

Abstract
Calcific aortic valve stenosis (CAVS) is common in the ageing population and set to become an increasing economic and health burden. Once present, it inevitably progresses and has a poor prognosis in symptomatic patients. No medical therapies are proven to be effective in holding or reducing disease progression. Therefore, aortic valve replacement remains the only available treatment option. Improved knowledge of the mechanisms underlying disease progression has provided us with insights that CAVS is not a passive disease. Rather, CAVS is regulated by numerous mechanisms with a key role for calcification. Aortic valve calcification (AVC) is actively regulated involving cellular and humoral factors that may offer targets for diagnosis and intervention. The discovery that the vitamin K-dependent proteins are involved in the inhibition of AVC has boosted our mechanistic understanding of this process and has opened up novel avenues in disease exploration. This review discusses processes involved in CAVS progression, with an emphasis on recent insights into calcification, methods for imaging calcification activity, and potential therapeutic options.
Keywords: Review, Aortic valve stenosis, Calcification
Introduction
Degenerative calcific aortic valve stenosis (CAVS) is the most common type of valvular disease in the Western world, representing a substantial and increasing disease burden in the ageing population.1 Upon mild valve obstruction, disease progression with increasing haemodynamic severity is inevitable. Once symptomatic severe CAVS has developed, the prognosis without intervention is dismal. Despite growing knowledge, experience, and technological developments, the only treatment for (symptomatic) severe CAVS is surgical or transcatheter aortic valve replacement (AVR), to which not all patients are suited.2 Pharmacological interventions have thus far failed to alter the course of CAVS. Therefore, an unmet clinical need exists to develop new treatment strategies delaying CAVS progression.
We still lack precise molecular insight in to the pathophysiological underlying CAVS, although calcification is well known to play a fundamental role in progressive valvular narrowing. Today calcification is no longer considered a passive consequence of ageing but an active process involving cellular and molecular pathways. The exact processes underlying the initiation and progression of valvular calcification remain unresolved.3 Understanding the biomolecular mechanisms related to the genesis of calcification in CAVS will propel our knowledge and open novel avenues for diagnosis and treatment. In this review, we summarize the latest research progress in the pathophysiology of CAVS and offer novel targets holding potential for pharmacological interventions and imaging.
Aortic valve cusp function
Aortic valve cusps (or leaflets) must be both strong and flexible to withstand the considerable mechanical stress and strain associated with valve closure. To maintain cusp function, the specialized cusp microarchitecture is crucial and consists of three layers: fibrosa, spongiosa, and ventricularis4 (Figure 1). Valvular endothelial cells (VECs) are located at valvular blood-contacting surfaces, constituting a barrier that regulates valve permeability, the adhesion of inflammatory cells and paracrine signalling. Valvular interstitial cells (VICs), the major cell type, are present throughout all valvular layers. Valvular interstitial cells are key in valve remodelling, regulating both the synthesis and degradation of extracellular matrix components. Physiologically, VICs exist in a quiescent state, with similar characteristics to fibroblasts.5 Stimulation of VECs and VICs by molecular and mechanical triggers including high blood pressure, altered shear stress, cytokines, and growth factors contributes to CAVS pathophysiology, altering the local valve environment and making it calcification prone.
Figure 1.

Aortic valve. Left panel: 3D reconstruction (from bottom to top): aortic valve with three cusps and proximal ascending aorta. Middle panel: 2D view. Right panel: valvular histology (bottom to top): ventricularis, spongiosa, and fibrosa.
Calcific aortic valve stenosis aetiology
Although the most common cause of aortic stenosis in the Western world is degenerative CAVS (referred to as ‘CAVS’ in this review), rheumatic heart disease remains common in developing countries. Rheumatic aortic stenosis is caused by an abnormal immune response to Group A streptococcal infections. Calcification is again a predominant feature, and although this is believed to relate to chronic inflammation, precise mechanisms remain poorly defined.6 Calcific aortic valve stenosis is accelerated in patients with congenitally bicuspid aortic valves (BAVs) with aortic stenosis developing several decades earlier than in patients with trileaflet valves. More than 50% of patients with severe aortic stenosis requiring aortic valve replacement have BAV.7
Calcific aortic valve stenosis pathophysiology
Initiation phase
Calcific aortic valve stenosis can be divided in two distinct phases; the initiation and propagation phase, each dominated by different mechanisms (Figure 2). The initiation phase shows similarities with atherosclerosis, both ignited by endothelial activation/damage and an inflammatory response8 and sharing common risk factors including age, male gender, body mass index, smoking, hypertension, and elevated lipid levels including Lp(a).9 Moreover, stenotic valves from animals fed a high-fat diet display similar lesions as found in early human atherosclerotic plaques.10,11
Figure 2.

