

Abstract
Dysregulated NOTCH1 signaling, by either gene mutations or microenvironment interactions, has been increasingly linked to chronic lymphocytic leukemia (CLL). Thus, inhibiting NOTCH1 activity represents a potential therapeutic opportunity for this disease. Using gene expression‐based screening, we identified the calcium channel modulator bepridil as a new NOTCH1 pathway inhibitor. In primary CLL cells, bepridil induced selective apoptosis even in the presence of the protective stroma. Cytotoxic effects of bepridil were independent of NOTCH1 mutation and other prognostic markers. The antitumor efficacy of bepridil was associated with inhibition of NOTCH1 activity through a decrement in trans‐membrane and activated NOTCH1 protein levels with unchanged NOTCH2 protein levels. In a CLL xenotransplant model, bepridil significantly reduced the percentage of leukemic cells infiltrating the spleen via enhanced apoptosis and decreased NOTCH1 activation. In conclusion, we report in vitro and in vivo anti‐leukemic activity of bepridil associated with inhibition of the NOTCH1 pathway in CLL. These data provide a rationale for the clinical development of bepridil as anti‐NOTCH1 targeted therapy for CLL patients.
Keywords: NOTCH1, chronic lymphocytic leukemia, targeted therapy
Short abstract
What's new?
In search of new inroads against chronic lymphocytic leukemia, these authors turned to the NOTCH1 signalling pathway. Could inhibiting NOTCH1 help treat CLL? To find out, they first identified the calcium channel blocker bepridil as a NOTCH1 inhibitor. Next, they showed that bepredil induced apoptosis in isolated CLL cells, regardless of whether they bore NOTCH1 mutations. In mice bearing CLL cells, bepredil inhibited the proliferation of the tumor cells with no overt signs of toxicity. Thus, it seems advisable to pursue bepredil as a possible candidate for treating CLL.
Introduction
The NOTCH pathway plays an important role in the modulation of cell proliferation, differentiation and apoptosis in various tissues.1 Alterations in NOTCH signaling are associated with multiple diseases, including hematological cancers.2 Emerging evidence suggests that the NOTCH signaling cascade is involved in survival and resistance to apoptosis in chronic lymphocytic leukemia (CLL).3, 4 Leukemic B cells constitutively express NOTCH1/2 receptors, as well as their ligands JAGGED1/2, which lead to the upregulation of the anti‐apoptotic protein Mcl‐1.5 Stimulation of CLL cells by NOTCH ligands increases cell survival through activation of the NFkB pathway, whereas NOTCH pathway inhibition accelerates the spontaneous apoptosis of CLL cells.6, 7
Mutations in the NOTCH1 gene emerged as one of the mechanisms leading to constitutive activation of NOTCH signaling in CLL.8, 9, 10 We were the first group to demonstrate recurrent mutations of the C‐terminal PEST domain of the protein resulting in impaired NOTCH1 degradation and deregulated signaling.11, 12, 13, 14, 15 NOTCH1 mutations represent a new biomarker for the identification of poor‐risk CLL as NOTCH1‐mutated patients have a significantly shorter survival compared to NOTCH1‐wild‐type.16, 17, 18, 19 The prevalence of NOTCH1 mutations increases with disease aggressiveness, in relapsed CLL and in patients whose CLL has transformed to Richter syndrome.20, 21 Thus, inhibiting NOTCH1 activity represents a potential therapeutic opportunity in CLL, and the incorporation of NOTCH1 pathway antagonists may improve standard treatment for this disease.
