

Abstract
Essentials.
FV‐Short, a normal splice isoform of Factor V, binds tissue factor pathway inhibitor (TFPIα) with high affinity.
FV‐Short functions as a synergistic TFPIα cofactor with protein S in inhibition of Factor Xa.
FV‐Short is much more efficient as TFPIα cofactor than full length FV.
TFPIα‐cofactor activity of FV‐Short is lost upon activation of coagulation by thrombin‐mediated cleavage.
Background
FV‐Short is a normal splice variant of Factor V (FV) having a short B domain, which exposes a high affinity‐binding site for tissue factor pathway inhibitor α (TFPIα). FV‐Short and TFPIα circulate in complex in plasma.
Objectives
The aim was to elucidate whether FV‐Short affects TFPIα as inhibitor of coagulation FXa and to test whether the TFPIα‐cofactor activity of protein S is influenced by FV‐Short.
Methods
Recombinant FV, wild‐type FV‐Short and a FV‐Short thrombin‐cleavage resistant variant were expressed and purified. The influence of FV and FV‐Short variants and/or protein S on the FXa inhibitory activity of TFPIα was monitored both in a purified system and in a plasma‐based thrombin generation assay.
Results
FV‐Short had intrinsically weak TFPIα‐cofactor activity but with protein S present, FV‐Short yielded efficient inactivation of FXa. Protein S alone did not promote full TFPIα‐activity. Intact FV was inefficient at low protein S concentrations and had 10‐fold lower activity compared to FV‐Short at physiological protein S levels. Activation of FV‐Short by thrombin resulted in the loss of the TFPIα‐cofactor activity. The synergistic TFPIα‐cofactor activity of FV‐Short and protein S was also demonstrated in plasma using a thrombin generation assay.
Conclusions
FV‐Short and protein S are highly efficient, synergistic cofactors to TFPIα in the regulation of FXa activity, whereas full length FV has lower activity. Our results suggest the formation of an efficient FXa‐inhibitory complex between FV‐Short, TFPIα and protein S on the surface of negatively charged phospholipids.
Keywords: coagulation factor, factor V, factor X, hemostasis, protease inhibitor, protein S, tissue factor
1. INTRODUCTION
Factor V (FV) circulates as a single chain 330 kDa procofactor at a concentration of ≈20 nmol L−1.1, 2 It is composed of multiple domains, A1‐A2‐B‐A3‐C1‐C2 and undergoes a series of thrombin‐ or factor Xa (FXa)‐mediated proteolytic cleavages, at R709, R1018, and R1545, during its activation. FVa comprises the non‐covalently linked heavy (A1‐A2‐A3) and light (C1‐C2) chains, whereas the large B domain is released. FXa binds with high affinity to FVa on the surface of negatively charged phospholipid membranes, thus forming the prothrombinase (PTase) complex that efficiently converts prothrombin to thrombin. The activity of FVa is regulated by activated protein C (APC) that cleaves and degrades FVa (at R306, R506, and R679) in a process stimulated by the vitamin K‐dependent protein S.3 Intact FV also serves as an anticoagulant APC cofactor in regulation of FVIIIa in the phospholipid membrane‐bound tenase complex (FIXa+FVIIIa), functioning in synergy with protein S.2 A point mutation in the FV gene replacing R506 with a Q (FV Leiden) is associated with the APC‐resistance phenotype, which is the most common inherited thrombophilia described.4, 5, 6
In its unactivated circulating form, FV has very low affinity for FXa and no procoagulant activity.7 This is in stark contrast to fully activated FVa that in the PTase complex binds FXa with very high (sub‐nM) affinity.8 The three cleavage sites in FV have different sensitivity to thrombin and FXa, R709 > R1018 > R1545, and intermediates are generated with partial FXa cofactor activity.7 Full activation requires the cleavage at R1545, which results in the release of the B domain and exposure of the FXa‐binding site.7, 8 The large B domain comprises two highly charged regions that are shown to be important for keeping FV in a procofactor state.1 A positively charged cluster (963‐1008) hypothetically binds to an acidic region (1493‐1537), an interaction that is crucial for keeping the procofactor state.1 Partial activation of FV, with cleavages at R709 and R1018, results in the release of the positive cluster and the generation of intermediate FXa affinity and FXa‐cofactor activity.7
FV‐deficiency is a rare autosomal recessive bleeding disease having variable severity and bleeding phenotype.9 It has been shown that FV‐deficiency is associated with low plasma levels of tissue factor pathway inhibitor (TFPIα), which regulates the initiation step of the extrinsic tissue factor (TF) pathway.10 Immune depletion of FV results in depletion of TFPIα, which demonstrates that TFPIα is circulating in complex with FV.10, 11
TFPIα, the full‐length isoform of TFPI, comprises three Kunitz domains and a highly charged C‐terminal extension that contains the binding site for the acidic region in the B‐domain of FV.11, 12 The full length TFPIα (≈0.2 nmol L−1 in plasma) constitutes around 20% of total TFPI in circulation.13 The major part of TFPI in plasma, which is complexed with lipoproteins, has only two Kunitz domains and an unclear functional significance.13 The first Kunitz domain of full length TFPIα inhibits FVIIa, the second inhibits FXa, whereas the third Kunitz domain interacts with protein S.13, 14, 15, 16 The interaction between TFPIα and protein S is associated with stimulation of the inhibition of FXa by TFPIα.17, 18 Full length FV has also been reported to potentiate the activity of TFPIα .19, 20 Recently, it was reported that the TFPIα stimulation of high concentrations of full length FV (30 nmol L−1) requires the presence of protein S (100 nM).21
Elucidation of the pathogenesis of the East Texas bleeding disorder revealed a mutation in the S756 codon of exon 13 that encodes the B domain. The mutation resulted in the activation of an existing splice donor site, which under normal conditions is only used to a small degree.2, 22, 23 The splicing results in the in‐frame deletion of 702 amino acid residues from the B domain and the creation of a short form of FV (FV‐Short). Family members carrying the mutation have high plasma concentrations of FV‐Short (≈2‐5 nmol L−1). The C‐terminus of the truncated B domain of FV‐Short exposes the negatively charged cluster to which TFPIα binds with high affinity.12, 23 As a result, the TFPIα concentration in circulation increases 10‐fold and causes a moderately severe bleeding phenotype. Under normal conditions, the FV‐Short is present in plasma at sub‐nM concentrations circulating in complex with TFPIα23. FV‐Short is thus an important carrier of TFPIα in the normal circulation. It is not yet known if FV‐Short affects the TFPIα function. The aim of the present investigation was to elucidate whether the complex formation between FV‐Short and TFPIα affects the function of TFPIα. We have used recombinant variants of FV‐Short to show that FV‐Short and protein S functions as efficient synergistic TFPIα cofactors in inhibition of FXa.
2. MATERIALS AND METHODS
2.1. Materials
Human prothrombin, FXa, thrombin, FV monoclonal antibodies AHV5146 and AHV5112, a polyclonal TFPI antibody (PAHTFPI), and FV‐deficient plasma were from Hematologic Technologies, Inc (Essex Junction, VT, USA). A rabbit anti‐protein S (A0384) from Dako (Glostrup, Denmark) was used. TFPIα expressed in eukaryotic cells was a kind gift from Dr. T. Hamuro at the Chemo‐Sero‐Therapeutic Research Institute, Japan. Protein S was purified as previously described.24 Human FV was purified from plasma as described.25, 26 Phosphatidylserine (PS), phosphatidyl ethanolamine (PE) and phosphatidyl choline (PC) were from Avanti Polar Lipids. Phospholipid vesicles were prepared using the LiposoFast basic extruder (Armatis, Germany) as previously described.27 The phospholipid vesicles were used within 2 days. Synthetic substrates S‐2238 and S2765 were kindly provided by Chromogenix Ltd., Milan, Italy.
