telomere length in specific FFPE cells.
Keywords: DNA double-strand breaks, FFPE, fluorescent in situ hybridization, formalin fixed paraffin embedded, gamma-H2AX, immunofluorescence, laser scanning confocal microscopy, poly(A)-specific ribonuclease, pulmonary fibrosis, telomere
Introduction
Telomeres are DNA–protein complexes at the ends of chromosomes, which are involved in preservation of genetic information by acting as a buffer in chromosome end shortening.1–3 In humans, telomeres consists of double-stranded TTAGGG repeats with a combined total length between 4 and 15 kb.4 In healthy subjects, mean telomere length declines approximately 20–30 base pairs per year as a result of incomplete replication during DNA synthesis. Initial telomere shortening has no consequences.5,6 However, when the remaining telomere length is insufficient, the termini of linear chromosomes are recognized as double-strand breaks, resulting in abnormal DNA repair or degradation and eventually in cell senescence.7–10
Telomere shortening is associated with telomeropathy-related diseases, such as dyskeratosis congenita, liver cirrhosis, and pulmonary fibrosis.11 Mutations in genes associated with telomere maintenance are found to be causal to congenital forms of these diseases. For instance, poly(A)-specific ribonuclease, encoded by the gene PARN,12–14 positively affects mRNA stability through deadenylation activity of the poly(A) tail. Deficiencies in the deadenylation process cause reduced production of proteins involved in telomere maintenance and induce telomere shortening.14 Although many telomeropathy-associated mutations have been discovered, the type of cells affected by telomere shortening and how these are related to disease pathogenesis must still be elucidated. Interestingly, PARN mutations initiate a p53-regulated early DNA damage response.14 To assess both telomere length and DNA double-strand breaks in specific cells of formalin-fixed paraffin-embedded (FFPE) lung tissue, DNA and protein staining techniques need to be combined in one assay.
Quantitative fluorescence in situ hybridization (Q-FISH) is widely used to visualize and measure relative DNA or RNA with fluorescently labeled probes containing sequences complementary to target DNA.15,16 For the analysis of relative telomere length, fluorescent signals per individual immunofluorescence (IF) marked cell can be obtained by Q-FISH as previously described by Meeker and coworkers.17 To visualize proteins, IF is a standard staining technique, using antibodies labeled with fluorescent tags.18 Moreover, IF is suitable for quantification.19,20 DNA double-strand breaks initially result in the phosphorylation of histon protein H2AX (gamma-H2AX).21,22 Therefore, gamma-H2AX staining is generally used in DNA damage assays.19,23 In case of telomeres and DNA damage, FISH and IF are mostly used for co-localization studies.24,25 However, no studies have quantified both telomere length and gamma-H2AX signals per cell type specifically. The telomere Q-FISH probe, gamma-H2AX, and specific cell markers must all be identified separately in one tissue specimen. Spectral overlap will occur when all stainings occur simultaneously. To circumvent fluorophore spectral overlap, heat-mediated antibody and FISH probe slide elution have been proposed to allow reuse of the same tissue for a different staining.26–28
In FFPE material, cell-specific antibody stains are essential in identifying different cell subsets in lung material. Lung cells are subdivided into three main compartments: alveolar cells, bronchial and bronchiolar epithelium cells, and pulmonary vascular cells.29 To account for these three groups, we selected alveolar type I- (AT1, CAV-1+) and type II (AT2, pro-Spc+) pneumocytes, club (CC10+) cells and smooth muscle (aSMA+) cells as proof of principle in the assessment of telomere length and gamma-H2AX.
A delicate way to study tissue biomolecules is laser scanning confocal microscopy (LSCM). Advantages include optical sectioning within a single-cell, three-dimensional imaging and high signal-to-background ratios,30,31 which makes the system ideal for quantification of fluorescent labeled cell structures in fixed tissue.32,33 In this study, the main challenge is to quantify FISH and IF signals simultaneously in multiple individually stained cell types in FFPE tissue. Because LSCM can be used to image multiple fluorescent targets at once,34 this is the method of choice.
Here, we describe a novel, accessible method combining Q-FISH and IF staining techniques to quantitatively analyze the relationship between telomere length and DNA double-strand breaks in different cell types of FFPE lung tissue. To our knowledge, the procedures used in this assay were never combined into one protocol before. Lung FFPE material obtained from a pulmonary fibrosis patient with a PARN mutation was included as proof of principle.
Materials and Methods
Tissue Inclusion and Study Approval
Residual tissue was obtained from FFPE lung tissue from patients with pulmonary fibrosis. An experienced lung pathologist reviewed all tissues to select the biopsies showing all features of a distinct pathological usual interstitial pneumonia (UIP) pattern. Lung control tissue was collected from residual donor organ. The patient has written biobank informed consent, and the study was approved by the Medical research Ethics Committees United (MEC-U) of the St Antonius Hospital (approval number R05-08A).