Targeting NOTCH1 has been a therapeutic strategy of great interest in many cancers. However, the use of gamma secretase inhibitors (GSIs) evaluated in clinical trials showed on‐target toxicities22 suggesting the need for the discovery of more selective NOTCH1 pathway antagonists that preferentially target NOTCH1 vs. NOTCH2 or that target mutated receptors compared to wild‐type. Some compounds directed against the NOTCH pathway are under preclinical development, including metalloproteinase inhibitors, antibodies directed against the extracellular domains of NOTCH1 and antagonists directly targeting the NOTCH transactivation complex.23 Recently, an expression‐based screen identified several calcium modulators as a potential strategy to target NOTCH1.24 Among many ion flux modulators validated to induce a NOTCH1 off signature, one of the top hits was the clinically relevant calcium channel blocker, bepridil, used to treat patients with cardiac disease. Bepridil demonstrated anti‐NOTCH1 modulating activity in T‐ALL by a mechanism unique from GSIs.25
Here, we evaluated the anti‐NOTCH1 activity and the sensitivity of primary CLL patients to bepridil in vitro and in vivo. Our data indicate that the NOTCH1 pathway and mutation as a new rational molecular therapeutic target in CLL.
Materials and Methods
Identification of NOTCH CLL signature
The GSEA software v2.0.1426, 27 was used to identify associations of the CLL NOTCH inhibition phenotype with inhibitory NOTCH gene signatures based on dataset previously described.24, 28, 29 GSEA the list of differentially expressed genes between two experimental classes, ranked by the correlation with the class distinction. GSEA identifies gene sets with common biological function that are distributed at the top or the bottom of the ranked list of differentially expressed genes.
The gene signatures for CLL NOTCH inhibition were created using previously published Affymetrix microarray expression profiling data sets (GEO: GSE51044) based on the Student's t‐test for differential expression and corrected for multiple hypotheses with the Bonferroni correction (significance cutoffs, p = 0.05 and FDR = 0.05).
Gene expression‐based high‐throughput screening and bepridil identification
We adapted a NOTCH off signature to an assay that uses ligation‐mediated amplification (LMA) and fluorescence bead‐based detection (FlexMap Technology, Luminex, Austin, TX). Full details of this protocol have been described previously.24 The signature performance was evaluated by calculating two scores that incorporate information about signature gene expression: the summed score and weighted summed score. The summed score metric combined expression ratios by summing them with a sign determined by the normal direction of regulation as determined from the GSI‐treated positive controls. The weighted summed score metric is a variant of the summed score that combines expression ratios by summing them with a weight and sign determined by the signal‐to‐noise ratio defined by GSI‐treated positive controls and the DMSO‐treated negative controls.
Patients
Peripheral blood samples from CLL patients were obtained after informed consent in accordance with Institutional Guidelines and the Declaration of Helsinki. Their clinical and biological characteristics are summarized in Supporting Information Table S1.
Isolation and culture of primary cells
B and T cells fractions were obtained from the blood of CLL patients using Ficoll density‐gradient centrifugation followed by sheep erythrocyte rosetting. This procedure allowed the simultaneous separation of highly purified rosetting T (91 ± 4.2% CD3+) from non rosetting B leukemic cells (94.6 ± 3.1% CD19+/CD5+). The mean starting fraction of T cells in CLL samples was 11.4%. Normal B and T cells were purified from the peripheral blood of healthy donors by using a B Cell Isolation Kit II and CD3+ microbeads, respectively (Miltenyi Biotec, GmbH, Bergisch Gladbach, Germany). The average purity of the isolated healthy CD19+ cells was 96.3 ± 3.1%. Normal T samples contained on average 94.2 ± 3.4% CD3+ cells. Isolated cells were incubated in RPMI 1640 media supplemented with 10% heat‐inactivated human serum (FBS, Gibco‐BRL, Gaithersburg, MD), 2 mM l‐glutamine, and 100 U/mL penicillin/100 µg/mL streptomycin and cultured with DMSO or bepridil, for 24 hr at 37°C in an atmosphere of 5% CO2. Bepridil (Sigma‐Aldrich, Saint Louis, MO) were dissolved in chloroform (0.0025%) and DMSO. We excluded cytotoxic effects of chloroform on CLL cells viability (Supporting Information Fig. S1). Experiments examining survival signals included stromal co‐cultures. The HS‐5 and OP‐9 stromal cell lines were obtained from ATCC; Nurse like cells and mesenchymal stem cells were generated as previously described.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 Stromal co‐culture was done by plating a 60.000 cells (80–100% confluent) per 24‐well plate, 24 hr before the addition of 106 CLL cells.
