

CD4+ helper T cells are immune cells that can specifically target cancer cells, but the antigens they recognize on tumor cells are mostly unknown. A new study shows that CD4+ T cells recognize peptides encoded by mutated genes in human melanoma, opening the way for new approaches to cancer immunotherapy.
Cancer immunotherapies based on blocking inhibitory immune signaling with anti-CTLA-4 and anti-PD-1 antibodies or adoptively transferring T cells are now curing patients1 with previously incurable cancers. These therapies rely on activated T cells to recognize and destroy cancer cells. CD8+ 'killer' T cells can directly kill cancer cells, whereas CD4+ 'helper' T cells exist in various functional subtypes that promote, inhibit, or regulate other immune cells, including CD8+ T cells. Both CD8+ and CD4+ T cells are initially activated when their unique T cell receptors recognize specific peptides on the surface of antigen presenting cells, such as dendritic cells or B cells. Once the T cells are activated, re-encounter of the same peptide on target cells, such as cancer cells, triggers T cell effector functions such as direct target cell killing and cytokine production.
In cancer, so-called neoepitope peptides are derived from proteins encoded by mutated genes. Recent advances in next-generation DNA and RNA sequencing now enable rapid mapping of all expressed mutated genes in an individual tumor, and it is possible to predict epitopes that are efficiently presented on the surface of cancer cells. Thus, it has been demonstrated that CD8+ T cells in human melanomas are able to recognize one or more neoepitopes from mutant proteins unique to that specific melanoma. However, efficient methods for studying CD4+ T cells that recognize neoepitopes arising from somatic mutations in cancer have been lacking. In this issue of Nature Medicine, Linnemann et al.2 describe a novel screening platform to systematically search for neoepitope-specific CD4+ T cells in five people with metastatic cutaneous melanoma, a cancer that is typically rich in mutations (Fig. 1). Using this assay, they detect neoepitope-specific CD4+ T cells in melanoma tumors from four of the five subjects they studied.
Figure 1. Identification of human CD4+ T cells that recognize neoepitopes.
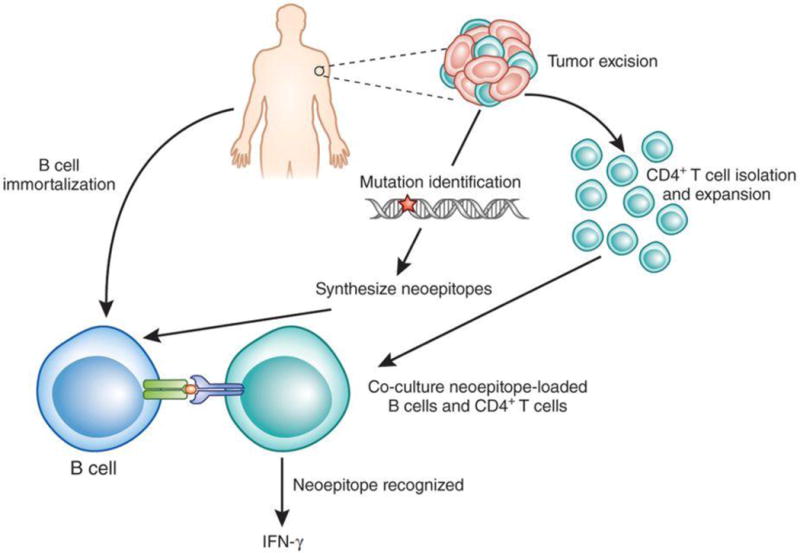
Linnemann et al.2 first purify CD4+ T cells from a person's excised melanoma tumor and expand them in culture. Part of the same tumor tissue is used for DNA/RNA sequencing to identify all protein-encoding mutations, and neoepitope peptides encoded by the mutated genes are synthesized. From the person's blood, B cells are isolated and immortalized by retroviral transduction of the genes encoding BCL-6 and BCL-XL. The CD4+ T cells are then incubated with B cells that are loaded with individual neoepitopes. CD4+ T cells that recognize a given neoepitope will produce cytokines (such as IFN-γ) that can be measured in the culture medium.
Linnemann et al.2 first purified and expanded CD4+ T cells from three melanoma tumors in vitro. They carried out DNA and RNA sequencing of each tumor, and synthesized all neoepitope peptides encoded by mutated sequences. The peptides were then incubated with B cells derived from the same patients as the tumors, which are able to efficiently present peptides to CD4+ T cells (Fig. 1). If a CD4+ T cell recognizes a neoepitope, it will secrete cytokines, particularly interferon (IFN)-γ, which can be detected at the single-cell level using sensitive ELISA and flow cytometry assays.
One of the innovations that was crucial for the success of this study is the manner in which B cells were expanded. Typically, human B cells are immortalized by infecting them with Epstein-Barr virus, the virus that causes infectious mononucleosis. The authors show that this approach causes IFN-γ secretion by some CD4+ T cells used in the screening assay, masking the IFN-γ signal from CD4+ T cells responding to the neoepitope2. To overcome this limitation, Linnemann et al.2 grew large numbers of B cells by retrovirally expressing the oncogene encoding BCL-6 and the anti-apoptotic gene encoding BCL-XL, and this prevented undue stimulation of CD4+T cells in the screening assay.
