

Abstract
Essentials.
Metalloproteinases regulate release/shedding of bioactive membrane proteins.
Shedding is critically important for normal cell function in the vasculature.
Levels of platelet receptors GPIb‐IX‐V and GPVI are regulated by metalloproteolytic shedding.
The premier platelet sheddases belong to the A Disintegrins And Metalloproteinase (ADAMs) family.
Modulating ADAM activity may alter platelet adheso‐signalling receptor density and function.
Platelets have a major role in hemostasis and an emerging role in biological processes including inflammation and immunity. Many of these processes require platelet adhesion and localization at sites of tissue damage or infection and regulated platelet activation, mediated by platelet adheso‐signalling receptors, glycoprotein (GP) Ib‐IX‐V and GPVI. Work from a number of laboratories has demonstrated that levels of these receptors are closely regulated by metalloproteinases of the A Disintegrin And Metalloproteinase (ADAM) family, primarily ADAM17 and ADAM10. It is becoming increasingly evident that platelets have important roles in innate immunity, inflammation, and in combating infection that extends beyond processes of hemostasis. This overview will examine the molecular events that regulate levels of platelet receptors and then assess ramifications for these events in settings where hemostasis, inflammation, and infection processes are triggered.
Keywords: glycoprotein, metalloproteinase, platelet, receptor, shedding
1. INTRODUCTION
Along with red cells, platelets feature as a predominant cell type in the bloodstream. Platelet numbers in a healthy individual are usually maintained at a stable level ranging from 150 to 400 × 106 per mL of blood.1, 2 These levels of platelets vastly exceed the numbers required to mount a normal hemostatic response, and so they are consistent with the idea that the role of platelets in biology extends beyond hemostasis.3, 4, 5, 6 Within the critical biological process of hemostasis, platelets play a pivotal role in identifying injured or disrupted endothelium lining the vasculature. Through a number of different but highly integrated processes, platelets transition from a rapidly moving, nonactivated state to a situation where they roll slowly, adhere, and activate at a site of injury.7 This enables recruitment of additional platelets to form a thrombus. This process requires the engagement of platelet receptors that mediate both the rolling and adhesion of platelets, as well as the intraplatelet signalling leading to platelet degranulation, Ca2+ flux, release of secondary agonists such as adenosine diphosphate (ADP) and thromboxanes, exposure of phosphatidylserine and upregulation of fibrinogen‐binding capacity by the platelet‐specific integrin αIIbβ3. This process has been described extensively in a number of recent reviews.8, 9, 10, 11
The process of primary hemostasis is governed by the platelet‐specific adhesion/signalling proteins glycoprotein (GP) Ib‐IX‐V and GPVI which predominantly bind von Willebrand factor (VWF) and collagen, respectively.12 Both receptors can engage with other ligands, however, engagement of these receptors by VWF and collagen coordinate the platelet response to exposed subendothelial matrix across a range of vascular flow rates. As GPVI and the GPIb‐IX‐V complex cooperate and coordinate the platelet adhesion‐signaling response, the relative densities of these receptors on the membrane are important for efficient and effective function.13, 14 This review will discuss molecular mechanisms that rapidly alter the densities of these primary platelet adhesion receptors and influence capacity of platelets to respond. The review will also consider how the same mechanisms modulating platelet and other vascular cell receptors may contribute to vascular inflammation.
1.1. GPIb‐IX‐V
The GPIb‐IX‐V complex consists of GPIbα disulphide linked to GPIbβ and noncovalently linked to GPIX and to GPV. All four subunits of the complex are members of the leucine‐rich repeat family of proteins however the N‐terminal portion of GPIbα is the major ligand‐binding region of the complex (Figure 1). Within the leucine‐rich repeat domain of GPIbα, repeats 2‐4 (amino acids 60‐128) play a crucial role in regulating adhesion to VWF under shear conditions.15, 16 The ectodomain of GPIbα is essential for thrombus formation17 and likely also for other aspects relating to the role of platelets in coagulation and innate immunity responses as this portion of the receptor complex is able to bind a number of key molecular players in these critical pathways.18 Apart from both plasma and extracellular matrix VWF, other GPIbα binding partners include coagulation proteins factors XI and XII, thrombin, thrombospondin, and high‐molecular‐weight kininogen, the leukocyte integrin αMβ2, and P‐selectin, found on activated platelets and endothelial cells. The ectodomain of GPIbα also associates with the extracellular portion of the platelet collagen receptor GPVI19 (see below) and this interaction influences how collagen engages GPVI.19, 20 Whilst the binding sites within GPIbα for these ligands remain to be fully described, all of these binding proteins engage the extracellular region of GPIbα. The consequences of VWF binding to the GPIb‐IX‐V complex has remained a matter of some debate; however, if the A1 domain (the GPIbα‐binding portion of VWF) is presented to the platelet in an appropriate context, this binding interaction can generate powerful intracellular phosphorylation events.21 The cytoplasmic tail portions of GPIbα and GPIbβ associate directly with components of the platelet cytoskeleton including actin, and α‐actinin, and engagement of GPIb‐IX‐V by VWF leads to actin polymerisation, an event that is sensitive to the level of shear stress to which the platelet surface is exposed.22 GPIb‐IX‐V also contains sequences which bind 14‐3‐3ζ at the GPIbα C‐terminus, as well as protein kinase A, tissue necrosis factor‐alpha receptor‐associated factor (TRAF)‐4, and calmodulin binding sites on GPIbβ.23, 24, 25 14‐3‐3ζ in association with phosphoinositol 3‐kinase26 regulates the VWF‐binding affinity of GPIb‐IX‐V and inhibiting 14‐3‐3ζ association blocks receptor signalling.23
Figure 1.