Tissue Preparation and Fluorescence In Situ Hybridization
Two serial sections of 4 μm were cut, air-dried for 10 min, and heated at 56C for 30 min. Slides were then placed at 4C until staining. Material was incubated at 56C for 4 hr. The sequential sections were deparaffinized and hydrated using a xylene and ethanol series, respectively (20 sec per step), after which they were rinsed in tab water (2 times 1 min). Next, the two slides were washed in phosphate-buffered saline (PBS) and boiled in a trisaminomethane-ethylenediaminetetraacetic acid solution (Tris-EDTA; 40-mM Tris, 1-mM EDTA, pH 9) for 20 min. The boiling pan containing the Tris-EDTA solution and the slides were cooled down to room temperature by placing the boiling pan in cold water. Samples were washed once with PBS and thereafter with demineralized (DEMI) water. Next, slides were air-dried and dry slides were incubated, using cover glasses, with a telomere-Cy3 peptide nucleotide acid (PNA) probe (2.70 µg/ml, F1002; Panagene, Daejeon, South Korea) which was diluted in a 2× saline-sodium citrate buffer (SSC) (0.3-M NaCl, 0.03-M sodium citrate), 5% dextran sulfate, 50% deionized formamide, and 0.5% Tween-20 hybridization mixture. The PNA probe consists of the sequence CCCTAACCCTAACCCTAA, which is complementary to the human telomere repeat sequence. Slides with the probe were heated for 5 min on a stretching table at 84C and subsequently placed on an ice-cooled metal plate (3–5 min). The slides were hybridized overnight at 37C in a moistened environment to prevent evaporation. To remove the excess probe, PNA wash solution (1% 1-M Tris, 29% Aquadest, and 70% Formamide) was used for 15 min while shaking. This step was repeated once. Subsequently, slides were washed 2 times 5 min in PBS and PBS-0.05% Tween (PBST) before starting the immunofluorescent staining protocol. The flow diagram of methodological steps is presented in Fig. 1.
Figure 1.
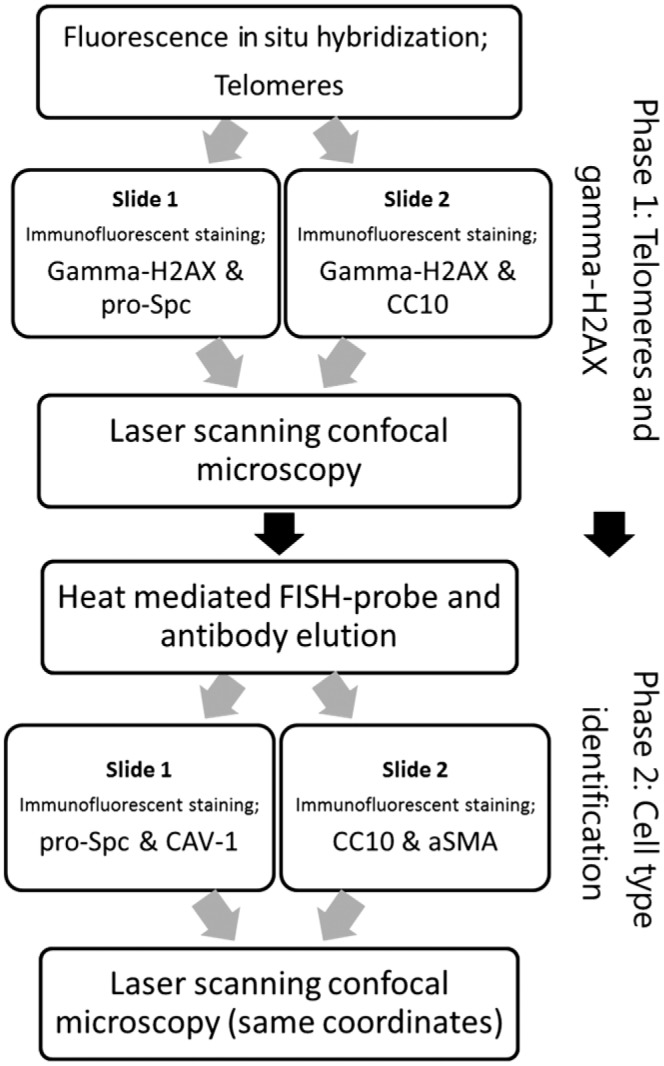
Flow chart of methodological steps. The methods are subdivided in a telomere and gamma-H2AX staining phase and a second, postelution phase, which include the IF staining of specific cell markers. Integration of data from phase 1 and 2 was done by scanning coordinates. Abbreviations: IF, immunofluorescence; CC10, club cell-10; FISH, fluorescence in situ hybridization; aSMA, smooth muscle actin.
Gamma-H2AX Immunofluorescent Staining
Both sequential slides were incubated with 1% bovine serum albumin (BSA) in PBS for 20 min. On slide 1, a predilution of mouse anti-human gamma-H2AX antibody (05-636-I; Merck Millipore, Darmstadt, Germany) and rabbit anti-human pro-Spc antibody (AB3786; Merck Millipore) was applied. The second slide was incubated with a prediluted combination of mouse anti-human gamma-H2AX antibody and rabbit anti-human CC10 antibody (sc-25554; Santa Cruz Biotechnology, Dallas, TX). All primary antibodies were diluted 1:100 in 1% BSA/PBS and incubated 1 hr at room temperature. Slides were washed 3 times 5 min in PBST after each antibody cycle. Subsequent secondary antibodies goat anti-mouse Alexa Fluor 647 (1:100 in 1% BSA/PBS, A21235; Thermo Fisher Scientific, Waltham, MA) and Goat anti-rabbit Alexa Fluor 488 (1:80 in 1% BSA/PBS, A11008, Thermo Fisher Scientific) were applied sequentially on both slides for 30 min at room temperature. Subsequently, tissue was washed in PBS for 5 min. Next, samples were incubated with 4′,6-diamidino-2-phenylindole (DAPI; 25 µg/ml), washed in PBS for 5 min, and rinsed in DEMI water. Samples were air-dried, Vectashield antifade mounting medium (Vector Laboratories, Burlingame, CA) was applied, and slides were stored at 4C until analysis with LSCM (see section “Laser Scanning Confocal Microscopy”).