Pathophysiology and potential treatment targets (schematic overview). Upper panel: Progressive calcific aortic valve stenosis stages from non-stenotic to severe stenosis (left–right). Progressive thickening and calcification result in valvular dysfunction, characterized by decreased cusp mobility and opening, altered haemodynamics and stress. Middle panel: Cellular involvement in calcific aortic valve stenosis. Endothelial damage triggers lipid infiltration and upon oxidation an inflammatory response involving macrophages, T-lymphocytes, and mast cells. Inflammation triggers phenotypic switching of valvular interstitial cells resulting in increased extracellular vesicle release, providing a nidus for calcification. Microcalcification provokes an inflammatory response, resulting in increased apoptosis and/or delayed phagocytosis thereby expanding calcium deposition. Upon propagation, pro-fibrotic and pro-calcific processes dominate. Pro-fibrotic changes leading to collagen deposition and facilitating progressive calcification are mediated by reduced nitric oxide expression and up-regulation of renin–angiotensin system. Calcification is the dominant process driving disease progression. valvular interstitial cell phenotype switching to an osteoblast phenotype is thought to play a role in the progression phase by multiple regulatory pathways including Notch, receptor activator of nuclear factor kappa B/receptor activator of nuclear factor kappa B ligand/osteoprotegerin, Wnt/b-catenin, and bone morphogenetic protein-2. Lower panel: Potential pharmacological interventions.
Classically, the initiation phase is triggered by mechanical stress in the valve causing endothelial damage and activation. This is perhaps best illustrated by the accelerated development of aortic stenosis in patients with BAV that are characterized by altered flow patterns, increased mechanical stress, and reduced shear stress.12 The endothelial damage results in lipid infiltration and subsequent oxidation, thereby initiating an inflammatory response within the valvular endothelium involving macrophages, T-lymphocytes, and mast cells.11 Within affected regions, microcalcifications colocalize with lipids. Formation of microcalcifications is mediated by the release of apoptotic bodies and extracellular vesicles, in a similar manner to vesicle-induced calcification in bone and the vasculature.8,13 These calcification-prone extracellular vesicles function as nucleating sites for calcium crystal deposition and facilitate formation of hydroxyapatite. Hydroxyapatite crystals in turn set the stage for CAVS progression by (i) expanding quickly (creating more nucleation sites for calcium deposition) and (ii) evoking additional pro-inflammatory responses.5,8,14
Propagation phase: fibrosis and calcification as hallmarks of disease progression
Although the initiation phase is mainly mediated by inflammatory responses, the role of inflammation and lipid deposition is less prominent in the propagation phase (Figure 2). Instead, it is characterized by fibrosis and accelerated calcification, leading to valvular dysfunction and changes in mechanical stress and flow, thereby creating self-sustaining mechanisms underlying CAVS progression. Pro-fibrotic processes are mediated by (i) reduced nitric oxide expression following endothelial injury15 and (ii) up-regulation of the renin–angiotensin system (RAS), and formation of angiotensin II (ANGII). Down-regulation of expression of the ANGII type 2 receptor has been shown to result in a predominant pro-fibrotic profile, resulting in collagen deposition and the facilitation of progressive calcification.16 Calcific aortic valve stenosis is viewed as a fibrocalcific disease; however, once calcification becomes abundant, pro-osteogenic mechanisms become overwhelming, ultimately leading to severe calcification and valvular dysfunction. The phenotypic switching of VICs into an osteoblast-like phenotype is thought to be the fundamental step in accelerating valvular calcification, initiated at least in part by inflammation. In the propagation phase, disease progression is driven by calcific regulatory pathways including Notch, receptor activator of nuclear factor kappa B(RANK)/receptor activator of nuclear factor kappa B ligand (RANKL)/osteoprotegerin (OPG), Wnt/b-catenin, and bone morphogenetic proteins (BMPs).16 Notch-1 is essential in the development of the aortic valve during embryology and a mutation in Notch-1 is associated with development of BAV (but multiple genetic factors associated with BAV and CAVS have been described17). Also, Notch-1 is associated with early valve calcification by stimulating BMP-2.18 Bone morphogenetic protein-2 is up-regulated through binding of RANKL to RANK. Activation of the RANK/RANKL pathway results in the formation of proteins involved in calcification such as alkaline phosphatase and osteocalcin16 and is involved in CAVS (Figure 2).