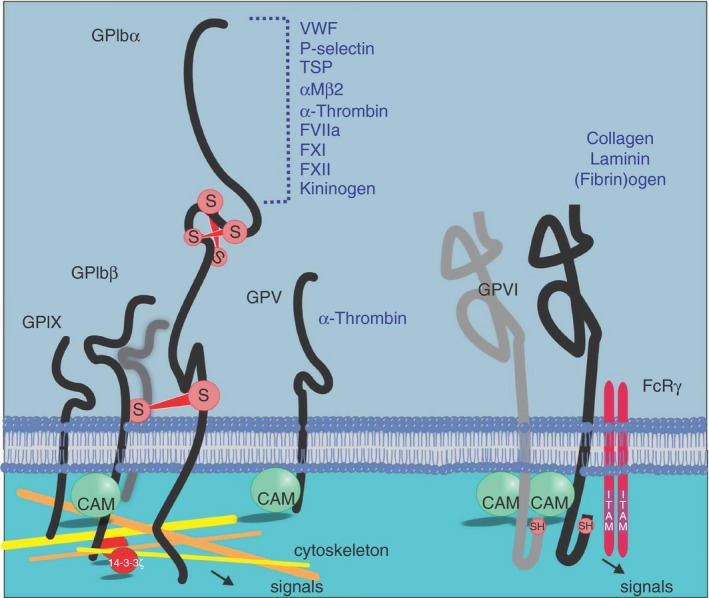
Platelet adhesion/signaling receptors and their ligands. Platelet glycoproteins GPIbα and GPVI can bind a variety of ligands, many of which trigger intracellular signals that lead to platelet activation, degranulation and increased affinity of αIIbβ3 for fibrinogen. Cytoskeletal proteins and 14‐3‐3ζ associate with the cytoplasmic tails of the GPIb‐IX‐V complex. GPIb‐IX‐V contains intersubunit and intrasubunit disulphide bonds, represented by S, symbolising the link between adjacent sulfhydryl moieties in GPIbα and between GPIbα and GPIbβ. Calmodulin (CAM) associates with the juxtamembrane cytoplasmic regions of GPIbβ, GPV, and GPVI. The penultimate residue of GPVI is cysteine, represented by a free sulfhydryl group, SH. Not all cytoplasmic components known to associate with cytoplasmic tails of each receptor are shown
1.2. GPVI
GPVI is one of a number of platelet membrane proteins that can bind collagen (others include GPV, CD36 and α2β1)27; however, it is regarded as the major receptor for collagen as this receptor rapidly triggers powerful intracellular signalling events and activating pathways that enable the platelet to respond and adhere to collagen28 (Figure 1). GPVI is a member of the immunoglobulin (Ig)‐like superfamily of adhesion proteins and contains two extracellular Ig‐like domains. Within the cytoplasmic tail region, GPVI has a calmodulin‐binding sequence29 and a sequence which binds TRAF‐4.25 Surface expression of GPVI requires the tandem expression of the Fc receptor (FcR) γ chain, an ~10‐kD protein that links with GPVI via a salt bridge within the plasma membrane. The cytoplasmic domain of FcRγ contains an immunoreceptor tyrosine activation motif (ITAM) and together the GPVI/FcRγ complex transmits ligand‐induced signalling events into the platelet by triggering phosphorylation of two tyrosine residues with the ITAM and subsequent activation of p72‐spleen tyrosine kinase (Syk).28 Along with collagen, GPVI can bind laminin,30 fibrin,31, 32 fibrinogen,33, 34 histones,35 adiponectin,36 and the extracellular matrix metalloproteinase inducer (EMMPRIN)37 expressed on monocytes and leukocytes. Intact GPVI is also essential for efficient generation of thrombin at the platelet surface.31 The best characterized GPVI interaction is with collagen. When engaged by collagen, both the intracellular38 and extracellular39 regions of GPVI/FcRγ can dimerize and this is likely to aid and enhance the clustering of the receptor,40 and bring ITAMs within the cytoplasmic tail of GPVI/FcRγ together. This triggers tyrosine phosphorylation of members of the Src family of kinases leading to upregulation of phosphoinositide (PI)‐3 kinase activity and integrin activation. As the ectodomains of GPVI and GPIbα are co‐associated on the platelet membrane,19 it is reasonable to believe that these two adheso‐signaling proteins display a level of functional cooperation and coordinated output across a range of shear and ligand exposure conditions. Interestingly, the ectodomain of GPIbα can modulate the rate and extent of activation of platelets by collagen20 and collagen‐related peptide.19 In particular, anti‐GPIbα monoclonal antibodies that target the anionic sulphated tyrosine region of GPIbα (amino acids 269‐282) interfere with platelet responsiveness to these GPVI ligands. This was not related to any specific antibody property as specific removal of the GPIbα ectodomain by treatment with the snake venom protein mocarhagin also ablated collagen‐related peptide‐induced aggregation.19 By altering one or both of the ligand binding regions of GPVI and GPIbα, platelet responsiveness particularly to collagen is likely to be modulated.
2. PLATELET ADHESION RECEPTORS FUNCTION UNDER FLUID SHEAR STRESS
The engagement of the GPIb‐IX‐V complex by VWF and VWF/collagen occurs in flowing blood and is an exquisite example of a shear‐sensitive interaction. The interaction occurs through immobilized VWF partially unfolding under fluid shear and enabling a region within the A1 domain of VWF to be accessible and interact with the N‐terminal portion of the GPIbα subunits within the complex.41 This complex interaction occurs and is sustained under a range of shear stress rates via specialized bonds that are sensitive to shear stress and this interaction directly impacts on the rate and extent of platelet activation.42, 43 GPIbα also senses and responds to changes in fluid shear stress and whilst the mechanisms by which this subunit of the GPIb‐IX‐V complex alters its affinity for VWF remain to be elucidated, regions within the GPIbα ectodomain that do not overlap with the VWF ligand binding region have been identified to influence both the affinity of the receptor for ligand and the ability of the receptor complex to maintain VWF binding under fluid shear stress. These include a region within leucine‐rich repeats 6 and 744 and a mechanosensing domain within the extracellular juxtamembrane region of GPIbα.45 The former was identified as binding a cyclic peptide termed OS‐1, identified by phage display to act as an allosteric inhibitor of VWF‐GPIbα interactions.44 The latter is a region spanning ~60 amino acids lying between the macroglycopeptide and transmembrane domain of GPIbα, which unwinds in response to pulling of prebound VWF A1 domain, as demonstrated in experimental systems using optical tweezers.45 Both studies illustrate the potential of nonligand binding ectodomain regions of the GPIbα subunit to influence and promote ligand binding capacity and affinity, and potentially stabilize receptor ligand interactions at fluid shear rates found in the vasculature.
GPVI plays an important role in hemostasis and thrombosis through integrin activation, supporting adhesion and the initial stages of platelet aggregation. However, patients and mice with platelets lacking GPVI show only a mild bleeding diathesis46, 47, 48 most likely due to the existence of compensatory pathways that generate thrombin and that lead to platelet activation independent of GPVI.49 However, the situation is different under thrombotic conditions where mice with platelets lacking GPVI are protected against arterial thrombosis and subsequent neointima formation50 and demonstrate an impaired thrombus formation at high shear rates.51 GPVI is likely to contribute significantly to stable thrombus formation as the ectodomain is important for efficient thrombin formation31, 52 and GPVI‐fibrin interactions are likely to stabilize a forming thrombus under shear stress.31, 32
Clinical therapies that target platelet responsiveness (antiplatelet therapy) can successfully reduce cardiovascular events, especially in people at higher risk; however, all current antiplatelet therapies carry an increased probability of bleeding. Because loss of GPVI does not result in major hemostatic complications, the therapeutic potential of targeting GPVI is an exciting area that is being actively explored.53, 54, 55 Anti‐GPVI antibodies, particularly single domain antibody clones and fragment antigen‐binding (Fab) fragments may be useful candidate antithrombotic reagents56, 57 as they could potentially interfere with collagen‐GPVI interactions and trigger metalloproteolytic GPVI shedding and/or internalization.
3. METALLOPROTEINASE‐MEDIATED RECEPTOR SHEDDING
Along with triggering fibrinogen binding to the major platelet integrin αIIbβ3, activation of pathways from both the GPIb‐IX‐V complex and GPVI leads to a rapid and irreversible metalloproteinase‐mediated cleavage of the ligand‐binding ectodomains of GPIbα, GPV, and GPVI58, 59, 60 (Figure 2). In a process that was initially characterized in murine platelets,61, 62, 63 the ectodomains of these receptors are cleaved within extracellular juxtamembrane regions resulting in the release of an ~110‐130‐kDa fragment of GPIbα (termed glycocalicin) and an ~55‐kDa GPVI fragment59 from human platelets. The extracellular region of GPV is also released by the action of thrombin to produce an ~20‐kDa platelet‐associated fragment59, 64 and by metalloproteolytic cleavage of the complete extracellular region to leave an ~5‐kDa remnant fragment.59 This process is clearly different from other forms of receptor removal which involve either the export of receptors from the plasma membrane via packaging in extracellular vesicles65 such as occurs with platelet and endothelial P‐selectin, or internalization processes whereby receptors are either moved to ligand‐inaccessible surface‐connected canalicular storage pools or degraded.66, 67 In contrast to GPIbα which appears to be constitutively shed,18, 68 GPVI is stable on the surface of circulating nonactivated platelets59, 60 with no evidence of a platelet‐associated 10‐kDa remnant fragment. This supports the use of intact and soluble GPVI as platelet‐specific markers of activation.69
Figure 2.