2.2. Recombinant FV‐variants
FV‐Short was created as previously described.23 Quikchange site directed mutagenesis (Stratagene, San Diego, CA, USA) was used to replace R709 and R1545 (numbering as in full length FV) with Q and the nucleotide sequence of the whole FV‐Short coding region was checked using Sanger sequencing.28 The FV‐Short variant is denoted FV‐Short QQ. Stable BHK cell clones expressing the FV‐Short variants and full length FV were created and the proteins purified as described.29, 30 On SDS‐PAGE, the purified proteins migrated as 250 kDa bands. After cleavage with thrombin, FV‐Short generated the expected heavy (≈115 kDa) and light (≈75 kDa) chains of FVa (not shown). The double mutant FV‐Short QQ was resistant to thrombin.
2.3. Electrophoretic techniques
SDS‐PAGE was performed using standard techniques. For western blotting, 4‐15% gradient SDS‐PAGE gels (Bio‐Rad, Hercules, CA, USA) were used.
2.4. Thrombin generation assay
Thrombin generation was performed as previously described with slight variations.23 Either 40 uL pooled normal citrated (50 ug mL−1 CTI) or FV‐deficient plasma were mixed with different combinations of FV‐Short (before and after activation with 1.6 nmol L−1 thrombin (0.5 units mL−1) for 10 minutes and then inhibition with 1 unit/ml hirudin), anti‐bodies against protein S or TFPI (100 ug mL−1 final concentration), TFPIα (40 uL) and 30 uL PL (10 umol L−1; 10/20/70 of PS/PE/PC)/TF (1.4 pmol L−1) mixture as indicated in each experiment. The concentrations given are final concentrations. Plasma was diluted 3.25 times in the assay and the contribution of intrinsic TFPIα (approximately 0.06‐0.08 nmol L−1) was disregarded. After 1 minute incubation at 37°C, the reaction was initiated by addition of 20 uL fluorescent substrate (Z‐Gly‐Gly‐Arg‐AMC*HCl, 300 umol L−1) containing Ca2+ (16.7 mmol L−1). The reaction was monitored for 30 minutes in a Tecan Infinite 200 and the results analyzed with GraphPad Prism and expressed as ΔFU/min a response of 300 ΔFU/min corresponding to ≈135 nM thrombin.
2.5. Inhibition of FXa by TFPIα in a purified system
Various concentrations of FV‐Short (before and after activation with 1 unit/ml thrombin followed by double concentration of hirudin to inhibit thrombin), were incubated with phospholipid (20:20:60 of PS:PE:PC, 25 umol L−1), protein S (0‐100 nmol L−1), and TFPIα (0‐1 nmol L−1) in HNBSACa2+ buffer (25 Hepes, 0.15 mol L−1 NaCl, 5 mmol L−1 CaCl2 pH 7.7, containing 0.5 mg mL−1 bovine serum albumin) for 10 minutes at 37°C. The reaction was initiated by addition of S2765 (0.8 mmol L−1) and FXa (0.3 nmol L−1) and followed for 15 minutes by monitoring absorbance at 405 nmol L−1 in a Tecan Infinite 200 system. The concentrations given in each experiment are the final concentrations.
3. RESULTS
3.1. Inhibition of FXa by TFPIα in presence of FV‐short and protein S
A system with purified components was used to investigate whether FV‐Short affects the activity of TFPIα as inhibitor or FXa. The FXa‐mediated cleavage of the synthetic substrate S2765 was monitored over 900 seconds (Figure 1A). TFPIα alone yielded a slight inhibition of FXa and the addition of either FV‐Short (0.5 nmol L−1) or protein S (3 nmol L−1) only marginally affected the inhibitory activity of TFPIα. However, when both FV‐Short and protein S were added, a strong inhibitory effect of TFPIα was obtained suggesting a synergistic cofactor activity of FV‐Short and protein S. Cleavage of FV‐Short with thrombin resulted in complete loss of the synergistic cofactor activity (Figure 1B).
Figure 1.
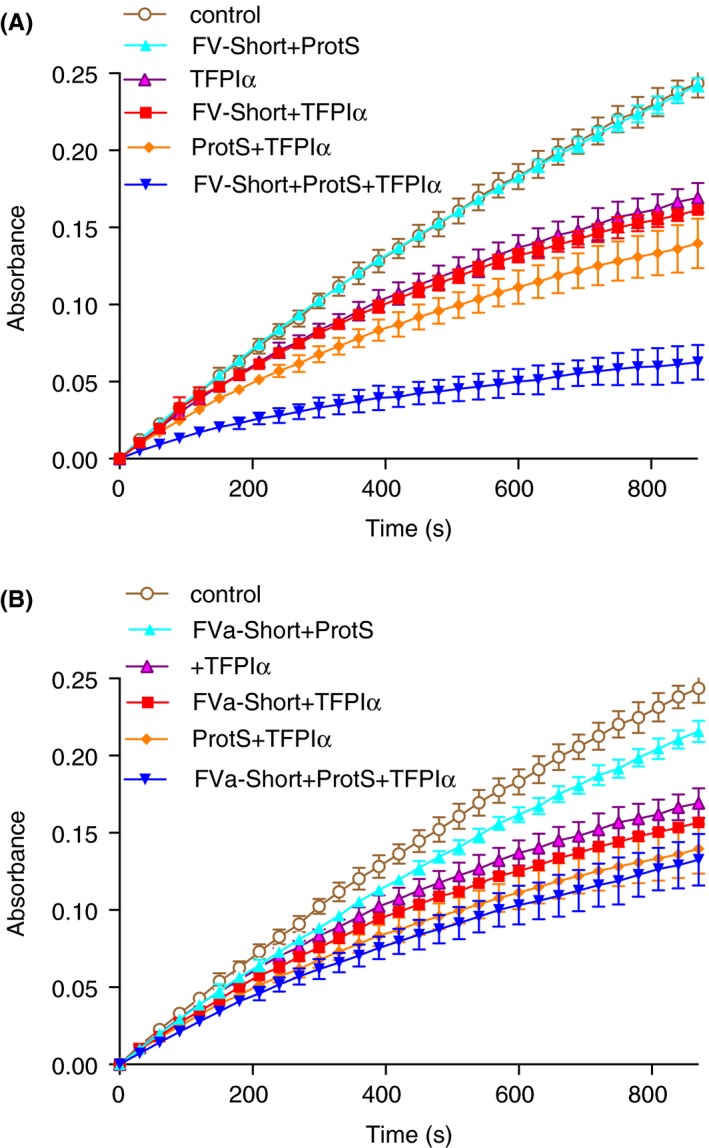
FV‐Short and protein S as synergistic cofactors to TFPIα in inhibition of FXa. (A) FV‐Short (0.5 nmol L−1) was incubated with FXa (0.3 nmol L−1) and 25 umol L−1 PL (20/20/60:PS/PE/PC) in the presence or absence of protein S (ProtS, 3 nmol L−1) and with or without TFPIα (0.25 nmol L−1). The inhibition of FXa amidolytic activity was monitored for 900 seconds with S2765. (B) the FV‐Short was cleaved with thrombin prior to the experiment. The composition of each mixture is denoted in the figure, controls contain with FXa and PL. Symbols and colors are explained in the figure. The experiment was performed three times and the mean ± SD are presented
The FXa‐inhibition system was used in titration experiments where the three components, FV‐Short, protein S and TFPIα were varied one at the time (Figure 2). In the presence of FV‐Short and TFPIα, maximal inhibition was observed at approximately 1.5 nmol L−1 protein S (Figure 2A), whereas in the absence of FV‐Short the TFPIα‐cofactor activity of protein S was lower and 25 nmol L−1 protein S did not yield maximum inhibition. When tested at 100 nmol L−1 (Figure 3C) and even up to 160 nmol L−1 (not shown), maximum inhibition was not achieved with protein S alone. After cleavage of FV‐Short with thrombin to FVa‐Short, the synergism with protein S was lost. However, it is noteworthy that the intrinsic TFPIα‐cofactor activity of protein S was unaffected by the presence of FVa‐Short. In the absence of added protein S, FV‐Short demonstrated at most weak cofactor activity. In contrast, in the presence of protein S, FXa inhibition was very efficient down to less than 0.5 nmol L−1 FV‐Short (Figure 2B). The TFPIα titration further demonstrated the synergistic cofactor activity of FV‐Short and protein S (Figure 2C). In the presence of both FV‐Short and protein S, maximum FXa‐inhibitory effect of TFPIα was observed at a concentration of 0.25 nmol L−1, which is approximately equimolar to the FXa concentration, suggesting formation of a 1:1 complex between FXa and TFPIα.