Belonging to the multifunctional TGF-β superfamily, BMP-2 is an important osteogenic differentiation factor. Bone morphogenetic protein-2 is a key protein in phenotypic switching of VICs and hence in the development of aortic valve calcification/calcium (AVC).16 Physiologically, BMP is inhibited by matrix Gla-protein (MGP).19 The vital role of MGP to inhibit vascular calcification was demonstrated in MGP-deficient mice, showing lethal rupture of severely calcified arteries >2 months after birth.20 Although the inhibitory function of MGP on BMP-2 and subsequent VIC differentiation in CAVS seems evident, MGP also exerts its effect via a second mechanism. Matrix Gla-protein interacts directly with hydroxyapatite, inhibiting the growth of hydroxyapatite crystals in vascular tissue.19 Since we hypothesize that hydroxyapatite crystals are involved in the early phase of CAVS, MGP is a potential target to inhibit microcalcification. Matrix Gla-protein is a vitamin K-dependent protein and is present in two distinct forms: uncarboxylated inactive (ucMGP) and carboxylated active (cMGP). Like all vitamin K-dependent proteins, MGP requires vitamin K-induced carboxylation to exert its function19 (Figure 3). Vitamin K antagonist (VKA) inhibits recycling of vitamin K, thereby inducing inactive vitamin K-dependent proteins. Although VKA is important for prophylaxis of thrombo-embolic events in certain patient populations, calcification should be acknowledged as a side effect. In animal models, warfarin treatment increased vascular and valvular calcification, similar to the MGP knock-out mouse.21 The detrimental effect of warfarin was also identified in humans, where patients using VKA demonstrated more vascular and valvular calcification.22,23 With our expanding knowledge of CAVS pathophysiology, possible treatment targets for pharmacological interventions become evident.
Figure 3.
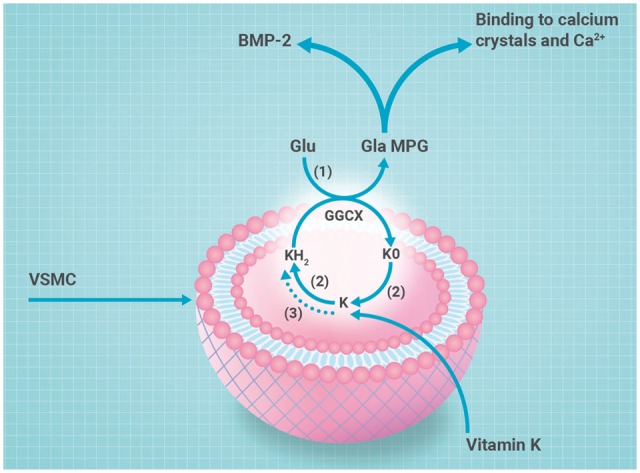
Synthesis of active matrix-Gla protein: schematic overview of vitamin K metabolism. The vitamin K cycle has a central role in the posttranslational carboxylation of glutamate (Glu) to γ-carboxyglutamate (Gla). Reduction of vitamin K to vitamin K hydroxyquinone (KH2) in presence of vitamin K epoxide reductase (VKOR) (2) or DDT-diaphorase (3). Vitamin K hydroxyquinone is oxidized during γ-glutamyl carboxylation by gamma-glutamyl carboxylase (GGCX) (1) into vitamin K epoxide (KO). Vitamin K epoxide is reduced to vitamin K by vitamin K epoxide reductase (2). Carboxylated matrix-Gla protein is active matrix-Gla protein that is secreted in the extracellular environment and inhibits calcification via binding to bone morphogenetic protein-2 or direct inhibition of calcium crystal formation.
Pharmacological treatment targets in calcific aortic valve stenosis
Current guidelines do not recommend pharmacological interventions to halt CAVS progression. However, the importance and need to reduce or even reverse progression of CAVS is evident. Therefore, multiple observational studies and randomized controlled trials (RCTs) have attempted to repurpose commonly used pharmacological interventions to slow CAVS progression.
Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers
Hypertension affects the stenotic aortic valve and increases afterload, thereby accelerating left ventricular (LV) hypertrophy. Both LV hypertrophy and high valvuloarterial impedance are associated with adverse events in patients with CAVS.24,25 Therefore, current guidelines recommend treatment of concomitant hypertension.2
Renin–angiotensin system (RAS) is an important player in cardiovascular disease, being involved in pathological processes in both the valve and myocardium in CAVS. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are well-known attenuators of RAS effects. However, observational retrospective studies investigating the ACE inhibitor effects on CAVS progression provided conflicting results. Treatment with ACE inhibitors was associated with less AVC26 but did not appear to slow haemodynamic progression.27 In principle, ARBs might have superior effects on both valve fibrosis and calcification,28 but prospective RCTs are lacking. With respect to the LV hypertrophic response, the RIAS RCT showed a modest but significant reduction of myocardial hypertrophy in patients with CAVS treated with ramipril.29 Finally, clinical observational studies have suggested that ACE inhibitors and ARBs are associated with favourable effects on symptoms (dyspnoea and exercise tolerance) and improved survival in patients with CAVS.30 Again RCT data are lacking.