Tissue Slide Elution and Immunofluorescent Staining to Identify Cell Types
Pro-Spc- and CC10-positive cells were identified during the assessment of telomeres and gamma-H2AX, and tissue coordinates of the locations were saved. However, to avoid spectral overlap due to the use of more than four colors, we introduced heat-mediated FISH-probe and antibody elution. After the first phase of LSCM analysis, cover glasses were removed, and slides were washed in PBS and subsequently boiled in Tris-EDTA (40-mM Tris, 1-mM EDTA, pH 9) for 20 min to elute previously used FISH probe and antibodies. The proceeding steps were as described above using the following premixed primary antibodies for slides 1 and 2. Slide 1: mouse anti-human caveolin-1 (1:50, 610407, BD Biosciences, Franklin Lane, NJ) and rabbit anti-human pro-Spc (1:100, AB3786; Merck Millipore). Slide 2: mouse anti-human αSMA (1:50; Sigma-Aldrich, Darmstadt, Germany) and rabbit anti-human CC10 antibody (1:100 sc-25554; Santa Cruz Biotechnology).
Secondary antibodies were the same for both slides: goat anti-mouse Alexa Fluor 546 (1:50 in 1% BSA/PBS, A11030; Thermo Fisher Scientific) and goat anti-rabbit Alexa Fluor 488 (1:80 in 1% BSA/PBS, A11008; Thermo Fisher Scientific). Afterward, LSCM pictures were taken of the same slide location, using previously saved coordinates.
Laser Scanning Confocal Microscopy
Telomere and gamma-H2AX pictures were taken at the end of phase 1 for quantification (Fig. 1). Digital images of resolution 1024 × 1024 were obtained on an LSM700 laser scanning confocal microscope (Zeiss, Jena, Germany) with a 63×/1.4 oil immersion objective (DIC, M27, Plan-Apochromat). For AT1, at least six cells, but on average 15 cells were obtained from five images scattered over the biopsy and analyzed for each patient. The average of AT2 cells was 20. The minimum amount of images required for the analysis was determined by analyzing 10 images of a control lung using the method of cumulative means/medians.35 Fluorescent probes were excited at 405 nm (DAPI), 488 nm (AF488), 555 nm (Cy3 and AF546), and 639 nm (AF647), using solid-state diode lasers. In addition, emission was detected between 406 and 479 nm (DAPI), 489 and 533 nm (AF488), 556 and 604 nm (Cy3 and AF546), and 640 and 700 nm (AF647), using corresponding emission filters. Pinholes were set equally between fluorescence channels. Z-stacking of nine focal planes with 0.5-µm intervals was used to maximize the coverage of cell nuclei. Tissue X- and Y-coordinates were saved. Next, Z-stack images were captured by a photo multiplier tube (PMT) detector and per laser channel merged into one image by LSM2011 software for further analysis. Microscope and software settings were the same for all experiments, in which fluorescent signal saturation was avoided to maintain detection linearity. After slide elution and IF restaining (see see “Phase 2” in Fig. 1), LSCM was used for identification of cell markers. Previously imaged sites were located using the saved coordinates. To save time capturing the images in phase 2 for identifying cell types using specific fluorescent markers, a maximum Z-stack of four focal planes (1.5 µm) was used.
Image Analysis
To quantify fluorescent signals of telomeres and gamma-H2AX, images were analyzed using the image analysis Telometer plugin (available at http://demarzolab.pathology.jhmi.edu/telometer/index.html) of ImageJ (http://rsb.info.nih.gov/ij/). First, images were converted into 16-bit output. Rolling ball radius and threshold settings were set to 3 and 30 for telomeres and 3 and 15 for gamma-H2AX measurements, respectively. To account for total fluorescence, maximum of pixels was configured at 1,000,000. Next, pictures were normalized for background. In addition, DAPI nuclei were cross-checked with images showing cell-specific markers, encircled, and manually saved as regions of interest (ROIs; Fig. 2J and K). The advantage of using ROIs is that cell selection is concordant between separate telomere and gamma-H2AX images. To avoid background fluorescence in an ROI, cells overlapping unidentified (auto)fluorescent structures were not selected for analysis. Telometer draw mode in the software was not utilized. Next, ImageJ calculated total fluorescence per cluster of positive pixels. Multiple clusters were manually summed up to calculate total telomere or gamma-H2AX fluorescence within a cell.
Figure 2.

Antibody and FISH probe elution procedures are essential for the use of multiple fluorophores. Representative images of a patient with pulmonary fibrosis. Z-stacked LSCM pictures of (A) telomeres (red), (B) gamma-H2AX (yellow), and (C) overlay stains containing the AT2 cell–specific pro-Spc marker (green). (D–F) Magnifications of boxed areas in images A–C, respectively. (G and H) Telomere and gamma-H2AX channels after FISH probe and antibody elution, showing complete loss of cell nucleus–specific signal and persistent presence of autofluorescent collagen and elastin fibers. (I) DAPI DNA staining (blue) and slide coordinates were used to determine the location. (J, K) ImageJ Telometer images showing manually encircled cells of telomere and gamma-H2AX analyzed pictures. Both images were mirrored to (L), indicating pro-Spc (AT2) and caveolin-1 (AT1) specific cell markers, to identify cell types. White arrows indicate autofluorescent collagen and elastin fibers. Scale bars represent 10 µm. Abbreviations: FISH, fluorescence in situ hybridization; LSCM, laser scanning confocal microscopy; AT2, alveolar type 2 pneumocyte; DAPI, 4′,6-diamidino-2-phenylindole; AT1, alveolar type 1 pneumocyte.