Figure 2.
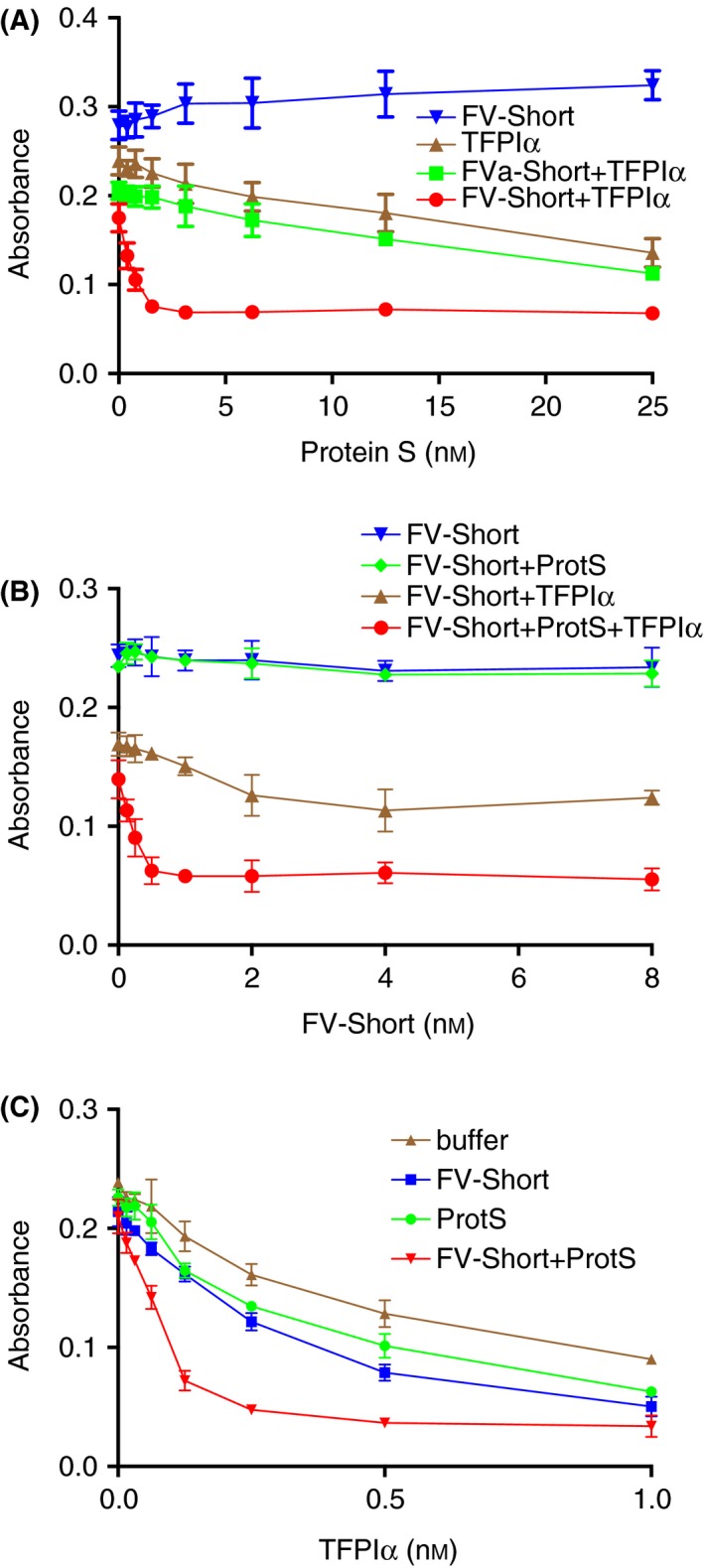
FV‐Short and protein S as efficient synergistic cofactors to TFPIα in inhibition of FXa. The inhibition of FXa (0.3 nmol L−1) was monitored for 900 seconds in the presence of increasing concentrations of FV‐Short (or thrombin activated FV‐Short), protein S (ProtS) or TFPIα. The absorbance reached at the end of the incubation was plotted versus the concentration of the varied protein. (A) protein S was varied and TFPIα was 0 or 0.25 nmol L−1 and FV‐Short (or FVa) was 0 or 2 nmol L−1; (B) FV‐Short was varied whereas TFPIα was 0 or 0.25 nmol L−1 and protein S 0 or 3 nmol L−1; C, TFPIα was varied, whereas protein S was 0 or 3 nmol L−1 and FV‐Short was 0 or 2 nmol L−1. Symbols and colors are explained in the figure. The experiment was performed three times and the means ± SD are presented
Figure 3.
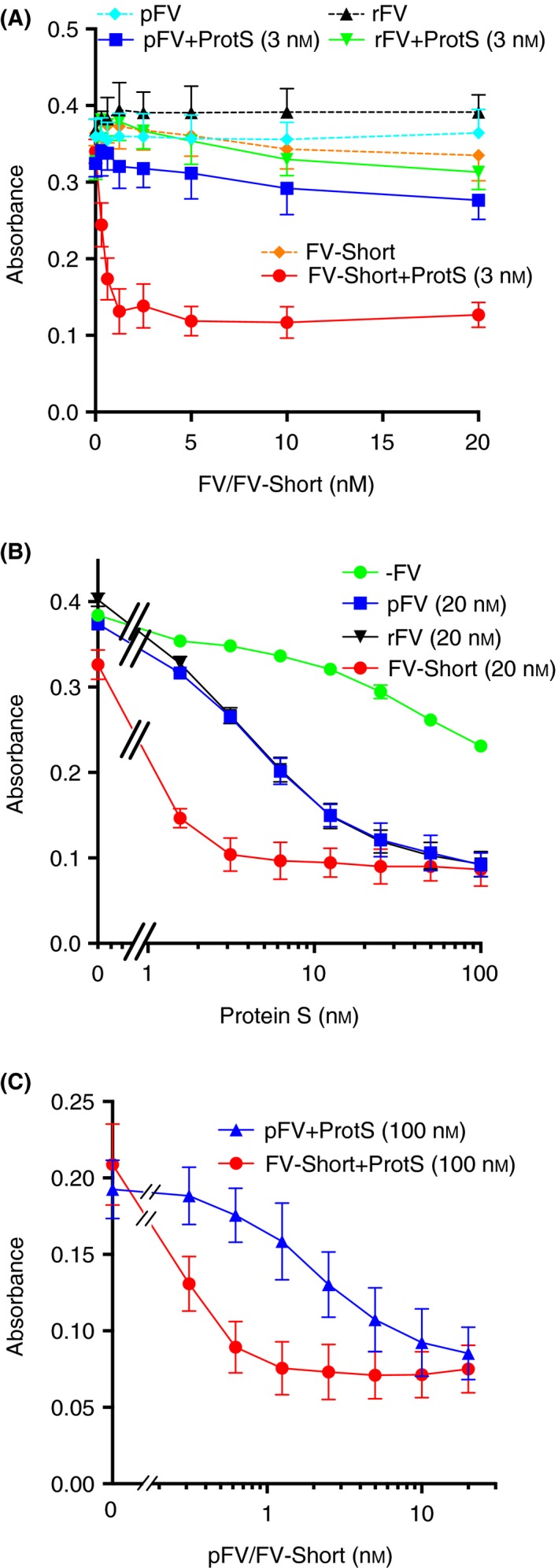
TFPIα cofactor activity of full‐length Factor V requires high protein S concentrations. (A) The inhibition of FXa (0.3 nmol L−1) was monitored for 900 seconds as described in Figure 1 in the presence of 0.25 nmol L−1 TFPIα and increasing concentrations of either full length plasma‐derived FV (pFV) or recombinant FV (rFV) or FV‐Short. The experiment was done in the presence or absence of 3 nmol L−1 protein S (ProtS). (B) Inhibition of FXa (0.3 nmol L−1) by 0.25 nmol L−1 TFPIα was monitored in the presence or absence of 20 nmol L−1 pFV, rFV or FV‐Short and increasing concentrations of protein S. (C) Inhibition of FXa (0.3 nmol L−1) by 0.25 nmol L−1 TFPIα was monitored in the presence of 100 nmol L−1 protein S (ProtS) and increasing concentrations of FV‐Short or pFV. The absorbance reached at the end of the incubation was plotted vs. the concentration of the varied protein. Symbols and colors are explained in the figure. Each experiment was performed three‐four times and the means ± SD are presented
Plasma‐derived intact FV and recombinant intact FV up to a concentration of 20 nmol L−1 did not demonstrate any TFPIα‐cofactor activity alone or in the presence of a low concentration of protein S (3 nmol L−1) (Figure 3A). In a protein S titration experiment using 20 nmol L−1 FV or FV‐Short, FV‐Short required approximately 10‐fold lower protein S to achieve full TFPIα‐cofactor activity than did FV (Figure 3B). A FV‐Short/FV titration performed at 100 nmol L−1 protein S demonstrated that FV‐Short was approximately 10‐fold more efficient than FV but that full TFPIα‐cofactor activity was achieved at the highest FV level (Figure 3C). Similar results as those in Figure 3C were obtained when protein S was raised to 150 nmol L−1 protein S (not shown).