Flow cytometry analysis
Cell viability/apoptosis were assessed by flow cytometry (EPICS‐XL‐MCL; Beckman Coulter, Fullerton, CA) after Annexin V‐FITC/propidium iodide staining, performed using a commercial kit (Immunotech, Beckman Coulter, Marseille, FRANCE). Results were expressed as the percentage of viable (AnV‐/PI‐) or late apoptotic (AnV+/PI+) over the vehicle‐treated control. The analysis of NOTCH1 surface expression (EC) was performed using an anti‐human NOTCH1‐PE antibody (clone 527425) obtained from R&D Systems (Minneapolis, MN) on 7AAD negative (Beckman Coulter). For intracellular cleaved NOTCH1 detection we used the primary antibody to NOTCH1 cleaved at Val1744 (Cell Signaling Technology, Boston, MA) and the secondary Alexa fluor 488 as previously described.31 CD20 expression was evaluated by a PC7‐coniugated anti‐CD20 antibody (clone B9E9, Beckman Coulter) calculating the mean fluorescence intensity in CD19 + CD5+ CLL cells. Ca2+ fluxes were measured using the Fluo‐4 DirectTM calcium Assay Kit (Life Technologies, Thermo Scientific, Eugene, Oregon, USA) following the manufacturer's instructions. Analysis was performed according to methods described previously.15
Chemotaxis assay
The chemotaxis assay across polycarbonate Transwell inserts was performed as described.15 Briefly, CLL samples (107 cells/mL) were incubated in complete RPMI medium (control) or in complete medium supplemented with bepridil 2.5 µM at 37°C in 5% CO2. After 24 hr, CLL cells were suspended to a concentration of 107 cells/mL in RPMI 1640 with 0.5% bovine serum albumin, and a total of 100 μL, containing 106 cells, was added to the top chamber of a Transwell culture insert (Costar, Kennebunk, ME, USA) with a diameter of 6.5 mm and a pore size of 5 μm. Filters then were transferred to wells containing medium with or without 200 ng/mL CXCL12 (Sigma). The chambers were incubated for 3 hr at 37°C in 5% CO2. After this incubation, the cells in the lower chamber were suspended and divided into aliquots for counting with a FACSCanto flow cytometer (BD Biosciences) for 30 sec in duplicates. Migration index was calculated as the ratio between cells transmigrating in presence of the chemokine and the cells transmigrated in absence of the chemokine.15
Western blotting
Western blot was performed as described.3 We used primary antibodies to Caspase‐3, PARP and cleaved NOTCH1(Val1744) (Cell Signaling Technology); MCL‐1 (Santa Cruz Biotechnology, Santa Cruz, CA); GAPDH (Sigma‐Aldrich); NOTCH1 (clone bTAN20) and NOTCH2 (clone C651.6DbHN) obtained from DSHB‐Iowa University; Densitometric analysis was performed using Quantity One software (Bio‐Rad, Hercules, CA).
Real‐time quantitative PCR
RNA was extracted using RNeasy Plus Kits (Qiagen, Hilden, Germany), and cDNA was obtained using Prime Script RT Master Mix (Takara Bio, Dalian, China). Real‐time qPCR was performed with PCR Master Mix Power SYBER Green (Applied Biosystem, Warrington, UK) using the 7900HT fast Real‐Time PCR System (Applied Biosystem). The primer sequences are included in Supporting Information Table S2. Relative fold change was normalized to GAPDH and calculated using the 2−ΔΔCt method.
Xenograft model
A total of 1.0 × 108 CLL cells were i.v. transplanted into 8–15 weeks old NSG mice after sublethal irradiation (3.5Gy).32 After 3 days, mice were injected intraperitoneally with DMSO (vehicle) or bepridil (5 mg/kg) daily over a 20‐day period. Mice were killed 4 weeks after transplant. The engraftment was evaluated by flow cytometry analysis of the percentage of human CD45 + CD5 + CD19+ CLL cells.