Results
Antibody and FISH Probe Elution Procedures Allow for the Use of Multiple Fluorophores
For the analysis of multiple fluorescent probes in one FFPE tissue slide, we used antibody elution to avoid spectral overlap of different fluorophores. In phase 1, we obtained multiple fluorescent pictures from telomere (red) and gamma-H2AX (yellow) stained tissue (Fig. 2A, B, D, and E). For localization analysis, tissue was co-stained with a type 2 cell–specific pro-Spc antibody (green, Fig. 2C and F). To remove fluorescent signals, a Tris-EDTA heat-mediated elution procedure was used, resulting in a complete loss of telomere FISH probe and gamma-H2AX signals (Fig. 2G and H). This was confirmed by incubating tissue solely with the AF647-conjugated secondary antibody (data not shown). After obtaining telomere and gamma-H2AX pictures, antibodies against caveolin-1 (red) and pro-Spc (green) were applied for identification of, respectively, AT1 and AT2 cells at the same coordinates (Fig. 2L).
Telomere Length and Gamma-H2AX Quantification by the Telometer Plugin of ImageJ
To locate and select cells of interest, telomere and gamma-H2AX pictures were mirrored to pictures showing cell-specific markers. All antibodies were checked for cell specificity by experienced lung pathologists using consecutive hematoxylin and eosin (HE)–stained slides for comparison. The DAPI-stained nuclei of selected cells were encircled as ROIs and used in both telomere and gamma-H2AX channels to achieve an exact overlap of cell location between the channels. Image analysis was conducted using the ImageJ Telometer plugin, which was originally designed to quantify telomere fluorescence. In this study, visualization and detection of telomere fluorescence were verified (Fig. 2J), similarly to the work of Meeker and coworkers.17 To investigate whether gamma-H2AX signals could be reliably assessed, staining was performed on adenocarcinomic human alveolar basal epithelial (A549) cell cytospins which were treated with a concentration series of hydrogen peroxide (H2O2: 0–100–250–1000 µM) for 1 hr at 37C to induce DNA double-strand breaks. Recovery of cells was done in Roswell Park Memorial Institute 1640 medium (52400-041; Thermo Fisher Scientific) + 10% fetal calf serum (758093; Greiner Bio-One, Alphen a/d Rijn, the Netherlands) for 30 min at 37C, after which cytospins were produced. These data show that in treated cells, elevated concentrations of H2O2 resulted in significantly higher gamma-H2AX intensity (Fig. 3). Results were dose-dependent. In FFPE lung, gamma-H2AX signal clusters were predominantly found in the AT2 cell. In contrast, the AT1 cell contained no gamma-H2AX signal (Fig. 2K). Furthermore, to assess a variety of lung cell types, pictures were taken in bronchus and artery areas to assess telomere length and gamma-H2AX in club cells (CC10 positive, Fig. 4C) and smooth muscle cells (aSMA positive, Fig. 4F), respectively.
Figure 3.
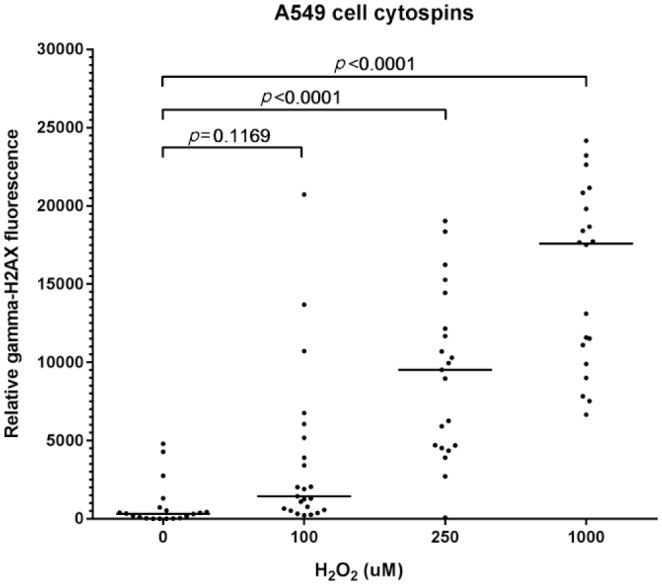
Elevated concentrations of H2O2 result in dose-dependent increase in gamma-H2AX intensity. Adenocarcinomic human alveolar basal epithelial (A549) cells were treated in four batches with 0–100–250–1000 µM H2O2 respectively. Cells for cytospins were spun down on glass slides and stained using a gamma-H2AX antibody. Quantification of fluorescence showed that cells treated with 250- and 1000-µM H2O2 had significant stronger gamma-H2AX signals than null-treated cells. Each dot represents a cell. Medians are indicated with horizontal bars. Values of p<0.05 indicate significant differences (Kruskal–Wallis multiple comparison test). Abbreviation: H2O2, hydrogen peroxide.
Figure 4.