3.2. Effects on thrombin generation of TFPIα and FV‐Short in FV‐deficient plasma
Increasing concentrations of FV‐Short, or thrombin‐cleaved FV‐Short, were added to FV‐deficient plasma and a thrombin generation assay was performed with initiation of the reactions using either TF or FXa (Figure 4). Approximately 2 nmol L−1 FV‐Short was selected for further experiments as this concentration gave a strong thrombin generation after a short lag phase. In this system, increasing concentrations of TFPIα were added. In the TF‐initiated thrombin generation, the addition of TFPIα yielded a dose‐dependent prolongation of lag phase and decrease in amounts of thrombin formed (Figure 4A). Addition of anti‐protein S antibodies inhibited the efficiency of TFPIα with only the highest concentrations inhibiting thrombin generation (Figure 4C). After thrombin‐cleavage of FV‐Short, TFPIα was considerably less efficient and only the highest TFPIα concentrations strongly inhibited the thrombin generation (Figure 4B). Addition of anti‐protein S decreased the TFPIα efficiency further (Figure 4D). Initiation of the thrombin generation with FXa in presence of FV‐Short yielded a similar pattern of TFPIα‐mediated inhibition as that observed in the TF‐initiated reactions (Figure 4E). The inhibitory effect was less efficient in incubations containing thrombin‐cleaved FV‐Short (Figure 4F). Addition of anti‐protein S strongly inhibited the TFPIα effect in reactions containing intact FV‐Short (Figure 4G), whereas there was almost no inhibitory effect in incubations containing thrombin‐cleaved FVa‐Short (Figure 4H). The results are in agreement with the conclusion from the experiments using purified components that FV‐Short can function as TFPIα cofactor in synergy with protein S and that the cofactor activity is lost after cleavage of FV‐Short by thrombin.
Figure 4.
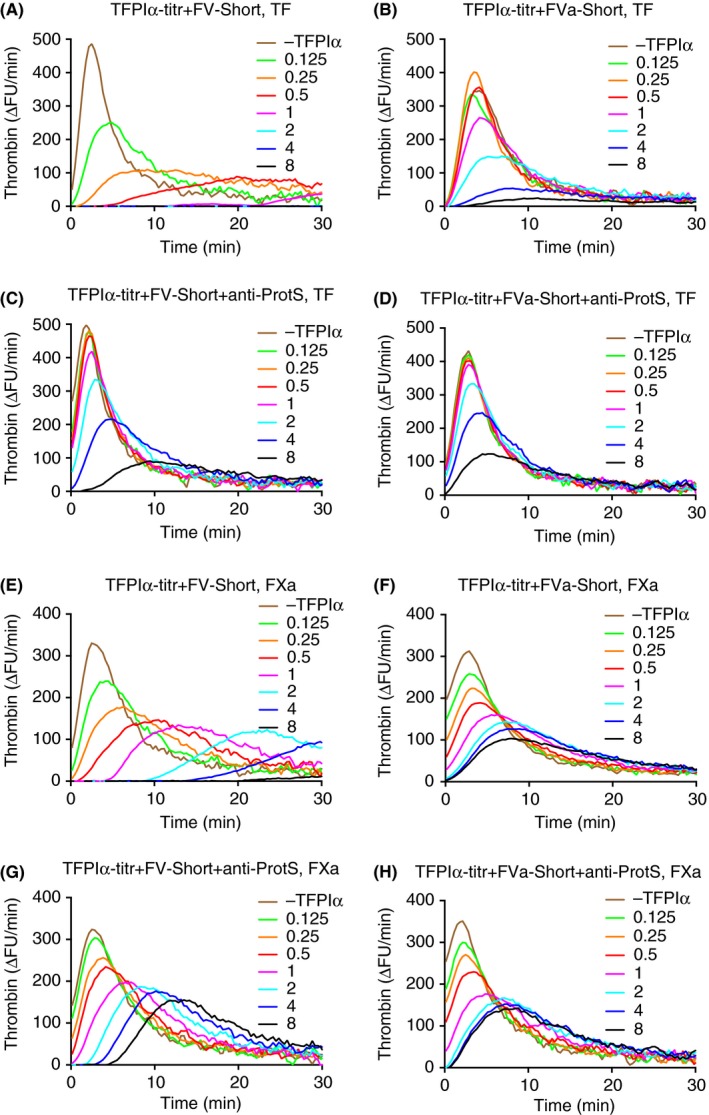
Thrombin‐generation assay using Factor V‐deficient plasma with added intact or thrombin‐cleaved FV‐Short. Increasing concentrations of TFPIα (as indicated in figure) were included and thrombin generation was initiated with either TF (1.4 pmol L−1, A–D) or FXa (62.5 pmol L−1, E–H). The final concentrations of added FV‐Short (A, C, E, and G) or thrombin‐cleaved FVa‐short (B, D, F and H) were 2 nmol L−1. The importance of protein S for the TFPIα response was investigated by running each experiment in the absence (A, B, E, and F) or presence (C, D, G, and H) of anti‐protein S (100 ug mL−1 final concentration). Each experiment was performed three times and the figure represents the means of the three. A response of 300 ΔFU min−1 corresponded to ≈135 nmol L−1 thrombin
3.3. FV‐Short added to normal plasma–TFPIα‐dependent effects on thrombin generation
Addition of FV‐Short (2 nmol L−1) to normal plasma decreased the thrombin peak but had no effect on the lag phase in the illustrated experiment (Figure 5). However, in many of the thrombin generation experiments, addition of FV‐Short, in particular at concentrations >2 nmol L−1, resulted in shortening of the lag phase, as is evident in later figures. Addition of anti‐TFPI increased the thrombin peak both in the absence and presence of FV‐Short but had no effect on the lag phase. This illustrates that the decrease in thrombin generation observed upon the addition of FV‐Short was at least partially TFPIα dependent even though FV‐Short had a small TFPIα independent anticoagulant effect, the mechanism of which is unknown. When TFPIα was included at a concentration of 0.25 nmol L−1, the thrombin generation was strongly inhibited both in presence and absence of FV‐Short, the inhibitory effect being more pronounced in the presence of FV‐Short. Addition of anti‐TFPI increased thrombin generation both in the presence and the absence of FV‐Short.
Figure 5.