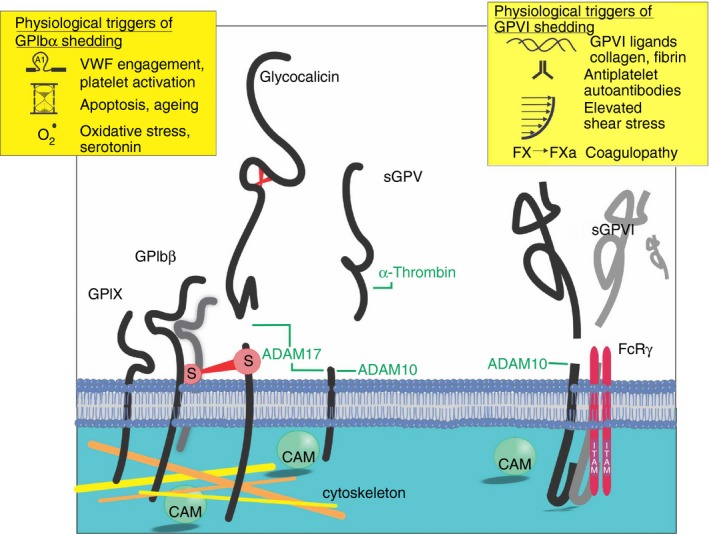
Ectodomains of GPIbα, GPV, and GPVI are shed under conditions of platelet activation. Calmodulin dissociation from the juxtamembrane cytoplasmic region of GPIbβ, GPV and GPVI is a common event also observed in metalloproteolytic shedding of a number of cell receptor ectodomains across vascular biology. GPIbα and GPIbβ intersubunit disulphide bonds which remain intact after shedding of the glycocalicin ectodomain are represented as S symbols
4. PLATELET METALLOPROTEINASES
The receptor and bioactive protein shedding process is mediated by members of the A Disintegrin And Metalloproteinase (ADAM) family with prominent roles for ADAM10 and ADAM17 across biology.70, 71, 72 The ADAMs family of metalloproteinases (Figure 3) has more than 40 members and most members share a basic domain structure consisting of an N‐terminal prodomain followed by a catalytic, a disintegrin, and a cysteine‐rich domain. Most family members contain epidermal growth factor‐like domains (although ADAM10 and ADAM17 do not) followed by a single pass transmembrane domain and a short cytoplasmic tail.72, 73 ADAM10 and ADAM17 are both found on the membrane of resting platelets and these enzymes mediate the cleavage of GPVI and GPIbα, respectively. In murine platelets, shedding of these receptors may involve contributions from both ADAM10 and ADAM17.74 ADAMs proteinases, particularly ADAM10 and ADAM17 are broadly expressed across a variety of cell types, both at the cell surface and in intracellular granules as zymogens. The prodomain is removed from immature ADAMs prior to being brought to the cell surface as mature catalytically active proteins.72 However, on platelets, mature ADAM10 and ADAM17 both seem to be constitutively present at the platelet surface, and in the case of ADAM10 at least, have detectable proteolytic activity.75 The crystal structure of the ADAM10 ectodomain was recently solved76 and revealed a compact arrangement of the domains permitting intrinsic autoinhibition of the catalytic domain within the mature protein by the disintegrin and cysteine‐rich domains and preventing substrate access to the metalloproteinase active site. This suggests that there is a level of control of ADAM10 activity at a membrane surface, under resting conditions.
Figure 3.
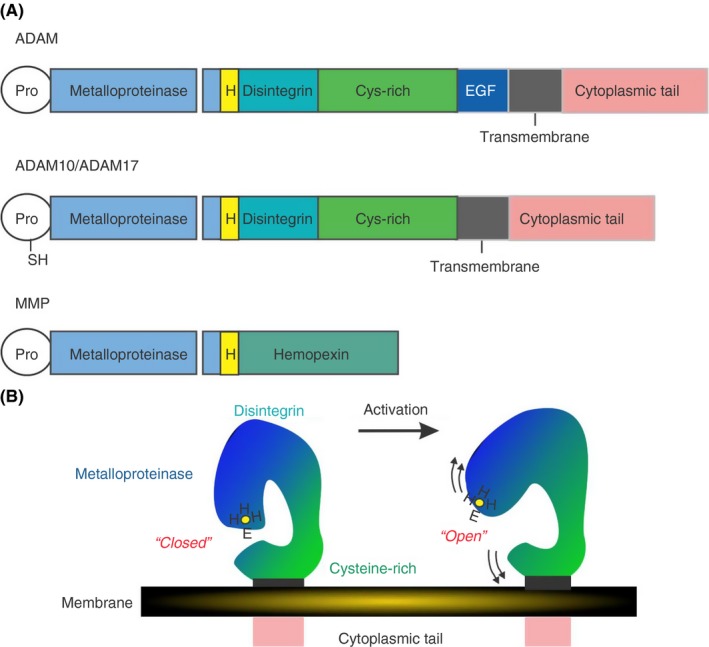
Domain structure of ADAMs. (A) ADAMs have a uniform domain structure with a prodomain that is removed prior to transport to the cell membrane, followed by a metalloproteinase, disintegrin,cysteine‐rich and epidermal growth factor‐like domains domains, a transmembrane domains and a cytoplasmic tail. ADAM10 and ADAM17 do not have EGF‐like domains and do have a free sulfhydryl (SH) group within the prodomain which may coordinate with the HEHH Zn2+ binding sequence within the metalloproteinase domain. (B) Analysis of the crystal structure of human ADAM10 [76] suggests a closed conformation of the enzyme under resting conditions where the cysteine‐rich domain occludes the metalloproteinase active site. Under conditions of activation, the metalloproteinase domain is freed and a substrate can gain access to the catalytic site
Platelet granules also contain a number of members of the matrix metalloproteinase (MMP) family.77 These metalloproteinases generally do not have a transmembrane domain and so are released from storage granules of platelets and many other cell types where they are able to diffuse into extracellular and interstitial spaces. As their name suggests, MMPs cleave many different types of matrix proteins including collagens, laminins, and fibronectin. Platelet‐associated MMP‐1, MMP‐2, MMP‐9, and MMP‐14 have been shown to differentially modulate and at times inhibit thrombus formation by exerting collagenolytic activity.78 MMPs are also able to act at the platelet surface where, for example, MMP‐1 cleaves the thrombin receptor protease activated receptor (PAR)‐1 at a distinct site that strongly activates Rho‐GTP pathways, signalling cell shape change and motility.79 Similarly, MMP‐2 engages with αIIbβ3 and is able to cleave PAR‐1 at a noncanonical site resulting in the activation of phosphatidylinositol 3‐ kinase, enhanced aggregation, and a contribution to arterial thrombosis.80 Dual roles for ADAMs and MMPs in platelet biology are likely, and it will be of great interest to examine how these metalloproteinase superfamily members cooperate and coordinate their respective activities to fully enable platelet function.
5. REGULATORY MECHANISMS THAT MAY INFLUENCE PLATELET RECEPTOR SHEDDING
How platelet receptor levels are regulated on circulating platelets remains an open question. As the process is largely driven by metalloproteinases, control of receptor cleavage events is likely to be provided either by direct inhibition of the catalytic process or by controlling access of the enzyme to the substrate. In the case of GPIbα, roles for a membrane‐proximal region of the GPIbβ cytoplasmic domain81 and a 28‐amino acid mechanosensory domain within the extracellular juxtamembrane region of GPIbα82 in maintenance of stable surface levels of the GPIbα subunit have been identified. Both of these regions regulate the availability of the ADAM17 cleavage site within GPIbα to metalloproteases such as ADAM17 and so aid in control of GPIbα levels.
The endogenous inhibitors of both ADAMs proteins and the MMPs are members of the tissue inhibitors of metalloproteinase (TIMP) family.83 There are four members of the TIMP family and studies have shown that megakaryocytes and platelets have mRNA transcripts and detectable levels of protein for all TIMPs.84 Interestingly, the TIMP‐2 transcript is actively transcribed in thrombin‐stimulated platelets.85 ADAM10 is primarily inhibited by TIMP‐1 and ADAM17 by TIMP‐3 although there is a significant amount of cross‐inhibition amongst the family.86 TIMPs are found in the plasma as well as in intracellular storage granules of most cell types including platelets. TIMPs are able to compete with endogenous ADAM substrates for binding sites within the catalytic and disintegrin‐like domains of ADAMs, and so disrupt access of the catalytic domain for the substrate. However, little is known about the regulatory role of TIMPs in platelet ADAMs and MMP biology.