Statistical analysis
Statistical analyses were performed with GraphPad (GraphPad Software, Inc., La Jolla, CA). In the text, data are presented as mean ± SD. Statistical differences between mean values were evaluated using the Student's t‐test or non‐parametric tests as Wilcoxon for paired data and Mann–Whitney for non paired data.
Results
Identification of bepridil as a NOTCH inhibitor
The recent observation that NOTCH signaling is aberrantly activated in CLL raised the possibility that some of the NOTCH pathways targeting molecules, identified in other hematological malignancies (e.g., T‐cell acute lymphoblastic leukemia (T‐ALL)), might be active in this disease. To test the idea of a subset genes commonly deregulated in NOTCH driven cancers we established a CLL NOTCH off signature from a publically available gene expression data. Here, primary NOTCH1‐mutated CLL cells were exposed to the γ‐secretase inhibitor PF‐03084014 for 48 hr and profiled by using the HT HG‐U219 GeneChip developed by Affymetrix.33 Next, we compared this CLL NOTCH off signature with transcriptional profiling of DSD41 T‐cell leukemia cells in which NOTCH was turned off with DAPT and Compound E. Gene set enrichment analysis (GSEA) (Fig. 1 a) revealed a statistically significant enrichment across three different signatures. This result suggests that a subset of genes (Fig. 1 a column) is similarly regulated by NOTCH in CLL and T‐ALL leukemia.
Figure 1.
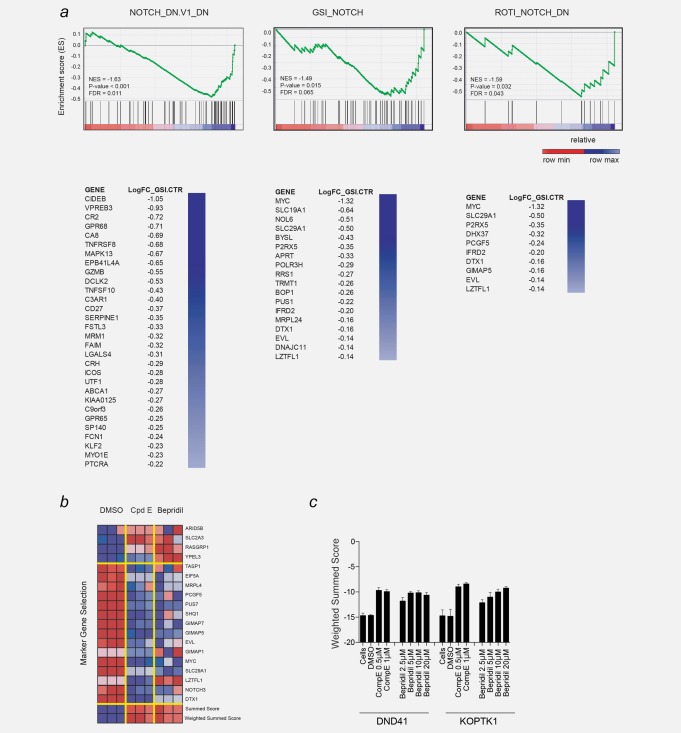
A transcription‐based screens identified bepridil as a NOTCH modulator. (a) GSEA plot showing the enrichment of the genes downregulated by gamma secretase inhibitor knockdown in the three NOTCH abrogation signature. p < 0.001, computed using the permutation test for gene set enrichment implemented in the GSEA v2.0.14 platform. Significantly enriched genes are ranked in columns. (b) Following treatment of DND41 cells for 72 hr with either DMSO (0.08%), GSI (compound E, 1 µM) or bepridil (20 µM) the expression levels of 19 genes that define a T‐ALL‐specific NOTCH1 signature were measured with a ligation‐mediated amplification/fluorescent bead‐based detection system. Each column represents an independent experimental replicate. Dark red indicates high gene expression and dark blue low gene expression. NOTCH marker gene expression is depicted as a ratio of the expression of the marker gene above or on the mean of four control genes. The summed score combines expression ratios by summing them with a sign determined by the expected direction of regulation as determined from the positive controls (GSI‐treated). The weighted summed score metric is a variant of the summed score metric that combines expression ratios by summing them with weight and sign determined by the signal‐to‐noise ratio of the positive control (GSI‐treated) and negative controls (DMSO‐treated). (c) Induction of NOTCH1 off abrogation signature (weighted summed score) measured by GE‐HTS in DND41 cells treated with the indicated concentrations of bepridil for 72 hr. Error bars denote the mean ± SD of eight replicates for untreated cells, and the mean ± SD of four replicates for the DMSO, bepridil and compound E (GSI)‐treated cells.