Bronchial club and arterial smooth muscle cells identification using CC10- and aSMA-selective immunofluorescence stains. White arrows point to the (A1–A3) row of bronchus epithelial cells and to (B1–B3) cells of the arterial wall. Z-stacked LSCM pictures of (A1 and B1) telomere (red dots) and (A2 and B2) gamma-H2AX (yellow dots). DAPI DNA staining (blue) and cell-type identification of (A3) CC10-positive club cells (white) and (B3) aSMA-positive smooth muscle cells (purple). Scale bar is valid for all images and represents 10 µm. Abbreviations: CC10, club cell-10; aSMA, smooth muscle actin; LSCM, laser scanning confocal microscopy; DAPI, 4′,6-diamidino-2-phenylindole.
Adjusting for the Amount of DNA Using Nuclear Surface Area Is Preferable Over DAPI Intensity
Telomere and gamma-H2AX signal in the nucleus is quantified, but requires adjustment for the amount of nuclear DNA captured in the LSCM images. To investigate whether either DAPI intensity or nucleus surface area is optimal for the adjustment of the telomere and gamma-H2AX signal, they were compared between two consecutive slides of the same tissue block. The consecutive slides were stained on different time points, using a 30-min incubation step with the same DAPI solution. Correlations between DAPI intensity and nucleus surface area in healthy controls showed a positive correlation (r = 0.8521, p<0.0001, Fig. 5A). In patients, we show that AT2 DAPI intensity is significantly higher in one slide compared with the other slide (p<0.0001), while nucleus surface area was constant between the two slides (Fig. 5B and C). Similar results were found for AT1, club cells, and smooth muscle cells (data not shown). Furthermore, telomere fluorescence was quantified and did not differ between slides (Fig. 5D).
Figure 5.

Nucleus surface area but not DAPI intensity is stable between experiments. (A) Significant spearman correlation in AT2 cells of healthy controls between DAPI intensity and nucleus surface area (r = 0.8521, p<0.0001). (B) Examples of one patient where AT2 DAPI intensity significantly differs between two independent experiments on two consecutive tissue sections. (C) Nucleus surface area and (D) unadjusted telomere fluorescence of the same AT2 cells indicate no significant differences between experiments. DAPI intensity, nucleus surface area, and telomere intensity data were obtained from the Telometer plugin of ImageJ. Each dot represents one cell. Medians are indicated with horizontal bars. The p values indicate significant differences (Mann–Whitney tests; ns = not significant). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; AT2, alveolar type 2 pneumocyte.
Specific Association of Telomere Shortening With DNA Double-strand Breaks in AT1 and AT2 Cells of a Familial Pulmonary Fibrosis Patient With a Poly(A)-Specific Ribonuclease (PARN) Mutation
To put the combined staining procedures into practice, we assessed telomere length and double-strand breaks of 25 AT1 and 27 AT2 cells in a case of familial pulmonary fibrosis with a PARN mutation (NM 001134477:c.98C>T; NP 001127949:p.P33L). This mutation segregated in a family in which two first-degree family members were diagnosed with pulmonary fibrosis. The biopsy contained extensive subpleural fibrosis, consistent with the UIP pattern. Multiple fibroblasts foci were present. In this subject, AT1 cells show significant higher telomere fluorescence (Fig. 6A) and lower gamma-H2AX (Fig. 6B) signals compared with AT2 cells. The correlation analysis between telomere length and DNA double-strand breaks on alveolar epithelial cells resulted in a significant negative correlation (r = −0.5202, p<0.0001, Fig. 5C), implying an increase in DNA double-strand breaks when telomeres shorten.
Figure 6.
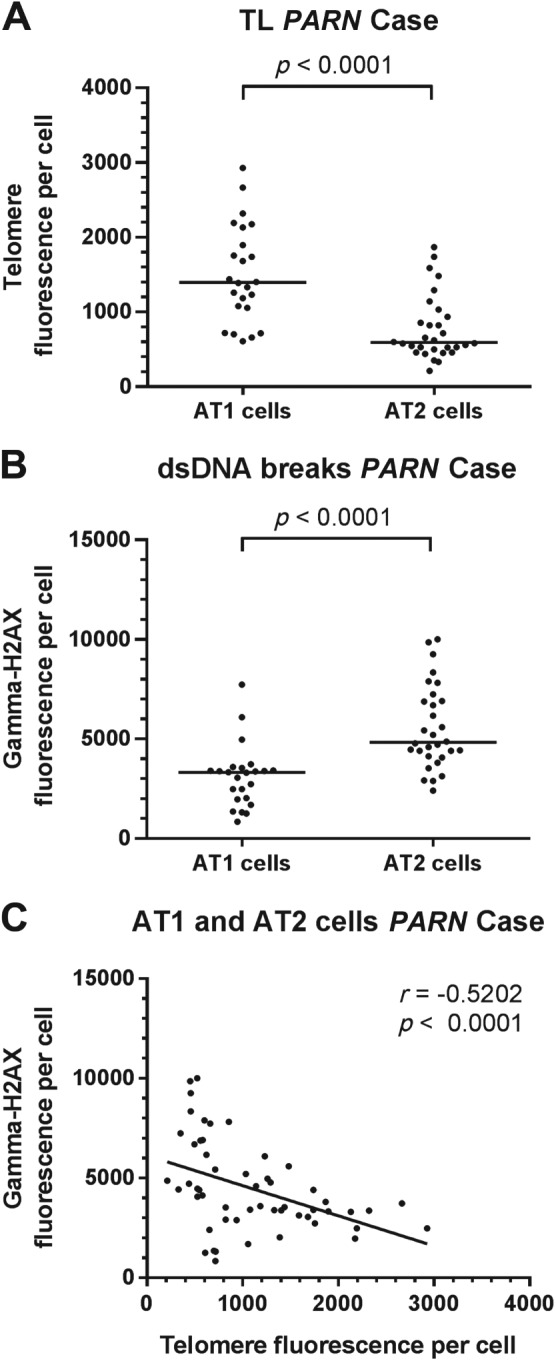
Telomere and gamma-H2AX fluorescence measurements in AT1 and AT2 lung cells of a patient with a PARN mutation. (A) FISH-stained telomere signals are significantly higher in AT1 than in AT2 cells (p<0.0001). (B) Immunofluorescent staining of gamma-H2AX showed significantly lower signals in AT1 cells compared with AT2 cells (p<0.0001). Medians are indicated with horizontal bars. Mann-Whitney tests were used for comparisons. (C) Significant negative spearman correlation in AT1 and AT2 cells between telomere and gamma-H2AX signals (r = −0.5202, p<0.0001). Each dot represents one cell. Abbreviations: AT1, alveolar type 1 pneumocyte; AT2, alveolar type 2 pneumocyte; PARN, poly(A)-specific ribonuclease; FISH, fluorescence in situ hybridization; DAPI, 4′,6-diamidino-2-phenylindole.