Tetraspanins featuring the TspanC8 subgroup (Tspan5, 10, 14, 15, 17, and 33)87, 88 and the iRhom subgroup of protease‐inactive rhomboids (iRhom1 and 2)89, 90 have emerged as important regulators of ADAM10 and ADAM17, respectively. In nucleated cells, members of the TspanC8 subgroup are required for correct enzymatic maturation and trafficking of ADAMs to the cell surface. In certain cell types, there is evidence that cells can target the ADAMs to distinct substrates91 and this may involve different TspanC8s and iRhoms.88, 92, 93, 94, 95, 96 Roles for iRhoms and tetraspanins (in particular Tspan14)97 in regulating platelet ADAMs activity are exciting avenues of research enquiry that are likely to explain differential cleavage of GPVI and GPIbα in circulating platelets.
In response to ligand engagement, exposure to elevated shear or during coagulopathy, GPVI is proteolytically cleaved from the platelet surface. In a system that is reminiscent of the classical ADAM17‐mediated shedding of leukocyte L‐selectin,98 detachment of calmodulin from the cytoplasmic juxtamembrane binding site, either by ligand engagement or by treatment of platelets with an inhibitor of calmodulin, triggers the release of the GPVI ectodomain.60 In the following sections, the mechanisms most relevant to physiological shedding of platelet receptors will be discussed, with a focus on the regulation of platelet GPVI levels.
6. TRIGGERS OF PLATELET RECEPTOR SHEDDING
6.1. Laboratory approaches
There are various ways that proteolytic release of GPVI can be triggered involving either physiological or experimental tools and reagents that act either in intracellular and/or extracellular spaces. The standard means of activating ADAMs across cell biology involves treatment of cells with phorbol myristyl acetate (PMA) which crosses the plasma membrane and serves to activate protein kinase C and either trigger passage of mature ADAMs proteins to a membrane surface, or (as in the case of platelets) enhance the proteolytic activity of ADAMs present at the cell surface.91, 99 ADAMs activity can also be upregulated by treatment of cells with thiol‐modifying reagents100 such as N‐ethyl maleimide (NEM) which is a very effective means to trigger almost complete release of GPVI from platelets.59, 101 While the mechanism of action of NEM is not clearly defined, this reagent may react with a cysteinyl group present within the prodomain of all ADAMs. This reactive “cysteine switch” sits within a divalent cation binding site and coordinates the binding of Zn2+ which is essential for catalytic activity of the metalloproteinase.100 NEM and other thiol‐modifying reagents may modify this cysteine group to release any inhibitory mechanism and drive the enzyme into a high affinity enhanced catalytic state. The calmodulin inhibitor N‐(6‐aminohexyl)‐5‐chloro‐1‐naphthalenesulfonamide hydrochloride (W7) is a cell‐permeable competitive antagonist which competes with intracellular calmodulin binding sequences for calmodulin binding. Dissociation of calmodulin from receptor cytoplasmic juxtamembrane sequences forms part of the ligand‐mediated signalling process,98 and so treatment with W7 circumvents the need to provide a ligand. In platelets, treatment with W7 results in detachment of receptor‐bound calmodulin and triggers shedding of GPVI, GPV, and GPIbα.102 Interestingly, the W7 mechanism of action does not increase the endogenous platelet ADAM10 activity,75 suggesting that calmodulin dissociation alters the availability of the ADAM10 cleavage site within GPVI. These reagents are highly useful laboratory tools that have enabled rapid evaluation of ADAMs structure and catalytic potential for a huge range of substrates in both primary cell culture and in cell lines. In platelets, these reagents have broad utility, particularly NEM where treatment of washed platelets or platelet‐rich plasma for 15‐30 minutes with 5 mmol L−1 NEM is sufficient to liberate greater than 90% of GPVI and so permit an assessment of GPVI shedding potential across blood donor populations, as well as create a GPVI‐deficient platelet to aid in the assessment of platelet‐activating plasma components.103
An additional means to remove GPVI from the surface of platelets that has been put to excellent use in mouse models of thrombosis and hemostasis is the use of anti‐GPVI antibodies.104, 105 Injection of mice with intact antibodies or Fab fragments that bound to GPVI epitopes either within the collagen‐binding domain or outside, induced a transient thrombocytopenia and a down regulation of platelet GPVI. Antibody treatment in vitro did not achieve the same loss, and additional work demonstrated the existence of a pathway downstream of GPVI that efficiently led to internalization and irreversible loss of murine GPVI.67 Whether the same process of GPVI internalization occurs in human platelets treated with anti‐GPVI antibodies in vitro, or with anti‐GPVI autoantibodies in vivo has not been reported, however, treatment of human platelets treated with anti‐GPVI antibodies can induce GPVI shedding in vitro. In one study with eight monoclonal antibodies, this loss was independent of engagement of FcγRIIa (present on human but not mouse platelets) by the Fc portion of the antibody106 indicating antibody binding to GPVI could directly trigger metalloproteolysis.
6.2. Exposure to GPVI ligands
Fibrillar collagen type 1 is the major collagen type that engages GPVI. Together with collagen type 3, it is the predominant collagen found in the subendothelium in the blood vessel wall.107 An assessment of GPVI binding of other collagen types has been made; however, the majority of studies of GPVI shedding induced by collagen exposure have utilized the type 1 form. Collagen and the chemically crosslinked collagen‐related peptide (CRP), a GPVI‐specific agonist, both induce shedding of GPVI in suspension assays and require activation of intracellular signalling events including phosphorylation of Src family kinases and Syk as well as activation of PI‐3 kinase but do not require engagement or outside‐in signalling from the integrin αIIbβ3. Ligand‐induced GPVI shedding can proceed in the absence of integrin engagement.60 Shedding triggered by other GPVI ligands which may engage and cluster GPVI through sites other than the collagen‐binding site remains a field of discovery. At the International Society on Thrombosis and Haemostasis meeting in Berlin, a number of new GPVI interactions were discussed in both oral and poster presentations. Fibrin is a more recently described ligand for GPVI and studies have demonstrated that while fibrin–GPVI interaction will generate intracellular signals,32, 108 this signalling is not required for fibrin‐induced GPVI shedding.109 The fibrin interaction with GPVI is mediated by the D‐dimer region of fibrin33, 108 and for GPVI shedding to occur, fibrin must be polymerized.109 Whether fibrin can bind platelet GPVI monomer or dimer33, 108 remains a matter for debate; however, dimeric GPVI‐Fc fusion proteins do not engage fibrin.110 Similarly, the fibrin‐binding site within GPVI is contentious. In one study, the GPVI‐fibrin interaction occurred only with GPVI in dimeric form and could be abrogated by pretreatment with collagen or CRP, implying at least partial overlap of the binding site for these ligands,33 however, fibrin‐GPVI monomer interactions and separate CRP and fibrin binding sites were proposed in another study.108 Under certain experimental conditions and in collaboration with αIIbβ3, the fibrin monomer component fibrinogen also can engage GPVI.33, 34 Understanding how these two GPVI ligands intersect and contribute to GPVI function is important, as selective disruption of one type of GPVI‐ligand interaction, either through competitive inhibition at the ligand‐binding site, or at the level of GPVI dimerization represents an enticing new approach to develop antiplatelet agents with minimal effects on hemostasis.
6.3. Activation of coagulation
Through comparison of sGPVI levels in matched plasma and serum samples from healthy donors, it emerged that GPVI shedding can be triggered by coagulation.111 Through the use of direct inhibitors of thrombin and active factor X (FXa), together with other inhibitors of the coagulation pathway, a major role for thrombin in triggering the release of GPVI either directly by acting on GPVI or indirectly through activation of thrombin receptors on platelets has been ruled out.60, 109, 111 Generation of FXa either through recalcification in the presence of thrombin inhibitors, or by treatment of platelet‐rich plasma with Russell viper venom, a direct FX activator, resulted in the rapid release of GPVI that could be blocked by broad spectrum metalloproteinase inhibitors, and partially blocked by a specific inhibitor of ADAM10. Similar to fibrin‐mediated GPVI shedding,109 this mechanism of shedding did not require platelet activation, degranulation, or aggregation, implying that FXa can directly trigger ADAM10‐mediated cleavage of GPVI. In the absence of a consensus sequence within GPVI that is recognized by FXa, FXa may either directly act on ADAM10 to enhance substrate cleavage or indirectly modulate an intermediary factor that is involved in GPVI stability at the platelet surface. Coagulation‐induced shedding of platelet GPVI in human plasma via a metalloproteinase‐mediated FXa‐dependent mechanism may serve to down‐regulate GPVI expression under procoagulant conditions independent of GPVI ligands. Monitoring levels of sGPVI in plasma from patients with high levels of FXa and/or fibrin deposition who are at risk of developing disseminated intravascular coagulation,111 or sepsis109 may be useful for clinical management of these complex patients.