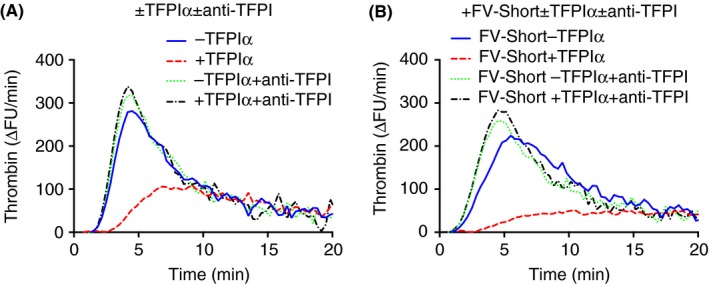
FV‐Short stimulates TFPIα in normal plasma. Thrombin‐generation assay using normal plasma with (B) or without (A) added FV‐Short (2 nmol L−1) and with or without added TFPIα (0.25 nmol L−1) as indicated in the figure. To elucidate the effect of TFPIα, the assay was performed in presence or absence of polyclonal anti‐TFPI (PAHTFPI) (100 ug mL−1). Thrombin generation was initiated with 1.4 pmol L−1 TF. All given concentrations are final. The experiment was performed multiple times with slight variations and a representative example is shown
3.4. Protein S and FV‐Short as synergistic TFPIα‐cofactors in normal plasma
TFPIα was added at increasing concentrations to normal plasma without or with FV‐Short (Figure 6A and C). To determine the importance of protein S for the inhibitory effect of TFPIα, polyclonal antibodies to protein S were added to block the contribution of protein S (Figure 6B and D). In the illustrated experiment, addition of FV‐Short to plasma resulted in a shortening of the lag phase from around 2 minutes to less than 1 minute. Despite the earlier activation of the thrombin generation, the response to added TFPIα, as monitored by thrombin peak height, was more pronounced in the presence of FV‐Short. When the experiment was performed in the presence of anti‐protein S (Figure 6B and D), TFPIα was less efficient in inhibiting the thrombin generation, in particular in the plasma containing added FV‐Short. This demonstrates that in the absence of protein S, the addition of FV‐Short to normal plasma results in early activation of coagulation (short lag phase) and low TFPIα efficiency (high thrombin peak).
Figure 6.
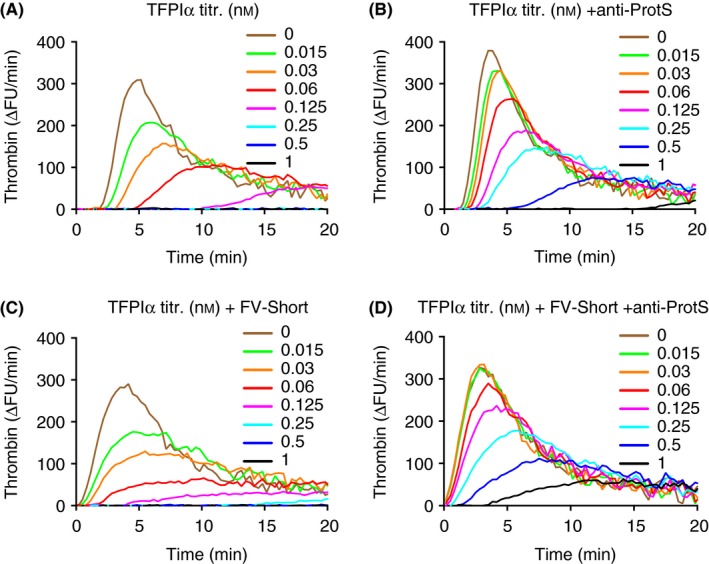
The importance of protein S for the anticoagulant effects of TFPIα and FV‐Short added to normal plasma. Thrombin generation was performed after addition of increasing concentrations of TFPIα to normal plasma without (A and B) or with (C and D) added FV‐Short and anti‐protein S (anti‐ProtS) (B and D). Thrombin generation was initiated with 1.4 pmol L−1 TF. The final concentrations of added TFPIα (nmol L−1) in the assay are indicated by the different colors in the figure. Note that the anticoagulant effects of TFPIα were dependent on protein S in particular in the presence of added FV‐Short. The experiment was performed multiple times with slight variations and a representative example is shown
3.5. Titration of FV‐Short to normal plasma demonstrating its synergistic TFPIα‐cofactor activity with protein S
Addition of increasing concentrations of FV‐Short to plasma resulted in a slight shortening of the lag phase and the highest concentrations added slightly reduced the height of the thrombin peak (Figure 7A). When TFPIα was included in the incubation mixtures, thrombin generation was strongly reduced with drop in thrombin peak height (Figure 7B). The height of the thrombin peak was lowest in the reaction mixtures containing the highest FV‐Short concentrations. The lag phase was shortened in the reactions containing added FV‐Short demonstrating early thrombin generation. The strong inhibition of thrombin generation observed in the presence of FV‐Short plus TFPIα was dependent on protein S and addition of protein S antibodies completely eliminated the inhibitory effect of added FV‐Short (Figure 7C) Moreover, the intrinsic procoagulant effect of FV‐Short was evident when protein S was blocked, with shortening of the lag phase and increased thrombin peak height (Figure 7C).
Figure 7.
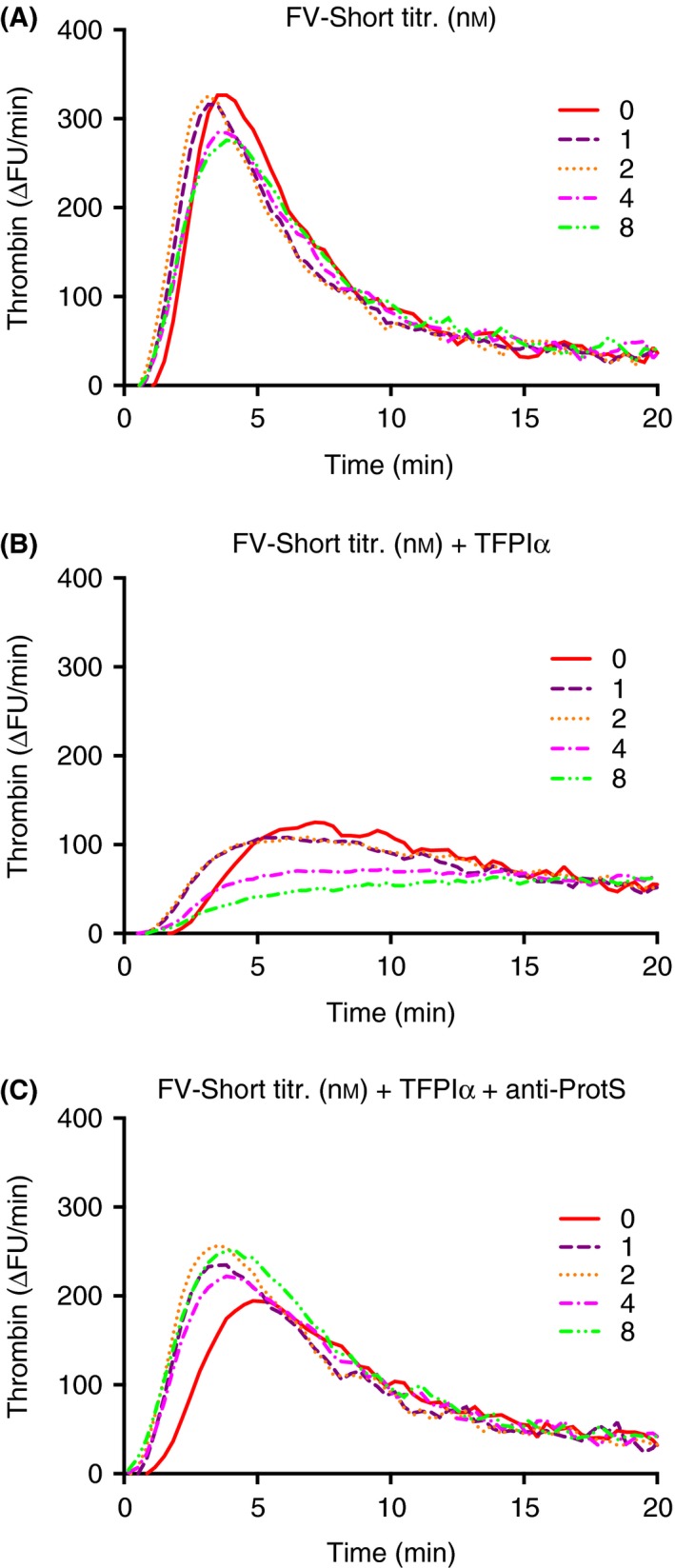
Anti‐coagulant effect in thrombin‐generation assay of FV‐Short plus TFPIα is dependent on the presence of protein S. Increasing amounts of FV‐Short (0–8 nmol L−1) as indicated in the figure were included in the thrombin generation assay in absence (A) or presence (B and C) of added TFPIα (0.0625 nmol L−1). In C, anti‐protein S (anti‐ProtS) (100 ug mL−1) was included. TF (1.4 pmol L−1) was used to initiate the reaction. The experiment was performed five times and the figure presents the mean
3.6. Thrombin‐resistant FV‐Short QQ as synergistic TFPIα‐cofactor with protein S
To investigate how much thrombin‐mediated cleavage of FV‐Short affected the results, increasing concentrations of FV‐Short QQ were added together with TFPIα. Despite being resistant to activation, FV‐Short QQ decreased the lag phase from 5 minutes to around 2.5 minutes (Figure 8A). The two highest concentrations of FV‐Short QQ decreased the thrombin generation, the peak dropping from 400 ∆FU/min to less than 100 ∆FU/min. That this was indeed dependent on the interaction with TFPIα was evident from the reaction mixtures containing anti‐TFPI that demonstrated very short lag phases and high thrombin peaks (Figure 8B). In a TFPIα‐titration performed with and without added FV‐Short WT (Figure 9B) or FV‐Short QQ (Figure 9C), the inhibitory effect of TFPIα was stronger in presence of FV‐Short QQ. Anti‐protein S blocked a large fraction of the inhibitory effect of TFPIα (Figure 9D). These results show that FV‐Short WT was activated during the progression of thrombin generation and this was associated with decreased TFPIα‐cofactor activity.