Statins
Statins are widely used for lipid lowering in atherosclerosis and inflammation, being a specific inhibitor of hydroxymethylglutarylco-enzyme A-reductase (HMG-CoA-reductase). Although retrospective studies suggested that statins might also be of benefit in CAVS, subsequent RCTs demonstrated that statins in fact have no effect on CAVS progression or clinical outcomes. This conclusion was confirmed by a subsequent meta-analysis.31 The most plausible explanation for this failure is that whilst statins might intervene with inflammation and lipid deposition in the initiation phase, they have little effect once the propagation phase has become established when fibrosis and calcification are the dominant pathological processes.
Lipoprotein(a)
Lipoprotein(a), the preferential plasma carrier of oxidized phospholipids, is an LDL-like particle, containing additional apolipoprotein(a) and apolipoprotein-B100. A causal relationship between AVC and a single nucleotide polymorphism in the LPA locus was suggested.32 Although the precise mechanisms of action of Lp(a) require further elucidation, there is considerable interest in investigating whether Lp(a) is a modifiable target in CAVS. Statins are ineffective in reducing Lp(a);33 however, several other therapeutic agents are currently in different stages of investigation. IONIS-APO(a)Rx and IONIS-APO(a)-LRx ([Ligand-conjugated] antisense oligonucleotides targeting hepatic apolipoprotein(a) mRNA) have been investigated in Phase 1 and 2 trials, demonstrating an ability to reduce Lp(a) concentrations.34 Other promising Lp(a) lowering alternatives are proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and Niacin.35 The effects of Niacin/PCSK9 on aortic stenosis are currently being investigated (‘EAVaLL’. Clinicaltrials.gov identifier: NCT02109614 and ‘PCSK9 inhibitors in the progression of aortic stenosis’, Clinicaltrials.gov identifier: NCT03051360).
Bisphosphonates and denosumab
The calcification paradox implies that treatments for bone diseases (f.i. bisphosphonates or denosumab) might have a beneficial effect on vascular and valvular calcification while maintaining bone health.36 Bisphosphonates inhibit osteoclast-mediated bone resorption, resulting in decreased bone loss.37 The inhibitory effect of bisphosphonates on vascular calcification was demonstrated in animals.38 Retrospectively, a delay in CAVS progression was confirmed,39 whereas a more recent study failed to show a positive effect on haemodynamic CAVS progression or survival.40 These data are, however, confounded by the disease accelerating effects of osteoporosis. The ongoing SALTIRE 2 (Clinicaltrials.gov identifier: NCT02132026) RCT will help determine the true impact of bisphosphonates. Denosumab, a human monoclonal antibody targeting RANKL, has been investigated in pre-clinical models. Its binding prevents the interaction between RANK and RANKL, resulting in inhibition of vascular calcification in mice.41 It is being investigated as part of SALTIRE 2.
Vitamin K
Vitamin K is a fat-soluble vitamin consisting of two forms, namely phylloquinone (vitamin K1, VK1) present in green leafy vegetables and menaquinones (vitamin K2, VK2) present in fermented food. Long-chain menaquinones (i.e. MK7) are transported more efficiently to extrahepatic tissues.42 However, dietary intake of vitamin K is not sufficient to ensure full activation of MGP.19 Vitamin K supplementation is an attractive option to replenish vascular vitamin K stores to ensure optimal calcification inhibition. Vitamin K supplementation in rats showed regression of warfarin-induced vascular calcification.43 The prospective Rotterdam study was the first to report that dietary intake of VK2 showed an inverse relation with vascular calcification and mortality.44 Furthermore, low vitamin K status was shown to be associated with increased ucMGP levels and coronary artery calcification.19 Although promising, these studies were limited by the short-term follow-up, precluding measurable effects on clinical endpoints. Recently, the first in-man RCT demonstrated that vitamin K supplementation decelerated valvular calcification on computed tomography (CT) in a small group of patients with CAVS.45 The effectiveness of vitamin K supplementation to reduce or hold calcification progression is currently subject of investigation in multiple trials (‘iPACK-HD’. Clinicaltrials.gov identifier: NCT01528800, ‘VitaVasK’. ClinicalTrials.gov identifier: NCT01742273, ‘VitaK-CAC trial’. ClinicalTrials.gov identifier: NCT01002157,‘BASIK2’. ClinicalTrials.gov identifier: NCT02917525).
Imaging in calcific aortic valve stenosis: from assessment of haemodynamics to disease progression
The development of potential CAVS therapies creates a need for novel imaging techniques to assess their efficacy in Phase 2 clinical trials. These would select the most promising agents to proceed to larger and more expensive Phase 3 trials incorporating clinical endpoints. Several of such imaging approaches are discussed below and are listed in Figure 4.
Figure 4.