6.4. Exposure to elevated fluid shear stress
Human platelets normally circulate in a resting state and are exposed to shear rates within a physiologic range (~20‐2000 s−1).11, 112, 113 Platelets may encounter shear rates well beyond 10 000 s−1 under pathologic conditions, for example, in a stenosed atherosclerotic artery or within mechanocirculatory support devices such as left ventricular assist devices (LVADs) or extra‐corporeal membrane oxygenation (ECMO) devices, and become activated and begin to aggregate. Shear‐dependent platelet activation is initiated by binding of plasma VWF to platelets primarily through GPIbα, leading to platelet activation, secretion of ADP, and other agonists, and αIIbβ3‐dependent aggregation.113, 114 Additionally, when exposed to elevated fluid shear stress, metalloproteolytic shedding of GPVI is triggered.101 In experimental systems shear‐induced GPVI shedding was not as a consequence of VWF engagement of GPIb‐IX‐V or platelet activation as shear‐induced shedding occurred in washed platelets where VWF engagement was blocked by anti‐GPIbα or anti‐VWF monoclonal antibodies, and in platelet‐rich plasma isolated from a patient with Type 3 von Willebrand disease (where VWF was absent). Shear‐induced shedding did not require platelet signalling pathways or activation of αIIbβ3 and appeared to be a direct effect of exposure to fluid shear stress. This shear‐dependent instability of the platelet adhesion receptors is likely to be of paramount importance in patient groups where risk of both thrombosis and of bleeding are heightened. Deployment of devices such as LVADs or ECMO necessitates the use of significant antiplatelet and anticoagulant medication however in cohorts of people in receipt of mechanocirculatory support, exposure to fluid shear stress levels approaching 50 000 s−1 was associated with loss of platelet adhesion receptors in conjunction with loss of VWF multimers.115, 116, 117 This loss may combine with other disease‐related vascular factors and contribute to the high rate of serious bleeding seen in this patient cohort.118 Whether measurement of sGPVI levels in plasma samples taken prior to implantation will enable stratification of patients into low‐ and high‐risk bleeding groups115 and the opportunity to tailor antiplatelet and anticoagulant therapy is the subject of ongoing research.
6.5. Antiplatelet antibodies and autoantibodies
In primary immune thrombocytopenia (ITP) and in heparin‐induced thrombocytopenia (HIT), patients generate antibodies that are reactive with antigens on the surface of platelets and megakaryocytes. In ITP, these antibodies disrupt megakaryocytopoiesis, induce platelet apoptosis or opsonise the surface of the platelet enhancing the rate of clearance of platelets by the liver and spleen.119 In at least a subset of patients, antibody binding to platelet surface antigens including GPIbα, αIIbβ3, and α2β1 leads to engagement of platelet FcγRIIa by the Fc portion of the autoantibody. In ITP patients with anti‐GPVI autoantibodies, the loss of responsiveness to collagen by light transmission aggregometry, loss of platelet GPVI by flow cytometry, or enhanced GPVI shedding has been demonstrated.103, 120, 121, 122 In this pathological scenario, autoantibody‐mediated GPVI loss may involve signalling contributions from both GPVI and FcγRIIa. In HIT, autoantibodies that recognise platelet factor‐4 in combination with heparin, form immune complexes which also engage FcγRIIa.123 FcγRIIa is a second ITAM‐containing signalling receptor, and this binding can trigger significant platelet activation and platelet clearance124 as well as activation of GPVI shedding pathways.125
7. CONCLUSION
Metalloproteolysis of receptor ectodomains is a regulatory mechanism that is common to many cell types across cell biology (Figure 4). In some cases, this mechanism liberates a bioactive portion of a latent factor, while in others cases, it is a means of controlling the reactive or adhesive properties of a cell or enabling the cell to sense its surroundings. In platelets, the release of the ligand binding portions of GPVI and GPIbα are likely to modulate the densities of each of these cooperating receptors, parameters that are important for the adhesive properties of the platelet. When using platelets from mice deficient in their subtle receptor density changes act to limit thrombus growth and propagation of coagulation at the site of thrombus formation. However, beyond these outcomes that are critical for hemostasis, and in keeping with the burgeoning roles for platelets in innate immunity and inflammation, loss of these ectodomains are also likely to influence how platelets engage with other cells such as leukocytes and endothelial cells as well as tumor cells.126 Indeed, modulation of receptor levels on the surface of platelets is likely to be critical for new avenues of research where platelets are demonstrated to undergo diapedesis127 and in the utility of platelets for delivery of therapies to critical sites of injury, inflammation, and metastasis.128, 129
Figure 4.
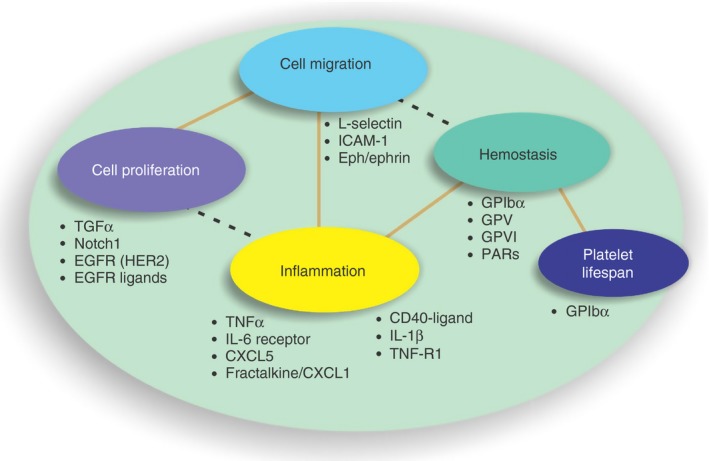
Extracellular proteolysis controls the active levels of a range of growth factors, chemokines and adhesion receptors. Levels of a range of active biological compounds are controlled by the action of members of the ADAMs family of metalloproteinases. Only a selection of these proteins are shown here. These bioactive proteins cooperate and influence a broad spectrum of critical biological processes. EGFR, epidermal growth factor receptor; GP glycoprotein; ICAM, intercellular adhesion molecule; IL, interleukin; TGF, transforming growth factor; TNF, tissue necrosis factor
RELATIONSHIP DISCLOSURE
The author has no conflicts of interest to declare.
Gardiner EE. Proteolytic processing of platelet receptors. Res Pract Thromb Haemost. 2018;2:240–250. 10.1002/rth2.12096
Funding information
This work was supported by funds from the National Health and Medical Research Council of Australia, the National Blood Authority and ACT Health.