By using this approach, the authors identified neoepitope-specific CD4+ T cells in two out of three melanoma patients2. The patients' T cells only recognized neoepitopes from the host's tumor, and they preferentially or exclusively recognized the neoepitope over the corresponding native, nonmutated peptide, demonstrating the exquisite specificity of the CD4+ T cells. The same approach was then applied to two additional individuals who had experienced a partial and complete cancer regression, respectively, after infusion of in vitro–expanded T cell cultures2. CD4+ T cells from these cultures also recognized neoepitopes. The neoepitope-specific CD4+ T cells were detectable in the blood of these patients for weeks and months after, but not before, T cell infusion.
The five tumors studied encoded between 99 and 582 neoepitopes, but they contained detectable numbers of CD4+ T cells specific for only 0–3 neoepitopes, suggesting that a substantive CD4+ T cell response to a given neoepitope is rare (∼0.5% of neoepitopes), as observed for CD8+ T cells3. In addition, the number of unique T cell clones—bearing a unique T cell receptor recognizing a specific neoepitope—was fewer than ten per recognized neoepitope, indicating that only very few of each person's naive CD4+ T cells spontaneously responded to each neoepitope. Recognized neoepitopes were unique to each individual, and the tumor from which no neoepitope-specific CD4+ T cells could be isolated had the lowest number of mutations. This suggests a possible correlation between mutational load and the probability of inducing neoepitope-specific CD4+ T cells, a notion recently supported by data on neoepitope-specific CD8+ T cells4.
CD4+ T cells can cause cancer regression through direct killing of cancer cells, by altering the tumor-promoting function of cells in the surrounding tumor microenvironment, and by facilitating the induction, function and tumor infiltration of cancer-specific CD8+ T cells5. Indeed, infusion of CD4+ T cells that recognized NY-ESO-1, a protein encoded by a nonmutated gene frequently overexpressed in cancer cells, was followed by the complete cure of a patient with metastatic melanoma6. Similarly, CD4+ T cells that recognize a neoepitope encoded by mutated ERBB2IP (encoding ERBB2-interacting protein) were isolated from metastatic cholangiosarcoma, grown to large numbers in vitro and returned to the patient, resulting in dramatic tumor shrinkage7. These studies show that cancer-specific CD4+ T cells can cause human tumor regression, adding to the importance of the findings by Linnemann et al.2.
The ability to detect neoepitope specific CD4+ T cells now allows validation of the hypothesis that the presence of neoepitope-specific CD4+ T cells in human tumors correlates with overall clinical outcome. This could be used to predict response to immunotherapies that are thought to be mediated at least in part by CD4+ T cells, such as interleukin-2, anti-CTLA-4, and possibly anti-PD-1. However, CD4+ T cells can assume many different phenotypes, each with distinct pro- or anti-tumor functions, and so better characterization of these cells, freshly isolated and without culturing, is needed. To be applied therapeutically, neoepitope-specific CD4+ T cells could be isolated from tumors or peripheral blood, grown to large numbers in vitro by stimulation with relevant epitopes and returned to the patient for personalized tumor therapy. In addition, the genes encoding the neoepitope-specific TCR could be isolated and expressed in normal, blood-derived CD4+ T cells to create large numbers of genetically engineered, cancer-specific CD4+ T cells for therapy. Finally, person-specific neoepitopes could be formulated into vaccines for the activation of the neoepitope-specific CD4+ T cells already present. Such vaccines will be less cumbersome to manufacture than cell-based therapies, but owing to their modest potency thus far they may be particularly useful in settings of minimal residual disease, for example after surgery8.
A remaining question is whether a tumor's mutational burden correlates with its probability to induce neoepitope-specific CD4+ T cells. Cutaneous melanoma, the tumor type studied by Linnemann et al.2, appears to be the most mutated of all human cancers, mainly as a result of UV-radiation–induced mutations. It is possible that the intensity of immune responses to mutations will correlate with mutational burden, but it remains unclear to what extent T cell immunity to neoepitopes is influenced by the biology of individual tumors, including tumor-induced immunosuppression, and by immune perturbations caused by many conventional cancer therapies. Furthermore, can neoepitope-specific T cells be reliably identified in peripheral blood? How often and how powerfully do T cells recognize neoepitopes versus peptide epitopes derived from nonmutated proteins? Also, which of these responses is better at shrinking tumors without attacking normal tissues? And how frequently do CD4+ T cells recognize peptides containing post-translational modifications9?
Notwithstanding its modest sample size of five individuals, the finding of Linnemann et al.2 that neoepitope-specific CD4+ T cells are commonly present in people with melanoma opens a gateway to a deeper understanding of the interaction between cancer and the immune system, and to harnessing this interaction into novel cancer therapies.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Rosenberg SA. Nat Rev Clin Oncol. 2014;11:630–632. doi: 10.1038/nrclinonc.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linnemann C, et al. Nat Med. 2015;2:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 3.Robbins PF, et al. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder A, et al. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, Cantor H. Cancer Immunol Res. 2014;2:91–98. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- 6.Hunder NN, et al. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran E, et al. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hailemichael Y, et al. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cobbold M, et al. Sci Transl Med. 2013;5:203ra125. doi: 10.1126/scitranslmed.3006061. [DOI] [PMC free article] [PubMed] [Google Scholar]