Role of imaging techniques in displaying calcification in stages of disease progression. Molecular imaging using 18F-sodium fluoride enables to visualize microcalcification and active calcification from beginning stages onwards. Computed tomography displays macrocalcification and anatomical changes in latter phases. Echocardiography visualizes anatomical changes and haemodynamic changes (2).
Echocardiography
Echocardiography is the most commonly used imaging technique to assess patients with aortic stenosis, providing detailed information on aortic stenosis severity, LV wall thickness, and function.2 Despite slow annual rate of CAVS progression and relatively high scan–rescan variability, echocardiography is the most commonly used method for assessing aortic stenosis progression.
Another application of echocardiography is quantification of valve calcification, using a semi quantitative four-point scale. Although echo-assessed calcification is an independent predictor of events (death or AVR) and disease progression,46 it is not widely used largely because of poor reproducibility and repeatability.
Computed tomography
Non-contrast multislice CT provides a more detailed and reproducible calcification scoring system. Computed tomography aortic valve calcification/calcium (CT-AVC) scoring enables quantification of mass, density, and volume of macroscopic valvular calcification, expressed in Agatston units (AU), similar to the approach developed for the coronary arteries. Computed tomography aortic valve calcification/calcium correlates well with haemodynamic parameters on echocardiography.47 Interestingly, women require less calcification to develop severe CAVS than men, resulting in gender-specific CT-AVC thresholds for severe CAVS (1275AU/2065AU for females/males), with additional prediction of subsequent disease progression and clinical events.48 Furthermore CT-AVC demonstrates relatively large annualized changes and specific calcification patterns provide additional insight for surgical and TAVI planning.49 Computed tomography aortic valve calcification/calcium is therefore appealing as an alternative method to assess disease severity and progression and was recommended in the recent ESC guidelines for this purpose.2 Although CT-AVC provides excellent quantification of the established valve calcific burden, it does not inform about disease activity or the biological mechanisms underlying CAVS.
Positron emission tomography
In contrast to echocardiography and CT, positron emission tomography (PET) is an imaging technique that informs about the activity of specific biological processes. Inflammation and calcification can both be targeted using the PET tracers 18F-fluorodeoxyglucose (18 F-FDG) and 18F-sodium fluoride (18 F-NaF), respectively. 18F-fluorodeoxyglucose has been applied to quantify vascular inflammation in the carotid arteries, correlating with macrophage infiltration.50 Increased valvular 18F-FDG uptake was demonstrated recently in CAVS and associated with faster subsequent disease progression.51–53 However, assessment of valvular 18F-FDG activity is frequently obscured by uptake in the adjacent myocardium54 and may reflect glucose utilization by a range of different cells or stimulating mechanisms.5518 F-sodium fluoride has been used for many decades for the detection of bone metastases and primary osteoblastic tumours.56 In the vasculature, it has been used to image developing microcalcification in carotid, coronary, and aortic atheroma57,58 and in CAVS,59 providing complementary information to CT-AVC. Indeed, a striking mismatch has consistently been observed between the localization of the macroscopic calcium deposits on CT and the developing microcalcification identified by 18F-NaF. 18F-sodium fluoride preferentially adsorbs to the available surface area of active hydroxyapatite crystal growth in areas of microcalcification, while uptake is low in regions with established areas of macroscopic calcification.60 Histological validation of 18F-NaF uptake in the valve in CAVS has been provided, demonstrating a close correlation with proteins involved in active calcification.59
Prospective longitudinal studies have demonstrated that areas of microcalcification on 18F-NaF PET develop with time into novel areas of macroscopic calcification. Thus 18F-NaF PET acts as a good predictor of early disease progression in CAVS.52,59 On this basis, 18F-NaF serves as a marker of calcification activity in CAVS and holds major potential as a surrogate endpoint to test the efficacy of novel pharmacological interventions.
Conclusion and future perspectives
Calcific aortic valve stenosis represents an increasing health care burden, leading to either adverse events or the requirement for major heart surgery. The pathophysiological mechanisms involved in CAVS initiation and progression are being rapidly elucidated and include inflammation, fibrosis, and calcification. With this advancing knowledge, we have identified novel therapeutic targets like vitamin K and new imaging techniques such as 18F-NaF PET that can be used to test the efficacy of novel agents and further inform our pathophysiological understanding. Indeed, several potential pharmacological treatments are under current investigation to achieve the ultimate goal, i.e. the inhibition of disease progression in CAVS.
Funding
National Institutes of Health [R01HL114805, R01HL136431, and R01HL119798 to E.A.]; the British Heart Foundation [FS/14/78/31020 to M.R.D.]. M.R.D is the recipient of the Sir Jules Thorn Award for Biomedical Research 2015.
Conflict of interest: none declared.