REFERENCES
- 1. Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Regulating billions of blood platelets: glycans and beyond. Blood. 2015;126:1877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quach ME, Chen W, Li R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood. 2018; 131:1512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li JL, Zarbock A, Hidalgo A. Platelets as autonomous drones for hemostatic and immune surveillance. J Exp Med. 2017;214:2193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jenne CN, Kubes P. Platelets in inflammation and infection. Platelets. 2015;26:286–92. [DOI] [PubMed] [Google Scholar]
- 5. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franco AT, Corken A, Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood. 2015;126:582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berndt MC, Metharom P, Andrews RK. Primary haemostasis: newer insights. Haemophilia. 2014;20:15–22. [DOI] [PubMed] [Google Scholar]
- 8. Holinstat M. Normal platelet function. Cancer Metastasis Rev. 2017;36:195–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tomaiuolo M, Brass LF, Stalker TJ. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv Cardiol Clin. 2017;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Welsh JD, Poventud‐Fuentes I, Sampietro S, Diamond SL, Stalker TJ, Brass LF. Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature. J Thromb Haemost. 2017;15:526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coenen DM, Mastenbroek TG, Cosemans JMEM. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. 2017;130:2819–28. [DOI] [PubMed] [Google Scholar]
- 12. Gardiner EE, Andrews RK. Structure and function of platelet receptors initiating blood clotting. Adv Exp Med Biol. 2014;844:263–75. [DOI] [PubMed] [Google Scholar]
- 13. Kruss S, Erpenbeck L, Amschler K, et al. Adhesion maturation of neutrophils on nanoscopically presented platelet glycoprotein Ibα. ACS Nano. 2013;7:9984–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen H, Locke D, Liu Y, Liu C, Kahn ML. The platelet receptor GPVI mediates both adhesion and signaling responses to collagen in a receptor density‐dependent fashion. J Biol Chem. 2002;277:3011–9. [DOI] [PubMed] [Google Scholar]
- 15. Shen Y, Cranmer SL, Aprico A, et al. Leucine‐rich repeats 2‐4 (Leu60‐Glu128) of platelet glycoprotein Ibα regulate shear‐dependent cell adhesion to von Willebrand factor. J Biol Chem. 2006;281:26419–23. [DOI] [PubMed] [Google Scholar]
- 16. Shen Y, Romo GM, Dong JF, et al. Requirement of leucine‐rich repeats of glycoprotein (GP) Ibα for shear‐dependent and static binding of von Willebrand factor to the platelet membrane GP Ib‐IX‐V complex. Blood. 2000;95:903–10. [PubMed] [Google Scholar]
- 17. Bergmeier W, Piffath CL, Goerge T, et al. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci USA. 2006;103:16900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrews RK, Gardiner EE. Metalloproteolytic receptor shedding…platelets “acting their age”. Platelets. 2016;27:512–8. [DOI] [PubMed] [Google Scholar]
- 19. Arthur JF, Gardiner EE, Matzaris M, et al. Glycoprotein VI is associated with GPIb‐IX‐V on the membrane of resting and activated platelets. Thromb Haemost. 2005;93:716–23. [DOI] [PubMed] [Google Scholar]
- 20. Ruan CG, Du XP, Xi XD, Castaldi PA, Berndt MC. A murine antiglycoprotein Ib complex monoclonal antibody, SZ2, inhibits platelet aggregation induced by both ristocetin and collagen. Blood. 1987;69:570–7. [PubMed] [Google Scholar]
- 21. Gardiner EE, Arthur JF, Shen Y, et al. GPIbα‐selective activation of platelets induces platelet signaling events comparable to GPVI activation events. Platelets. 2010;21:244–52. [DOI] [PubMed] [Google Scholar]
- 22. Mistry N, Cranmer SL, Yuan Y, et al. Cytoskeletal regulation of the platelet glycoprotein Ib/V/IX‐von willebrand factor interaction. Blood. 2000;96:3480–9. [PubMed] [Google Scholar]
- 23. Mu F‐T, Andrews RK, Arthur JF, et al. A functional 14‐3‐3ζ‐independent association of PI3‐kinase with glycoprotein Ibα, the major ligand‐binding subunit of the platelet glycoprotein Ib‐IX‐V complex. Blood. 2008;111:4580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andrews RK, Munday AD, Mitchell CA, Berndt MC. Interaction of calmodulin with the cytoplasmic domain of the platelet membrane glycoprotein Ib‐IX‐V complex. Blood. 2001;98:681–7. [DOI] [PubMed] [Google Scholar]
- 25. Arthur JF, Shen Y, Gardiner EE, et al. TNF receptor‐associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J Thromb Haemost. 2011;9:163–72. [DOI] [PubMed] [Google Scholar]
- 26. Mu FT, Cranmer SL, Andrews RK, Berndt MC. Functional association of phosphoinositide‐3‐kinase with platelet glycoprotein Ibα, the major ligand‐binding subunit of the glycoprotein Ib–IX–V complex. J Thromb Haemost. 2010;8:324–30. [DOI] [PubMed] [Google Scholar]
- 27. Kehrel B. Platelet receptors for collagens. Platelets. 1995;6:11–6. [DOI] [PubMed] [Google Scholar]
- 28. Nieswandt B, Watson SP. Platelet‐collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–61. [DOI] [PubMed] [Google Scholar]
- 29. Andrews RK, Suzuki‐Inoue K, Shen Y, Tulasne D, Watson SP, Berndt MC. Interaction of calmodulin with the cytoplasmic domain of platelet glycoprotein VI. Blood. 2002;99:4219–21. [DOI] [PubMed] [Google Scholar]
- 30. Inoue O, Suzuki‐Inoue K, McCarty OJT, et al. Laminin stimulates spreading of platelets through integrin α6β1‐dependent activation of GPVI. Blood. 2006;107:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mammadova‐Bach E, Ollivier V, Loyau S, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood. 2015;126:683–91. [DOI] [PubMed] [Google Scholar]
- 32. Alshehri OM, Hughes CE, Montague S, et al. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126:1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Induruwa I, Moroi M, Bonna A, et al. Platelet collagen receptor Glycoprotein VI‐dimer recognizes fibrinogen and fibrin through their D‐domains, contributing to platelet adhesion and activation during thrombus formation. J Thromb Haemost. 2018;16:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mangin PH, Onselaer MB, Receveur N, et al. Immobilized fibrinogen activates human platelets through GPVI. Haematologica. 2018; 10.3324/haematol.2017.182972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alshehri Osama M, Montague S, Watson S, et al. Activation of glycoprotein VI (GPVI) and C‐type lectin‐like receptor‐2 (CLEC‐2) underlies platelet activation by diesel exhaust particles and other charged/hydrophobic ligands. Biochem J. 2015;468:459–73. [DOI] [PubMed] [Google Scholar]
- 36. Riba R, Hughes CE, Graham A, Watson SP, Naseem KM. Globular adiponectin induces platelet activation through the collagen receptor GPVI‐Fc receptor γ chain complex. J Thromb Haemost. 2008;6:1012–20. [DOI] [PubMed] [Google Scholar]
- 37. Seizer P, Borst O, Langer HF, et al. EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI‐EMMPRIN interaction. Thromb Haemost. 2009;101:682–6. [DOI] [PubMed] [Google Scholar]
- 38. Arthur JF, Shen Y, Kahn ML, Berndt MC, Andrews RK, Gardiner EE. Ligand binding rapidly induces disulfide‐dependent dimerization of glycoprotein VI on the platelet plasma membrane. J Biol Chem. 2007;282:30434–41. [DOI] [PubMed] [Google Scholar]
- 39. Jung SM, Moroi M, Soejima K, et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J Biol Chem. 2012;287:30000–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poulter NS, Pollitt AY, Owen DM, et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J Thromb Haemost. 2017;15:549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruggeri ZM, Mendolicchio GL. Interaction of von Willebrand factor with platelets and the vessel wall. Hämostaseologie. 2015;35:211–24. [DOI] [PubMed] [Google Scholar]
- 42. Yago T, Lou J, Wu T, et al. Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. 2008;118:3195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor‐ligand flex‐bond important in the vasculature. Nature. 2010;466:992–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McEwan PA, Andrews RK, Emsley J. Glycoprotein Ibα inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism. Blood. 2009;114:4883–5. [DOI] [PubMed] [Google Scholar]