Discussion
In this article, we describe a novel method to semiquantitatively assess telomere length and gamma-H2AX–associated DNA damage in specific cell types in FFPE lung tissue using serial staining, elution, and restaining of slides with probes and antibodies. In our hands, gamma-H2AX was a suitable detector of DNA double-strand breaks. To obtain pictures, a standard 4-color detection confocal microscope was utilized. Furthermore, we showed that DAPI-positive nuclear cross-sectional surface area is preferable over DAPI intensity for the adjustment of DNA amount.
Previous approaches only quantified telomere length in one cell type using a cell identifying marker, or assessed cells only on histology features.17,36–38 However, most organs consist of many different cell types, and in some diseased tissues (e.g., fibrosis), histology alone is not sufficient to identify these different cells. Thus, to compare both telomere length and DNA double-strand breaks between multiple cell types, a more comprehensive approach is needed. Here, we identified multiple lung cells, for example, AT1 and AT2 pneumocytes, club cells, and smooth muscle cells, using appropriate antibodies. The locations of AT2 and club cells were selected by means of pro-Spc and CC10 staining, respectively. AT1 cells were identified juxtaposed AT2 cell coordinates, using a secondary staining after the elution procedure. The presence of artery smooth muscle cells was based on DAPI staining and checked afterward using aSMA positivity (Fig. 1). Obtained LSCM pictures often contained fluorescent structures, which were visible in all channels (Fig. 2, white arrows). These structures could include extracellular matrix components, of which elastin fibers and collagen possess autofluorescent features.39,40 Moreover, elastin is abundantly present in the lung.41 Obviously, cells overlaying these structures were not included in the analysis to avoid interfering fluorescence signals during analysis.
The percentage of a cell nucleus captured in a picture is dependent on the cutting procedure of the paraffin block and the Z-stack selection. Both telomere and gamma-H2AX are located in the nucleus. Therefore, measurements had to be adjusted for the amount of cell nucleus captured in a picture. In literature, DAPI intensity and nuclear surface area have been used to adjust telomere and gamma-H2AX measurements.17,36,42 However, we are not aware of any direct comparison of these procedures. Theoretically, we expect that the amount of DNA (DAPI) for each cell type is comparable. First, we have shown that in healthy controls, DAPI intensity correlated significantly with nuclear surface area (r = 0.8521, Fig. 4A), implying that in normal conditions factors are exchangeable. However, significant differences were found between patient DAPI intensity in AT2 cells of two separately stained consecutive slides of the same biopsy (Fig. 5B), while nucleus surface area was constant (Fig. 5C). Moreover, no significant differences were found in telomere signal between the consecutive slides, which implies that DNA is evenly preserved (Fig. 5D). This supports the idea that only DAPI intensity is fluctuating. Because nucleus surface area remained stable, we determined that this is a suitable factor to adjust for the amount of nuclear DNA present in an image.
Based on this methodological work, we analyzed telomere length and DNA double-strand breaks in AT1 and AT2 cells of a familial pulmonary fibrosis case with a rare c.98C>T; p.P33L PARN mutation. PARN is involved in the production of proteins related to telomere biology.14 Therefore, defects in PARN lead to telomere shortening. Previously, we showed that telomeres of surrounding cells of patients with a mutation-induced telomere maintenance problem were significantly longer than telomeres of AT2 cells. We concluded that short AT2 cell telomere length might play a role in the pathogenesis of idiopathic pulmonary fibrosis (IPF), because AT2 cells are the progenitor cells of lung alveoli, thereby preserving the distal lung structure. This requires active telomere maintenance. However, in fibrotic lungs, AT2 cell telomere conservation supposedly cannot be maintained.38 To study different types of surrounding cells, we therefore constructed a method to identify subsets of surrounding cells to investigate whether telomere length differs among these different types of surrounding cells. In a case of PARN mutation–associated pulmonary fibrosis, we showed that AT1 cell telomeres were significantly longer than AT2 cell telomeres. Whether this is a general feature of pulmonary fibrosis has to be investigated.