References
- 1. Bonow RO, Greenland P.. Population-wide trends in aortic stenosis incidence and outcomes. Circulation 2015;131:969–971. [DOI] [PubMed] [Google Scholar]
- 2. Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ, Iung B, Lancellotti P, Lansac E, Munoz DR, Rosenhek R, Sjogren J, Tornos Mas P, Vahanian A, Walther T, Wendler O, Windecker S, Zamorano JL.. 2017 ESC/EACTS Guidelines for the management of valvular heart disease: the Task Force for the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J 2017;38:2739–2791.28886619 [Google Scholar]
- 3. Dweck MR, Khaw HJ, Sng GK, Luo EL, Baird A, Williams MC, Makiello P, Mirsadraee S, Joshi NV, van Beek EJ, Boon NA, Rudd JH, Newby DE.. Aortic stenosis, atherosclerosis, and skeletal bone: is there a common link with calcification and inflammation? Eur Heart J 2013;34:1567–1574. [DOI] [PubMed] [Google Scholar]
- 4. Aikawa E, Libby P.. A rock and a hard place: chiseling away at the multiple mechanisms of aortic stenosis. Circulation 2017;135:1951–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutcheson JD, Aikawa E, Merryman WD.. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol 2014;11:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonini-Canterin F, Leiballi E, Enache R, Popescu BA, Roşca M, Cervesato E, Piazza R, Ginghină C, Nicolosi GL.. Hydroxymethylglutaryl coenzyme-a reductase inhibitors delay the progression of rheumatic aortic valve stenosis a long-term echocardiographic study. J Am Coll Cardiol 2009;53:1874–1879. [DOI] [PubMed] [Google Scholar]
- 7. Roberts WC, Ko JM.. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005;111:920–925. [DOI] [PubMed] [Google Scholar]
- 8. New SE, Aikawa E.. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res 2011;108:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM.. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol 1997;29:630–634. [DOI] [PubMed] [Google Scholar]
- 10. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R.. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007;115:377–386. [DOI] [PubMed] [Google Scholar]
- 11. Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD.. Characterization of the early lesion of ′degenerative′ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994;90:844–853. [DOI] [PubMed] [Google Scholar]
- 12. Sun L, Chandra S, Sucosky P, Aikawa E.. Ex vivo evidence for the contribution of hemodynamic shear stress abnormalities to the early pathogenesis of calcific bicuspid aortic valve disease. PloS One 2012;7:e48843.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E.. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater 2016;15:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc 1976;35:156–162. [PubMed] [Google Scholar]
- 15. El Accaoui RN, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, Lund DD, Brooks RM, Doshi H, Zimmerman KA, Kutschke W, Anseth KS, Heistad DD, Weiss RM.. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 2014;306:H1302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pawade TA, Newby DE, Dweck MR.. Calcification in aortic stenosis: the skeleton key. J Am Coll Cardiol 2015;66:561–577. [DOI] [PubMed] [Google Scholar]
- 17. Prakash SK, Bosse Y, Muehlschlegel JD, Michelena HI, Limongelli G, Della Corte A, Pluchinotta FR, Russo MG, Evangelista A, Benson DW, Body SC, Milewicz DM, Investigators BA.. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am Coll Cardiol 2014;64:832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nigam V, Srivastava D.. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol 2009;47:828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schurgers LJ, Uitto J, Reutelingsperger CP.. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med 2013;19:217–226. [DOI] [PubMed] [Google Scholar]
- 20. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G.. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997;386:78–81. [DOI] [PubMed] [Google Scholar]
- 21. Price PA, Faus SA, Williamson MK.. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol 1998;18:1400–1407. [DOI] [PubMed] [Google Scholar]
- 22. Schurgers LJ, Aebert H, Vermeer C, Bultmann B, Janzen J.. Oral anticoagulant treatment: friend or foe in cardiovascular disease? Blood 2004;104:3231–3232. [DOI] [PubMed] [Google Scholar]
- 23. Weijs B, Blaauw Y, Rennenberg RJ, Schurgers LJ, Timmermans CC, Pison L, Nieuwlaat R, Hofstra L, Kroon AA, Wildberger J, Crijns HJ.. Patients using vitamin K antagonists show increased levels of coronary calcification: an observational study in low-risk atrial fibrillation patients. Eur Heart J 2011;32:2555–2562. [DOI] [PubMed] [Google Scholar]