- 45. Zhang W, Deng W, Zhou L, et al. Identification of a juxtamembrane mechanosensitive domain in the platelet mechanosensor glycoprotein Ib‐IX complex. Blood. 2015;125:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arthur JF, Dunkley S, Andrews RK. Platelet glycoprotein VI‐related clinical defects. Br J Haematol. 2007;139:363–72. [DOI] [PubMed] [Google Scholar]
- 47. Nurden A, Nurden P. Advances in our understanding of the molecular basis of disorders of platelet function. J Thromb Haemost. 2011;9:76–91. [DOI] [PubMed] [Google Scholar]
- 48. Nurden AT, Freson K, Seligsohn U. Inherited platelet disorders. Haemophilia. 2012;18:154–60. [DOI] [PubMed] [Google Scholar]
- 49. Bynagari‐Settipalli YS, Cornelissen I, Palmer D, et al. Redundancy and interaction of thrombin‐ and collagen‐mediated platelet activation in tail bleeding and carotid thrombosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:2563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Konishi H, Katoh Y, Takaya N, et al. Platelets activated by collagen through immunoreceptor tyrosine‐based activation motif play pivotal role in initiation and generation of neointimal hyperplasia after vascular injury. Circulation. 2002;105:912–6. [DOI] [PubMed] [Google Scholar]
- 51. Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lecut C, Feijge MA, Cosemans JM, Jandrot‐Perrus M, Heemskerk JW. Fibrillar type I collagens enhance platelet‐dependent thrombin generation via glycoprotein VI with direct support of α2β1 but not αΙΙbβ3 integrin. Thromb Haemost. 2005;94:107–14. [DOI] [PubMed] [Google Scholar]
- 53. Chatterjee M, Gawaz M. Clinical significance of receptor shedding‐platelet GPVI as an emerging diagnostic and therapeutic tool. Platelets. 2017;28:362–71. [DOI] [PubMed] [Google Scholar]
- 54. Jamasbi J, Ayabe K, Goto S, Nieswandt B, Peter K, Siess W. Platelet receptors as therapeutic targets: past, present and future. Thromb Haemost. 2017;117:1249–57. [DOI] [PubMed] [Google Scholar]
- 55. Andrews RK, Arthur JF, Gardiner EE. Targeting GPVI as a novel antithrombotic strategy. J Blood Med. 2014;5:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zahid M, Loyau S, Bouabdelli M, Aubrey N, Jandrot‐Perrus M, Billiald P. Design and reshaping of an scFv directed against human platelet glycoprotein VI with diagnostic potential. Anal Biochem. 2011;417:274–82. [DOI] [PubMed] [Google Scholar]
- 57. Walker A, Pugh N, Garner SF, et al. Single domain antibodies against the collagen signalling receptor glycoprotein VI are inhibitors of collagen induced thrombus formation. Platelets. 2009;20:268–76. [DOI] [PubMed] [Google Scholar]
- 58. Andrews RK, Gardiner EE. Basic mechanisms of platelet receptor shedding. Platelets. 2017;28:319–24. [DOI] [PubMed] [Google Scholar]
- 59. Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb‐IX‐V by ADAM family metalloproteinases. J Thromb Haemost. 2007;5:1530–7. [DOI] [PubMed] [Google Scholar]
- 60. Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet‐derived metalloproteinase. Blood. 2004;104:3611–7. [DOI] [PubMed] [Google Scholar]
- 61. Bergmeier W, Piffath CL, Cheng G, et al. Tumor necrosis factor‐α‐converting enzyme (ADAM17) mediates GPIbα shedding from platelets in vitro and in vivo. Circ Res. 2004;95:677–83. [DOI] [PubMed] [Google Scholar]
- 62. Bergmeier W, Rabie T, Strehl A, et al. GPVI down‐regulation in murine platelets through metalloproteinase‐dependent shedding. Thromb Haemost. 2004;91:951–8. [DOI] [PubMed] [Google Scholar]
- 63. Bender M, Stegner D, Nieswandt B. Model systems for platelet receptor shedding. Platelets. 2017;28:325–32. [DOI] [PubMed] [Google Scholar]
- 64. Ravanat C, Freund M, Mangin P, et al. GPV is a marker of in vivo platelet activation – study in a rat thrombosis model. Thromb Haemost. 2000;83:327–33. [PubMed] [Google Scholar]
- 65. Arraud N, Linares R, Tan S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. 2014;12:614–27. [DOI] [PubMed] [Google Scholar]
- 66. Furman MI, Nurden P, Berndt MC, et al. The cleaved peptide of PAR1 results in a redistribution of the platelet surface GPIb‐IX‐V complex to the surface‐connected canalicular system. Thromb Haemost. 2000;84:897–903. [PubMed] [Google Scholar]
- 67. Rabie T, Varga‐Szabo D, Bender M, et al. Diverging signaling events control the pathway of GPVI down‐regulation in vivo. Blood. 2007;110:529–35. [DOI] [PubMed] [Google Scholar]
- 68. Coller BS, Kalomiris E, Steinberg M, Scudder LE. Evidence that glycocalicin circulates in normal plasma. J Clin Invest. 1984;73:794–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Al‐Tamimi M, Arthur JF, Gardiner EE, Andrews RK. Focusing on plasma glycoprotein VI. Thromb Haemost. 2012;107:648–55. [DOI] [PubMed] [Google Scholar]
- 70. Lisi S, D'Amore M, Sisto M. ADAM17 at the interface between inflammation and autoimmunity. Immunol Lett. 2014;162:159–69. [DOI] [PubMed] [Google Scholar]
- 71. Saftig P, Reiss K. The, “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527–35. [DOI] [PubMed] [Google Scholar]
- 72. Giebeler N, Zigrino P. A disintegrin and metalloprotease (ADAM): historical overview of their functions. Toxins. 2016;8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM‐mediated shedding in vascular biology. Eur J Cell Biol. 2012;91:472–85. [DOI] [PubMed] [Google Scholar]
- 74. Bender M, Hofmann S, Stegner D, et al. Differentially regulated GPVI ectodomain shedding by multiple platelet‐expressed proteinases. Blood. 2010;116:3347–55. [DOI] [PubMed] [Google Scholar]
- 75. Facey A, Pinar I, Arthur JF, et al. A‐Disintegrin‐And‐Metalloproteinase (ADAM) 10 activity on resting and activated platelets. Biochemistry. 2016;55:1187–94. [DOI] [PubMed] [Google Scholar]
- 76. Seegar TCM, Killingsworth LB, Saha N, et al. Structural basis for regulated proteolysis by the alpha‐secretase ADAM10. Cell. 2017;171:1638–48.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood. 2011;118:1903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mastenbroek TG, Feijge MA, Kremers RM, et al. Platelet‐associated matrix metalloproteinases regulate thrombus formation and exert local collagenolytic activity. Arterioscler Thromb Vasc Biol. 2015;35:2554–61. [DOI] [PubMed] [Google Scholar]
- 79. Trivedi V, Boire A, Tchernychev B, et al. Platelet matrix metalloprotease‐1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sebastiano M, Momi S, Falcinelli E, Bury L, Hoylaerts MF, Gresele P. A novel mechanism regulating human platelet activation by MMP‐2‐mediated PAR1 biased signaling. Blood. 2017;129:883–95. [DOI] [PubMed] [Google Scholar]
- 81. Mo X, Nguyen NX, Mu FT, et al. Transmembrane and trans‐subunit regulation of ectodomain shedding of platelet glycoprotein Ibα. J Biol Chem. 2010;285:32096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Deng W, Xu Y, Chen W, et al. Platelet clearance via shear‐induced unfolding of a membrane mechanoreceptor. Nat Commun. 2016;7:12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Villeneuve J, Block A, Le Bousse‐Kerdilès M‐C, et al. Tissue inhibitors of matrix metalloproteinases in platelets and megakaryocytes: a novel organization for these secreted proteins. Exp Hematol. 2009;37:849–56. [DOI] [PubMed] [Google Scholar]
- 85. Gresele P, Falcinelli E, Sebastiano M, Momi S. Matrix metalloproteinases and platelet function. Prog Mol Biol Transl Sci. 2017;147:133–65. [DOI] [PubMed] [Google Scholar]
- 86. Jackson HW, Defamie V, Waterhouse P, Khokha R. TIMPs: versatile extracellular regulators in cancer. Nat Rev Cancer. 2017;17:38–53. [DOI] [PubMed] [Google Scholar]