Telomere shortening plays a postulated role in pulmonary fibrosis according to the model of Chilosi and coworkers.10 In addition, in accordance with literature,12–14 we found that the mutation in PARN segregates with disease in a Dutch family of pulmonary fibrosis patients, implying the causative role of telomere shortening. However, the role of DNA double-strand breaks has not been elucidated in fibrogenesis. PARN mutations initiate a p53-regulated early DNA damage response.14 We found that in this one case, short telomeres correlated with elevated DNA double-strand breaks, specifically in AT2 cells. Crucial high numbers of DNA double-strand breaks result in degradation or abnormal DNA repair and eventually in cell senescence.7–10 A limitation of this study is that only one case with an inherited telomere shortening syndrome is evaluated. However, these data cautiously indicate that DNA double-strand breaks might bridge the gap between telomere shortening and AT2 cell dysfunction in pulmonary fibrosis and deserve further investigation.
In conclusion, this study presents an accessible way to investigate cell type–specific quantitative association of telomere attrition with DNA double-strand breaks in FFPE lung tissue.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: AAB performed the fluorescence in situ hybridization (FISH) and immunofluorescence, and carried out the analysis. AAB, KMK, RG, and CHMM designed the study. AAB wrote the initial manuscript. All authors drafted and approved the final manuscript.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: AA van Batenburg  https://orcid.org/0000-0002-2841-7911
https://orcid.org/0000-0002-2841-7911
Contributor Information
Aernoud A. van Batenburg, Department of Pulmonology, St Antonius ILD Center of Excellence, St Antonius Hospital, Nieuwegein, The Netherlands
Karin M. Kazemier, Department of Pulmonology, St Antonius ILD Center of Excellence, St Antonius Hospital, Nieuwegein, The Netherlands Division of Heart and Lungs, University Medical Center Utrecht, Utrecht, The Netherlands.
Ton Peeters, Department of Pathology, University Medical Center Utrecht, Utrecht, The Netherlands.
Matthijs F. M. van Oosterhout, Department of Pathology, St Antonius Hospital, Nieuwegein, The Netherlands
Joanne J. van der Vis, Department of Pulmonology, St Antonius ILD Center of Excellence, St Antonius Hospital, Nieuwegein, The Netherlands Department of Clinical Chemistry, St Antonius Hospital, Nieuwegein, The Netherlands.
Jan C. Grutters, Department of Pulmonology, St Antonius ILD Center of Excellence, St Antonius Hospital, Nieuwegein, The Netherlands Division of Heart and Lungs, University Medical Center Utrecht, Utrecht, The Netherlands.
Roel Goldschmeding, Department of Pathology, University Medical Center Utrecht, Utrecht, The Netherlands.
Coline H. M. van Moorsel, Department of Pulmonology, St Antonius ILD Center of Excellence, St Antonius Hospital, Nieuwegein, The Netherlands; Division of Heart and Lungs, University Medical Center Utrecht, Utrecht, The Netherlands.
Literature Cited
- 1. Chan SR, Blackburn EH. Telomeres and telomerase. Philos Trans R Soc Lond B Biol Sci. 2004;359(1441):109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maubaret CG, Salpea KD, Romanoski CE, Folkersen L, Cooper JA, Stephanou C, Li KW, Palmen J, Hamsten A, Neil A, Stephens JW, Lusis AJ, Eriksson P, Talmud PJ, Humphries SE. Simon Broome Research Group, EARSII consortium. Association of TERC and OBFC1 haplotypes with mean leukocyte telomere length and risk for coronary heart disease. PLoS ONE. 2013;8(12):e83122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wyatt HD, West SC, Beattie TL. InTERTpreting telomerase structure and function. Nucleic Acids Res. 2010;38(17):5609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Samassekou O, Gadji M, Drouin R, Yan J. Sizing the ends: normal length of human telomeres. Ann Anat. 2010;192(5):284–91. [DOI] [PubMed] [Google Scholar]
- 5. Nawrot TS, Staessen JA, Gardner JP, Aviv A. Telomere length and possible link to X chromosome. Lancet. 2004;363(9408):507–10. [DOI] [PubMed] [Google Scholar]
- 6. Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225(4):951–60. [DOI] [PubMed] [Google Scholar]
- 7. Prescott J, Kraft P, Chasman DI, Savage SA, Mirabello L, Berndt SI, Weissfeld JL, Han J, Hayes RB, Chanock SJ, Hunter DJ, De Vivo I. Genome-wide association study of relative telomere length. PLoS ONE. 2011;6(5):e19635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. d’Adda di, Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–8. [DOI] [PubMed] [Google Scholar]
- 9. Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569–74. [DOI] [PubMed] [Google Scholar]
- 10. Chilosi M, Poletti V, Rossi A. The pathogenesis of COPD and IPF: distinct horns of the same devil? Respir Res. 2012;13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, Weissler J, Fitzgerald J, Kershaw C, Klesney-Tait J, Mageto Y, Shay JW, Ji W, Bilguvar K, Mane S, Lifton RP, Garcia CK. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47(5):512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, Agarwal S. Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat Genet. 2015;47(12):1482–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, Powell J, Cooper N, Foster A, Mohammed S, Plagnol V, Vulliamy T, Dokal I. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cremer T, Landegent J, Bruckner A, Scholl HP, Schardin M, Hager HD, Devilee P, Pearson P, van der Ploeg M. Detection of chromosome aberrations in the human interphase nucleus by visualization of specific target DNAs with radioactive and non-radioactive in situ hybridization techniques: diagnosis of trisomy 18 with probe L1.84. Hum Genet. 1986;74(4):346–52. [DOI] [PubMed] [Google Scholar]