- 24. Gerdts E, Rossebo AB, Pedersen TR, Cioffi G, Lonnebakken MT, Cramariuc D, Rogge BP, Devereux RB.. Relation of left ventricular mass to prognosis in initially asymptomatic mild to moderate aortic valve stenosis. Circ Cardiovasc Imaging 2015;8:e003644; discussion e003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hachicha Z, Dumesnil JG, Pibarot P.. Usefulness of the valvuloarterial impedance to predict adverse outcome in asymptomatic aortic stenosis. J Am Coll Cardiol 2009;54:1003–1011. [DOI] [PubMed] [Google Scholar]
- 26. O’Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, Wu AH, Zhao XQ, Budoff MJ.. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med 2005;165:858–862. [DOI] [PubMed] [Google Scholar]
- 27. Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H.. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004;110:1291–1295. [DOI] [PubMed] [Google Scholar]
- 28. Cote N, Mahmut A, Fournier D, Boulanger MC, Couture C, Despres JP, Trahan S, Bosse Y, Page S, Pibarot P, Mathieu P.. Angiotensin receptor blockers are associated with reduced fibrosis and interleukin-6 expression in calcific aortic valve disease. Pathobiology 2014;81:15–24. [DOI] [PubMed] [Google Scholar]
- 29. Bull S, Loudon M, Francis JM, Joseph J, Gerry S, Karamitsos TD, Prendergast BD, Banning AP, Neubauer S, Myerson SG.. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril In Aortic Stenosis (RIAS trial). Eur Heart J Cardiovasc Imaging 2015;16:834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nadir MA, Wei L, Elder DHJ, Libianto R, Lim TK, Pauriah M, Pringle SD, Doney AD, Choy AM, Struthers AD, Lang CC.. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J Am Coll Cardiol 2011;58:570–576. [DOI] [PubMed] [Google Scholar]
- 31. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL.. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol 2011;27:800–808. [DOI] [PubMed] [Google Scholar]
- 32. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O’Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang SJ, Liu Y, Sjogren M, van der Pals J, Kalsch H, Muhleisen TW, Nothen MM, Cupples LA, Caslake M, Di Angelantonio E, Danesh J, Rotter JI, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O’Donnell CJ, Post WS; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thanassoulis G. Lipoprotein (a) in calcific aortic valve disease: from genomics to novel drug target for aortic stenosis. J Lipid Res 2016;57:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S.. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 35. Banach M. Lipoprotein (a)-we know so much yet still have much to learn. J Am Heart Assoc 2016;5:e003597.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Persy V, D’Haese P.. Vascular calcification and bone disease: the calcification paradox. Trends Mol Med 2009;15:405–416. [DOI] [PubMed] [Google Scholar]
- 37. Rodan GA, Fleisch HA.. Bisphosphonates: mechanisms of action. J Clin Investig 1996;97:2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Price PA, Faus SA, Williamson MK.. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler Thromb Vasc Biol 2001;21:817–824. [DOI] [PubMed] [Google Scholar]
- 39. Innasimuthu AL, Katz WE.. Effect of bisphosphonates on the progression of degenerative aortic stenosis. Echocardiography 2011;28:1–7. [DOI] [PubMed] [Google Scholar]
- 40. Aksoy O, Cam A, Goel SS, Houghtaling PL, Williams S, Ruiz-Rodriguez E, Menon V, Kapadia SR, Tuzcu EM, Blackstone EH, Griffin BP.. Do bisphosphonates slow the progression of aortic stenosis? J Am Coll Cardiol 2012;59:1452–1459. [DOI] [PubMed] [Google Scholar]
- 41. Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H, Kostenuik PJ, Erben RG, Hofbauer LC.. Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am J Pathol 2009;175:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schurgers LJ, Vermeer C.. Differential lipoprotein transport pathways of K-vitamins in healthy subjects. Biochim Biophys Acta 2002;1570:27–32. [DOI] [PubMed] [Google Scholar]
- 43. Schurgers LJ, Spronk HM, Soute BA, Schiffers PM, DeMey JG, Vermeer C.. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2007;109:2823–2831. [DOI] [PubMed] [Google Scholar]
- 44. Geleijnse JM, Vermeer C, Grobbee DE, Schurgers LJ, Knapen MH, van der Meer IM, Hofman A, Witteman JC.. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study. J Nutr 2004;134:3100–3105. [DOI] [PubMed] [Google Scholar]