Figure 8.
Anti‐coagulant effect of TFPIα plus FV‐Short QQ combination dependent on the presence of TFPIα. TFPIα (0.125 nmol L−1) was added together with increasing amounts of FV‐Short QQ (0–8 nmol L−1) to a mixture of normal and FV‐deficient plasma (2:8) and the thrombin generation was initiated with 5.6 pmol L−1 TF. The plasma mixing was done with the purpose to lower the contribution of normal full‐length FV present in the plasma. The assay was done in the presence (B) or absence (A) of anti‐TFPI (100 ug mL−1). The experiment was performed multiple times with variations and the figure illustrates a representative example
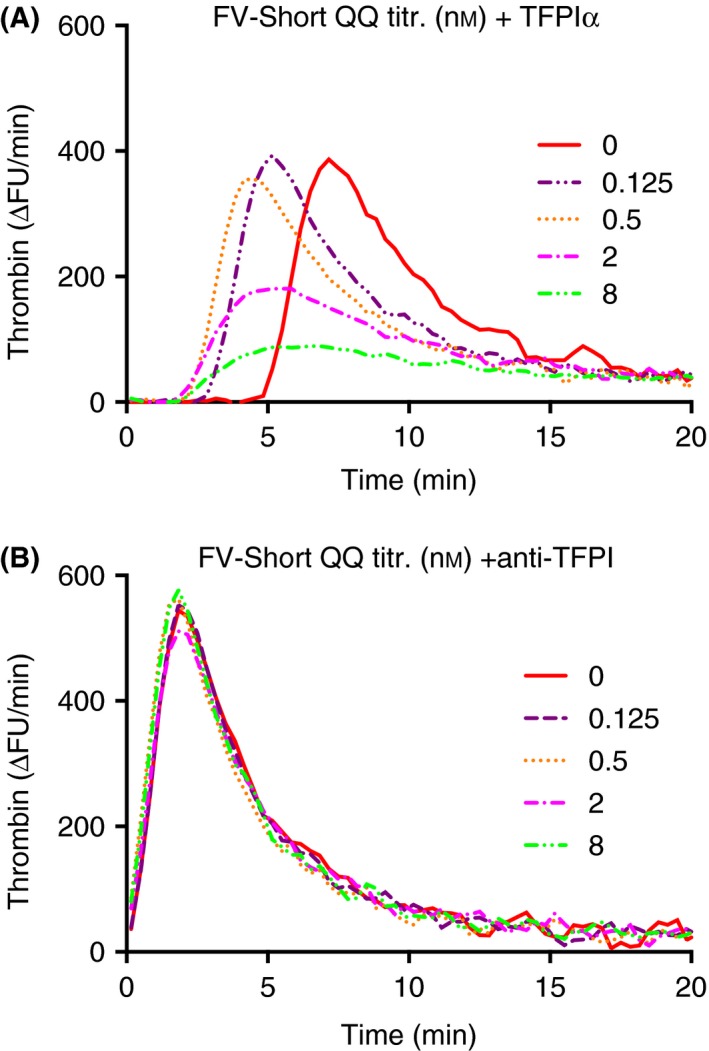
Figure 9.
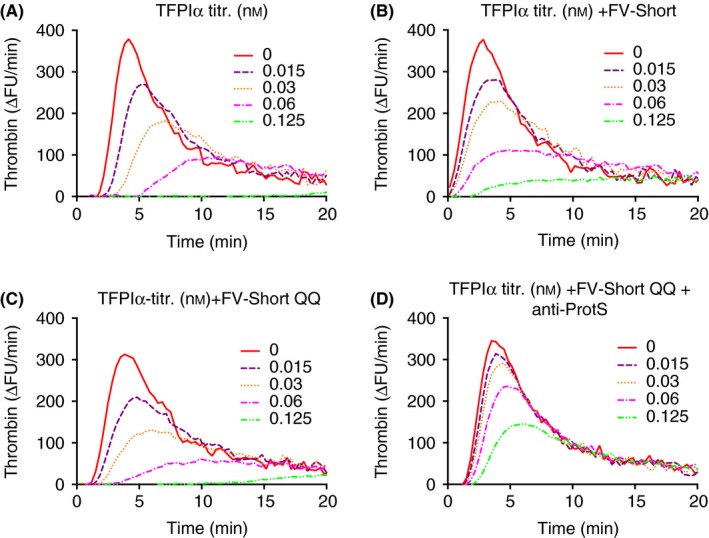
Anti‐coagulant effects of TFPIα plus FV‐Short QQ and dependence on protein S. (A) Increasing concentrations of TFPIα were added to normal plasma and thrombin generation was initiated with 1.4 pmol L−1 TF. B and C, as described in A but with either FV‐Short WT (B) or FV‐Short QQ (C) included (2 nmol L−1). D, the importance of protein S was investigated by adding anti‐protein S (anti‐ProtS) (100 ug mL−1) to an incubation also containing FV‐Short QQ (2 nmol L−1). The experiment was performed multiple times with variations and the figure illustrates a representative example
4. DISCUSSION
The recent elucidation of the molecular mechanisms of the East Texas Bleeding disorder revealed that FV‐Short, a splice variant of FV lacking 702 amino acid residues of the activation peptide B domain, is an important carrier of TFPIα in circulation.23 In this disease, a point mutation in the FV gene activates a normally existing but inefficient splice donor site in the codon for S756 and as a result affected individuals have greatly increased levels of circulating FV‐Short. The FV‐Short binds TFPIα with high affinity and through mechanisms that are yet to be elucidated, the concentration of FV‐Short/TFPIα complexes in circulation are more than 10‐fold increased as compared to unaffected individuals. In individuals with the wild‐type allele, the concentration of the circulating FV‐Short is not yet elucidated but can be estimated to be in the sub‐nM range (≈0.1‐0.2 nmol L−1), approximately 100‐fold lower than the concentration of full length FV.23 We now demonstrate that FV‐Short not only is an important carrier for TFPIα in circulation but also functions as an efficient synergistic TFPIα cofactor together with protein S in the inhibition of FXa.