- 87. Haining EJ, Yang J, Bailey RL, et al. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J Biol Chem. 2012;287:39753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dornier E, Coumailleau F, Ottavi JF, et al. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J Cell Biol. 2012;199:481–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science. 2012;335:225–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. McIlwain DR, Lang PA, Maretzky T, et al. iRhom2 regulation of TACE controls TNF‐mediated protection against Listeria and responses to LPS. Science. 2012;335:229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lorenzen I, Lokau J, Korpys Y, et al. Control of ADAM17 activity by regulation of its cellular localisation. Sci Rep. 2016;6:35067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Issuree PD, Maretzky T, McIlwain DR, et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J Clin Invest. 2013;123:928–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maretzky T, McIlwain DR, Issuree PD, et al. iRhom2 controls the substrate selectivity of stimulated ADAM17‐dependent ectodomain shedding. Proc Natl Acad Sci USA. 2013;110:11433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jouannet S, Saint‐Pol J, Fernandez L, et al. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell Mol Life Sci. 2016;73:1895–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Saint‐Pol J, Eschenbrenner E, Dornier E, Boucheix C, Charrin S, Rubinstein E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem Soc Trans. 2017;45:937–44. [DOI] [PubMed] [Google Scholar]
- 96. Reyat JS, Chimen M, Noy PJ, Szyroka J, Rainger GE, Tomlinson MG. ADAM10‐interacting tetraspanins Tspan5 and Tspan17 regulate VE‐cadherin expression and promote T lymphocyte transmigration. J Immunol. 2017;199:666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Noy PJ, Yang J, Reyat JS, et al. TspanC8 tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) interact via their extracellular regions: evidence for distinct binding mechanisms for different TspanC8 proteins. J Biol Chem. 2016;291:3145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kahn J, Walcheck B, Migaki GI, Jutila MA, Kishimoto TK. Calmodulin regulates L‐selectin adhesion molecule expression and function through a protease‐dependent mechanism. Cell. 1998;92:809–18. [DOI] [PubMed] [Google Scholar]
- 99. Blobel CP. ADAMS: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. [DOI] [PubMed] [Google Scholar]
- 100. Huovila AP, Turner AJ, Pelto‐Huikko M, Karkkainen I, Ortiz RM. Shedding light on ADAM metalloproteinases. Trends Biochem Sci. 2005;30:413–22. [DOI] [PubMed] [Google Scholar]
- 101. Al‐Tamimi M, Tan C, Qiao J, et al. Pathological shear triggers shedding of vascular receptors: a novel mechanism for downregulation of platelet glycoprotein (GP)VI in stenosed coronary vessels. Blood. 2012;119:4311–20. [DOI] [PubMed] [Google Scholar]
- 102. Gardiner EE, Arthur JF, Berndt MC, Andrews RK. Role of calmodulin in platelet receptor function. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:283–7. [DOI] [PubMed] [Google Scholar]
- 103. Rabbolini DJ, Gardiner EE, Morel‐Kopp M‐C, et al. Anti‐glycoprotein VI mediated immune thrombocytopenia: an under‐recognized and significant entity? Res Prac Thromb Haemost. 2017;1:291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nieswandt B, Schulte V, Bergmeier W, et al. Long‐term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193:459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schulte V, Rabie T, Prostredna M, Aktas B, Gruner S, Nieswandt B. Targeting of the collagen‐binding site on glycoprotein VI is not essential for in vivo depletion of the receptor. Blood. 2003;101:3948–52. [DOI] [PubMed] [Google Scholar]
- 106. Al‐Tamimi M, Mu FT, Arthur JF, et al. Anti‐glycoprotein VI monoclonal antibodies directly aggregate platelets independently of FcγRIIa and induce GPVI ectodomain shedding. Platelets. 2009;20:75–82. [DOI] [PubMed] [Google Scholar]
- 107. Kehrel B, Wierwille S, Clemetson KJ, et al. Glycoprotein VI is a major collagen receptor for platelet activation: it recognizes the platelet‐activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and von Willebrand Factor do not. Blood. 1998;91:491–9. [PubMed] [Google Scholar]
- 108. Onselaer M‐B, Hardy AT, Wilson C, et al. Fibrin and D‐dimer bind to monomeric GPVI. Blood Adv. 2017;1:1495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Montague SJ, Delierneux C, Lecut C, et al. Soluble GPVI is elevated in injured patients: shedding is mediated by fibrin activation of GPVI. Blood Adv. 2018;2:240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ebrahim M, Jamasbi J, Adler K, et al. Dimeric glycoprotein VI binds to collagen but not to fibrin. Thromb Haemost. 2018;118:351–61. [DOI] [PubMed] [Google Scholar]
- 111. Al‐Tamimi M, Grigoriadis G, Tran H, et al. Coagulation‐induced shedding of platelet glycoprotein VI mediated by Factor Xa. Blood. 2011;117:3912–20. [DOI] [PubMed] [Google Scholar]
- 112. Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88:1525–41. [PubMed] [Google Scholar]
- 113. Sakariassen KS, Orning L, Turitto VT. The impact of blood shear rate on arterial thrombus formation. Future Sci OA. 2015;1:FSO30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation‐independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Muthiah K, Connor D, Ly K, et al. Longitudinal changes in haemostatic parameters and reduced pulsatility contribute to non‐surgical bleeding in patients with centrifugal continuous flow left ventricular assist devices. J Heart Lung Transpl. 2016;35:745–51. [DOI] [PubMed] [Google Scholar]
- 116. Lukito P, Wong A, Jing J, et al. Mechanical circulatory support is associated with loss of platelet receptors glycoprotein Ibα and glycoprotein VI. J Thromb Haemost. 2016;14:2253–60. [DOI] [PubMed] [Google Scholar]
- 117. Chen Z, Mondal NK, Zheng S, et al. High shear induces platelet dysfunction leading to enhanced thrombotic propensity and diminished hemostatic capacity. Platelets. 2017; 10.1080/09537104.2017.1384542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Susen S, Rauch A, Van Belle E, Vincentelli A, Lenting PJ. Circulatory support devices: fundamental aspects and clinical management of bleeding and thrombosis. J Thromb Haemost. 2015;13:1757–67. [DOI] [PubMed] [Google Scholar]
- 119. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129:2829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gardiner EE, Al‐Tamimi M, Mu FT, et al. Compromised ITAM‐based platelet receptor function in a patient with immune thrombocytopenic purpura. J Thromb Haemost. 2008;6:1175–82. [DOI] [PubMed] [Google Scholar]
- 121. Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen‐induced aggregation and adhesion. J Clin Invest. 1989;84:1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Boylan B, Chen H, Rathore V, et al. Anti‐GPVI‐associated ITP: an acquired platelet disorder caused by autoantibody‐mediated clearance of the GPVI/FcRγ‐chain complex from the human platelet surface. Blood. 2004;104:1350–5. [DOI] [PubMed] [Google Scholar]
- 123. Greinacher A. Heparin‐induced thrombocytopenia. N Engl J Med. 2015;373:252–61. [DOI] [PubMed] [Google Scholar]
- 124. Qiao J, Al‐Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcγRIIa. Immunol Rev. 2015;268:241–52. [DOI] [PubMed] [Google Scholar]
- 125. Gardiner EE, Karunakaran D, Arthur JF, et al. Dual ITAM‐mediated proteolytic pathways for irreversible inactivation of platelet receptors: De‐ITAM‐izing FcγRIIa. Blood. 2008;111:165–74. [DOI] [PubMed] [Google Scholar]
- 126. Coupland LA, Hindmarsh EJ, Gardiner EE, Parish CR. The influence of platelet membranes on tumour cell behaviour. Cancer Metastasis Rev. 2017;36:215–24. [DOI] [PubMed] [Google Scholar]
- 127. Gaertner F, Ahmad Z, Rosenberger G, et al. Migrating platelets are mechano‐scavengers that collect and bundle bacteria. Cell. 2017;171:1368–82.e23. [DOI] [PubMed] [Google Scholar]
- 128. Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post‐surgical cancer immunotherapy. Nat Biomed Eng. 2017;1:0011. [Google Scholar]
- 129. Ziegler M, Wang X, Lim B, et al. Platelet‐targeted delivery of peripheral blood mononuclear cells to the ischemic heart restores cardiac function after ischemia‐reperfusion injury. Theranostics. 2017;7:3192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]