- 45. Brandenburg VM, Reinartz S, Kaesler N, Kruger T, Dirrichs T, Kramann R, Peeters F, Floege J, Keszei A, Marx N, Schurgers LJ, Koos R.. Slower progress of aortic valve calcification with vitamin K supplementation: results from a prospective interventional proof-of-concept study. Circulation 2017;135:2081–2083. [DOI] [PubMed] [Google Scholar]
- 46. Rosenhek R, Klaar U, Schemper M, Scholten C, Heger M, Gabriel H, Binder T, Maurer G, Baumgartner H.. Mild and moderate aortic stenosis. Natural history and risk stratification by echocardiography. Eur Heart J 2004;25:199–205. [DOI] [PubMed] [Google Scholar]
- 47. Cowell SJ, Newby DE, Burton J, White A, Northridge DB, Boon NA, Reid J.. Aortic valve calcification on computed tomography predicts the severity of aortic stenosis. Clin Radiol 2003;58:712–716. [DOI] [PubMed] [Google Scholar]
- 48. Clavel M-A, Pibarot P, Messika-Zeitoun D, Capoulade R, Malouf J, Aggarwal SR, Araoz PA, Michelena HI, Cueff C, Larose E, Miller JD, Vahanian A, Enriquez-Sarano M.. Impact of aortic valve calcification, as measured by MDCT, on survival in patients with aortic stenosis: results of an international registry study. J Am Coll Cardiol 2014;64:1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sturla F, Ronzoni M, Vitali M, Dimasi A, Vismara R, Preston-Maher G, Burriesci G, Votta E, Redaelli A.. Impact of different aortic valve calcification patterns on the outcome of transcatheter aortic valve implantation: a finite element study. J Biomech 2016;49:2520–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, Yates D, LaMuraglia GM, Furie K, Houser S, Gewirtz H, Muller JE, Brady TJ, Fischman AJ.. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol 2006;48:1818–1824. [DOI] [PubMed] [Google Scholar]
- 51. Abdelbaky A, Corsini E, Figueroa AL, Subramanian S, Fontanez S, Emami H, Hoffmann U, Narula J, Tawakol A.. Early aortic valve inflammation precedes calcification: a longitudinal FDG-PET/CT study. Atherosclerosis 2015;238:165–172. [DOI] [PubMed] [Google Scholar]
- 52. Dweck MR, Jones C, Joshi NV, Fletcher AM, Richardson H, White A, Marsden M, Pessotto R, Clark JC, Wallace WA, Salter DM, McKillop G, van Beek EJ, Boon NA, Rudd JH, Newby DE.. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 2012;125:76–86. [DOI] [PubMed] [Google Scholar]
- 53. Jenkins WS, Vesey AT, Shah AS, Pawade TA, Chin CW, White AC, Fletcher A, Cartlidge TR, Mitchell AJ, Pringle MA, Brown OS, Pessotto R, McKillop G, Van Beek EJ, Boon NA, Rudd JH, Newby DE, Dweck MR.. Valvular (18)F-fluoride and (18)F-fluorodeoxyglucose uptake predict disease progression and clinical outcome in patients with aortic stenosis. J Am Coll Cardiol 2015;66:1200–1201. [DOI] [PubMed] [Google Scholar]
- 54. Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, Fletcher AM, Richardson H, White A, McKillop G, van Beek EJ, Boon NA, Rudd JH, Newby DE.. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol 2012;59:1539–1548. [DOI] [PubMed] [Google Scholar]
- 55. Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, Di Carli MF, Libby P.. Hypoxia but not inflammation augments glucose uptake in human macrophages: implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. J Am Coll Cardiol 2011;58:603–614. [DOI] [PubMed] [Google Scholar]
- 56. Grant FD, Fahey FH, Packard AB, Davis RT, Alavi A, Treves ST.. Skeletal PET with 18F-fluoride: applying new technology to an old tracer. J Nucl Med 2007;49:68–78. [DOI] [PubMed] [Google Scholar]
- 57. Derlin T, Richter U, Bannas P, Begemann P, Buchert R, Mester J, Klutmann S.. Feasibility of 18F-sodium fluoride PET/CT for imaging of atherosclerotic plaque. J Nucl Med 2010;51:862–865. [DOI] [PubMed] [Google Scholar]
- 58. Derlin T, Wisotzki C, Richter U, Apostolova I, Bannas P, Weber C, Mester J, Klutmann S.. In vivo imaging of mineral deposition in carotid plaque using 18F-sodium fluoride PET/CT: correlation with atherogenic risk factors. J Nucl Med 2011;52:362–368. [DOI] [PubMed] [Google Scholar]
- 59. Dweck MR, Jenkins WS, Vesey AT, Pringle MA, Chin CW, Malley TS, Cowie WJ, Tsampasian V, Richardson H, Fletcher A, Wallace WA, Pessotto R, van Beek EJ, Boon NA, Rudd JH, Newby DE.. 18F-sodium fluoride uptake is a marker of active calcification and disease progression in patients with aortic stenosis. Circ Cardiovasc Imaging 2014;7:371–378. [DOI] [PubMed] [Google Scholar]
- 60. Irkle A, Vesey AT, Lewis DY, Skepper JN, Bird JL, Dweck MR, Joshi FR, Gallagher FA, Warburton EA, Bennett MR, Brindle KM, Newby DE, Rudd JH, Davenport AP.. Identifying active vascular microcalcification by (18)F-sodium fluoride positron emission tomography. Nat Commun 2015;6:7495.. [DOI] [PMC free article] [PubMed] [Google Scholar]