The B‐domain of FV contains a highly positively charged region (963‐1008) which is followed by a carbohydrate rich region comprising approximately 35 repeats each nine amino acid residues long and then a highly acidic region (1493‐1537) that is located in the C‐terminus of the B domain just prior to the Arg1545 cleavage site.1, 2, 30, 31 It has been proposed that the two highly charged regions in the B domain interact with each other, an interaction that is crucial for keeping FV in a procofactor state.1, 31, 32, 33 In a recombinant B‐domain truncated FV‐variant denoted FV‐810 (deletion of residues 810‐1492), the high affinity‐binding site for TFPIα has been localized to the acidic C‐terminal region of the B domain.12 It is therefore most likely that the same region in FV‐Short is the high affinity‐binding site for TFPIα.23 It is interesting to note that FV810 recently was reported to have no protein S‐dependent TFPIα‐cofactor activity despite having the binding site for TFPIα,20, 21 suggesting that FV‐Short (deletion of 756‐1458) contains structural elements important for the protein S interaction which FV810 is lacking. FV is also known to bind TFPIα but with much lower affinity (Kd estimated with Biacore to be 13 nmol L−1),10 which is compatible with the concept that the acidic region in intact FV is engaged in an interaction with the positively charged cluster in the B domain.
Protein S is a cofactor to TFPIα in the inactivation of FXa on the surface of negatively charged phospholipid vesicles and as TFPIα/FXa efficiently inhibits TF/FVIIa, protein S indirectly stimulates inhibition of TF/FVIIa.15, 18, 20, 34, 35 There is no consensus regarding the question whether protein S directly stimulates inhibition of TF/FVIIa. Ndonwi and Broze reported that protein S does not directly stimulate TFPIα inhibition of TF/FVIIa whereas Peraramelli et al. showed that protein S does directly stimulate inhibition of TF/FVIIa and thereby also inhibits the activation of FIX by TF/FVIIa.36, 37, 38 Whether FV‐Short affects these reactions remains to be elucidated. Protein S may also function as a carrier for TFPIα because protein S deficient individuals have low TFPIα and furthermore immune depletion of protein S is reported to results in decreased TFPIα.34 However, we could not detect any protein S in the immune precipitate of TFPIα from normal plasma, whereas FV‐Short and intact FV were readily observed.23 This may suggest that the protein S‐TFPIα interaction is of lower affinity, which is also compatible with the high molar concentration of protein S in plasma, ≈100 nmol L−1 free protein S and ≈200 nmol L−1 in complex with C4b‐binding protein.39
In a system with purified components that follow the inhibition of FXa by TFPIα, we demonstrate that FV‐Short functions as a TFPIα cofactor and that its activity is stimulated by protein S and vice versa that the TFPIα‐cofactor activity of protein S is stimulated by FV‐Short. FV‐Short has intrinsically weak TFPIα‐cofactor activity but the presence of protein S results in rapid inactivation of the FXa. Thus, the experimental data suggest that FV‐Short and protein S function as efficient synergistic TFPIα cofactors in the inhibition of FXa. In our experimental system, both pFV and rFV when tested at low protein S (3 nmol L−1) demonstrated little TFPIα cofactor activity. However, at physiological protein S (100‐160 nmol L−1), both pFV and rFV had TPFIα‐cofactor activity, which agrees with a recent report that demonstrates that the combination of 30 nmol L−1 FV and 100 nmol L−1 protein S result in a 8‐12‐fold increased efficiency of TFPIα in inhibition of FXa.21 However, even at high protein S concentration, the FV‐Short was several‐fold more efficient than both pFV and rFV. A word of caution is relevant in relation to interpretation of results obtained with FV, because even the best preparations of purified FV contains traces of poorly defined shorter FV molecules being the result of partial proteolysis or alternative splicing.21, 25, 26 Whether the TFPIα‐cofactor activity observed with FV at high protein S levels is truly due to intact FV or caused by traces of shorter FV‐molecules remains to be determined. If one assumes that circulating FV indeed has the ability to express full TFPIα‐cofactor activity in the presence of protein S, the question is what role FV‐Short plays. In this context it is noteworthy that FV‐Short circulates in a very high affinity complex with TFPIα,23 and it is unlikely that this tightly bound TFPIα dissociates from the FV‐Short to interact with FV. This argues in favor for FV‐Short being a physiologically important TFPIα‐cofactor in addition to keeping TFPIα in the circulation. The relative importance of the TFPIα‐cofactor activities of FV‐Short and FV remains to be determined. Interesting questions for future research include whether TFPIα bound to FV‐Short or to FV have different specificities and kinetics and whether flow affects the TFPIα activity differently when bound to FV‐Short or to FV.
The synergistic TFPIα cofactor activity of FV‐Short and protein S requires the presence of negatively charged phospholipid suggesting that a multi‐molecular inhibitory complex between FV‐Short, protein S, and TFPIα is formed on the membrane and that FXa interacts with several of the components of this complex. Multiple protein‐protein and protein‐phospholipid interactions are important in the inhibition by the fully assembled FV‐Short/protein S/TFPIα complex. Thus, FXa and protein S bind to the phospholipid via their Gla domains and FV‐Short with its C1‐C2 domains.2 The C‐terminus of TFPIα binds to the C‐terminus of the FV‐Short B domain and the third Kunitz domain interacts with protein S.11, 13, 16, 40 Possibly protein S interacts directly with FV‐Short. Whether there is a direct interaction between FXa and the FV‐Short moiety of the FV‐Short‐TFPIα‐complex or if the bound TFPIα prevents direct binding of FXa to FV‐Short remains to be elucidated. In any event, all the molecular interactions ensure that the active site of FXa and the second Kunitz domain are efficiently brought together.
In our plasma thrombin generation experiment, addition of recombinant FV‐Short (WT or the QQ mutant) to normal plasma to create a molar surplus of FV‐Short over TFPIα resulted in shortening of the lag phase but despite this in a decrease of the thrombin peak. This would argue that the FV‐Short/TFPIα‐complex does not affect the initial activation of FX by the TF/FVIIa complex but that the generated FXa is quickly inhibited. Hypothetically, FV‐Short/TFPIα/FXa efficiently inhibits TF/FVIIa in the creation of a TF/FVIIa/FXa/TFPIα complex – possibly also containing FV‐Short and protein S. This interpretation was further supported by the thrombin generation experiments using FV‐deficient plasma to which either FV‐Short or thrombin‐cleaved FV‐Short were added together with TFPIα. The FV‐Short/TFPIα combination resulted in greatly prolonged lag phase and decreased thrombin generation, whereas the thrombin‐cleaved FV‐Short/TFPIα combination was in comparison quite inefficient both in prolonging lag phase and in decreasing the thrombin peak.
In conclusion, we demonstrate FV‐Short and protein S are efficient synergistic cofactors to TFPIα primarily in the regulation of FXa activity and possibly indirectly in the inhibition of TF/FVIIa. This is the second anticoagulant activity associated with FV and it further emphasizes the central importance of FV as a regulator of blood coagulation. A detailed knowledge on the interactions between FV‐isoforms, protein S and TFPIα and the role this plays in regulation of blood coagulation may help develop strategies for manipulation of the system in pro‐ or anticoagulant direction aiming at developing therapeutic opportunities for bleeding and thrombotic disorders.
AUTHOR CONTRIBUTIONS
B. Dahlbäck designed and supervised the study, interpreted results and wrote the paper, L. Jun Guo performed experiments and analyzed results, R. Livaja‐Koshiar performed experiments and analysed results, S. Tran performed experiments and analyzed results.