- 16. Iourov IY. Quantitative fluorescence in situ hybridization (QFISH). Methods Mol Biol. 2017;1541:143–9. [DOI] [PubMed] [Google Scholar]
- 17. Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, March GE, De Marzo AM. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002;160(4):1259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramos-Vara JA. Technical aspects of immunohistochemistry. Vet Pathol. 2005;42(4):405–26. [DOI] [PubMed] [Google Scholar]
- 19. Tang MM, Mah LJ, Vasireddy RS, Georgiadis GT, El-Osta A, Royce SG, Karagiannis TC. Quantitation of gammaH2AX foci in tissue samples. J Vis Exp. 2010;40:2063. doi: 10.3791/2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toki MI, Cecchi F, Hembrough T, Syrigos KN, Rimm DL. Proof of the quantitative potential of immunofluorescence by mass spectrometry. Lab Invest. 2017;97(3):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–68. [DOI] [PubMed] [Google Scholar]
- 22. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8(12):957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rothkamm K, Horn S. Gamma-H2AX as protein biomarker for radiation exposure. Ann Ist Super Sanita. 2009;45(3):265–71. [PubMed] [Google Scholar]
- 24. Gonzalez-Garcia MP, Pavelescu I, Canela A, Sevillano X, Leehy KA, Nelson AD, Ibañes M, Shippen DE, Blasco MA, Caño-Delgado AI. Single-cell telomere-length quantification couples telomere length to meristem activity and stem cell development in Arabidopsis. Cell Rep. 2015;11(6):977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lan HY, Mu W, Nikolic-Paterson DJ, Atkins RC. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem. 1995;43(1):97–102. [DOI] [PubMed] [Google Scholar]
- 27. Summersgill BM, Shipley JM. Fluorescence in situ hybridization analysis of formalin fixed paraffin embedded tissues, including tissue microarrays. Methods Mol Biol 2010;659:51–70. [DOI] [PubMed] [Google Scholar]
- 28. Kitayama Y, Igarashi H, Kozu T, Nagura K, Ohashi Y, Sugimura H. Repeated fluorescence in situ hybridization by a microwave-enhanced protocol. Pathol Int. 2006;56(9):490–3. [DOI] [PubMed] [Google Scholar]
- 29. Franks TJ, Colby TV, Travis WD, Tuder RM, Reynolds HY, Brody AR, Cardoso WV, Crystal RG, Drake CJ, Engelhardt J, Frid M, Herzog E, Mason R, Phan SH, Randell SH, Rose MC, Stevens T, Serge J, Sunday ME, Voynow JA, Weinstein BM, Whitsett J, Williams MC. Resident cellular components of the human lung: current knowledge and goals for research on cell phenotyping and function. Proc Am Thorac Soc. 2008;5(7):763–6. [DOI] [PubMed] [Google Scholar]
- 30. Lichtman JW, Conchello JA. Fluorescence microscopy. Nat Methods. 2005;2(12):910–9. [DOI] [PubMed] [Google Scholar]
- 31. Pawley J. Handbook of biological confocal microscopy. Springer; Available from: http://www.springer.com/la/book/9780387259215 [Google Scholar]
- 32. Moerner WE, Orrit M. Illuminating single molecules in condensed matter. Science. 1999;283(5408):1670–6. [DOI] [PubMed] [Google Scholar]
- 33. Weiss S. Fluorescence spectroscopy of single biomolecules. Science. 1999;283(5408):1676–83. [DOI] [PubMed] [Google Scholar]
- 34. Garini Y, Gil A, Bar-Am I, Cabib D, Katzir N. Signal to noise analysis of multiple color fluorescence imaging microscopy. Cytometry. 1999;35(3):214–26. [DOI] [PubMed] [Google Scholar]
- 35. Roche WR. Pathology of the nucleus (current topics in pathology, vol. 82). J. C. E. Underwood (Ed.). Springer-Verlag, Heidelberg, 1990. No. of pages: 343. Price: DM 264 (approx. £90). ISBN: 3 540 51018 4. J Pathol. 1991;163(3):273–3. [Google Scholar]
- 36. Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, Hogan BLM, Mitzner W, Armanios M. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Heaphy CM, Yoon GS, Peskoe SB, Joshu CE, Lee TK, Giovannucci E, Mucci LA, Kenfield SA, Stampfer MJ, Hicks JL, De Marzo AM, Platz EA, Meeker AK. Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discov. 2013;3(10):1130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, van der Vis JJ, Goldschmeding R, Grutters JC, van Moorsel CHM. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE. 2017;12(12):e0189467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Banerjee B, Miedema BE, Chandrasekhar HR. Role of basement membrane collagen and elastin in the autofluorescence spectra of the colon. J Investig Med. 1999;47(6):326–32. [PubMed] [Google Scholar]
- 40. Baschong W, Suetterlin R, Laeng RH. Control of autofluorescence of archival formaldehyde-fixed, paraffin-embedded tissue in confocal laser scanning microscopy (CLSM). J Histochem Cytochem. 2001;49(12):1565–72. [DOI] [PubMed] [Google Scholar]
- 41. Pierce RA, Mariani TJ, Senior RM. Elastin in lung development and disease. Ciba Found Symp. 1995;192:199–212; discussion 212–4. [DOI] [PubMed] [Google Scholar]
- 42. Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1(14):e86704. [DOI] [PMC free article] [PubMed] [Google Scholar]