RELATIONSHIP DISCLOSURE
The authors declare that they have no conflicts of interest with the contents of this article.
ACKNOWLEDGMENTS
Dr T Hamuro at the Chemo‐Sero‐Therapeutic Research Institute, Japan is gratefully acknowledged for the gift of TFPIα. Dr Rodney Camire at University of Pennsylvania is gratefully acknowledged for the gift of purified FV‐Short and the FV/FV‐Short expression system. This work was supported by grants from the Swedish Research Council, Söderberg's foundation and the Swedish Heart‐Lung foundation.
Dahlbäck B, Jun Guo L, Livaja‐Koshiar R, Tran S. Factor V‐short and protein S as synergistic tissue factor pathway inhibitor (TFPIα) cofactors. Res Pract Thromb Haemost. 2018;2:114–124. 10.1002/rth2.12057
REFERENCES
- 1. Camire RM, Bos MH. The molecular basis of factor V and VIII procofactor activation. J Thromb Haemost. 2009;7:1951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dahlback B. Pro‐ and anticoagulant properties of factor V in pathogenesis of thrombosis and bleeding disorders. Int J Lab Hematol. 2016;38(Suppl 1):4–11. [DOI] [PubMed] [Google Scholar]
- 3. Dahlback B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure‐function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. 2005;25:1311–20. [DOI] [PubMed] [Google Scholar]
- 4. Castoldi E, Rosing J. Factor V Leiden: a disorder of factor V anticoagulant function. Curr Opin Hematol. 2004;11:176–81. [DOI] [PubMed] [Google Scholar]
- 5. Dahlback B. The discovery of activated protein C resistance. J Thromb Haemost. 2003;1:3–9. [DOI] [PubMed] [Google Scholar]
- 6. Dahlback B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–27. [DOI] [PubMed] [Google Scholar]
- 7. Steen M, Dahlback B. Thrombin‐mediated proteolysis of factor V resulting in gradual B‐domain release and exposure of the factor Xa‐binding site. J Biol Chem. 2002;277:38424–30. [DOI] [PubMed] [Google Scholar]
- 8. Krishnaswamy S. The transition of prothrombin to thrombin. J Thromb Haemost. 2013;11(Suppl 1):265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Asselta R, Peyvandi F. Factor V deficiency. Semin Thromb Hemost. 2009;35:382–9. [DOI] [PubMed] [Google Scholar]
- 10. Duckers C, Simioni P, Spiezia L, et al. Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood. 2008;112:3615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ndonwi M, Girard TJ, Broze GJ Jr. The C‐terminus of tissue factor pathway inhibitor alpha is required for its interaction with factors V and Va. J Thromb Haemost. 2012;10:1944–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor‐alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci U S A. 2013;110:17838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broze GJ Jr, Girard TJ. Tissue factor pathway inhibitor: structure‐function. Front Biosci (Landmark Ed). 2012;17:262–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. 2016;36:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peraramelli S, Rosing J, Hackeng TM. TFPI‐dependent activities of protein S. Thromb Res. 2012;129(Suppl 2):S23–6. [DOI] [PubMed] [Google Scholar]
- 16. Ahnstrom J, Andersson HM, Hockey V, et al. Identification of functionally important residues in TFPI Kunitz domain 3 required for the enhancement of its activity by protein S. Blood. 2012;120:5059–62. [DOI] [PubMed] [Google Scholar]
- 17. Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost. 2009;7(Suppl 1):165–8. [DOI] [PubMed] [Google Scholar]
- 18. Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci U S A. 2006;103:3106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mast AE, Broze GJ Jr. Physiological concentrations of tissue factor pathway inhibitor do not inhibit prothrombinase. Blood. 1996;87:1845–50. [PubMed] [Google Scholar]
- 20. Peraramelli S, Thomassen S, Heinzmann A, et al. Role of exosite binding modulators in the inhibition of fxa by TFPI. Thromb Haemost. 2016;115:580–90. [DOI] [PubMed] [Google Scholar]
- 21. Santamaria S, Reglinska‐Matveyev N, Gierula M, et al. Factor V anticoagulant cofactor activity that targets the early phase of coagulation. J Biol Chem. 2017;292:9335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Broze GJ Jr, Girard TJ. Factor V, tissue factor pathway inhibitor, and east Texas bleeding disorder. J Clin Invest. 2013;123:3710–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vincent LM, Tran S, Livaja R, Bensend TA, Milewicz DM, Dahlback B. Coagulation factor V(A2440G) causes east Texas bleeding disorder via TFPIalpha. J Clin Invest. 2013;123:3777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dahlback B. Purification of human vitamin K‐dependent protein S and its limited proteolysis by thrombin. Biochem J. 1983;209:837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dahlback B. Human coagluation factor V purification and thrombin‐catalyzed activation. J Clin Invest. 1980;66:583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki K, Dahlback B, Stenflo J. Thrombin‐catalyzed activation of human coagulation factor V. J Biol Chem. 1982;257:6556–64. [PubMed] [Google Scholar]
- 27. Happonen KE, Tran S, Morgelin M, et al. The Gas6‐Axl protein interaction mediates endothelial uptake of platelet microparticles. J Biol Chem. 2016;291:10586–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thorelli E, Kaufman RJ, Dahlback B. Cleavage requirements for activation of factor V by factor Xa. Eur J Biochem. 1997;247:12–20. [DOI] [PubMed] [Google Scholar]
- 29. Toso R, Camire RM. Removal of B‐domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J Biol Chem. 2004;279:21643–2150. [DOI] [PubMed] [Google Scholar]
- 30. Kalafatis M, Krishnaswamy S, Rand MD, Mann KG. Factor V. Methods Enzymol. 1993;222:224–36. [DOI] [PubMed] [Google Scholar]
- 31. Camire RM. Rethinking events in the haemostatic process: role of factor V and TFPI. Haemophilia. 2016;22(Suppl 5):3–8. [DOI] [PubMed] [Google Scholar]
- 32. Bos MH, Camire RM. A bipartite autoinhibitory region within the B‐domain suppresses function in factor V. J Biol Chem. 2012;287:26342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bunce MW, Bos MH, Krishnaswamy S, Camire RM. Restoring the procofactor state of factor Va‐like variants by complementation with B‐domain peptides. J Biol Chem. 2013;288:30151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Castoldi E, Simioni P, Tormene D, Rosing J, Hackeng TM. Hereditary and acquired protein S deficiencies are associated with low TFPI levels in plasma. J Thromb Haemost. 2010;8:294–300. [DOI] [PubMed] [Google Scholar]
- 35. Hackeng TM, Rosing J. Protein S as cofactor for TFPI. Arterioscler Thromb Vasc Biol. 2009;29:2015–20. [DOI] [PubMed] [Google Scholar]
- 36. Ndonwi M, Broze G Jr. Protein S enhances the tissue factor pathway inhibitor inhibition of factor Xa but not its inhibition of factor VIIa‐tissue factor. J Thromb Haemost. 2008;6:1044–6. [DOI] [PubMed] [Google Scholar]
- 37. Peraramelli S, Thomassen S, Heinzmann A, et al. Direct inhibition of factor VIIa by TFPI and TFPI constructs. J Thromb Haemost. 2013;11:704–14. [DOI] [PubMed] [Google Scholar]
- 38. Peraramelli S, Thomassen S, Heinzmann A, et al. Inhibition of tissue factor:factor VIIa‐catalyzed factor IX and factor X activation by TFPI and TFPI constructs. J Thromb Haemost. 2014;12:1826–37. [DOI] [PubMed] [Google Scholar]
- 39. Dahlback B. The tale of protein S and C4b‐binding protein, a story of affection. Thromb Haemost. 2007;98:90–6. [PubMed] [Google Scholar]
- 40. Ndonwi M, Tuley EA, Broze GJ Jr. The Kunitz‐3 domain of TFPI‐alpha is required for protein S‐dependent enhancement of factor Xa inhibition. Blood. 2010;116:1344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]