Although this result is partially expected, it supports the rationale of repurposing transcription‐based screens to the identification of NOTCH modulators in other diseases. We previously reported the results of Gene Expression‐based High‐throughput Screening (GE‐HTS), an approach using gene expression signatures as surrogates for cellular states. The premise of this innovation is that expression‐based signatures can be used for high‐throughput chemical screening. We applied GE‐HTS to modulating mutated NOTCH1 in T‐ALL.24, 34, 35 Expression signatures were used as surrogates for the expression vs. the loss of NOTCH1 (NOTCH1 on vs. off). Signature performance was evaluated by calculating two scores that incorporate information about signature gene expression: the summed score and weighted summed score. The summed score metric combined expression ratios by summing them with a sign determined by the expected direction of regulation as determined from the treated positive controls (GSI). The weighted summed score metric is a variant of the summed score metric that combines expression ratios by summing them with a weight and sign determined by the signal‐to‐noise ratio determined of GSI‐treated positive controls and the DMSO‐treated negative controls. Among various ion flux modulators that scored highly on our screen, we initially characterized thapsigargin in T‐ALL.24 Here, we chose to focus more closely on the FDA‐approved calcium channel modulator, bepridil, a drug previously used in the treatment of patients with cardiac disease. Bepridil demonstrates an ability to induce a NOTCH1 off signature (Fig. 1 b) with a concentration‐dependent effect (Fig. 1 c) in the range of plasma concentrations achievable in patients treated with this drug.36
Bepridil inhibits the NOTCH1 pathway in CLL cells
We have previously demonstrated that isolated CLL cells express transcriptionally active NOTCH1.3 Given the anti‐NOTCH1 modulating activity of bepridil in T‐ALL, we measured NOTCH1 expression and activation in primary CLL cells after bepridil treatment in vitro. Specifically, flow cytometric analysis of cultured CLL cells demonstrated that bepridil significantly reduced the surface expression of NOTCH1 compared to vehicle (33 ± 20% vs. 48.3 ± 20.5% respectively, N = 21; p < 0.05) (Fig. 2 ai and ii). Western blot analysis showed the capacity of bepridil to down regulate the trans‐membrane (TM) bound portion of NOTCH1, as previously reported in T‐ALL (Fig. 2 bi and ii). In addition, bepridil significantly decreased the expression of both the full‐length (FL) NOTCH1 precursor and the activated NOTCH1 intracellular domain (ICD), as assessed by an antibody against all forms of NOTCH1 (Fig. 2 bi and ii) and/or the anti‐NOTCH1(Val1744) antibody (Fig. 2 biii and iv), which specifically recognizes the cleaved NOTCH1‐ICD starting at Val1744. NOTCH2 protein levels remained unchanged in bepridil‐treated CLL samples compared to vehicle (Supporting Information Fig. S2), supporting a preferential effect of bepridil on the NOTCH1 pathway. To further test the ability of bepridil to inhibit NOTCH1 signaling activation, we analyzed the levels of NOTCH1 transcriptional target genes after the exposure to this compound. Using quantitative reverse transcription‐PCR, we found decreased mRNA levels of HES1 and c‐MYC after 6 hr bepridil treatment (Fig. 2 ci). The levels of NOTCH1 mRNA were not altered by bepridil (Supporting Information Fig. S3), suggesting non‐transcriptional effects that might interfere with NOTCH1 translation, degradation or stabilization.
Figure 2.
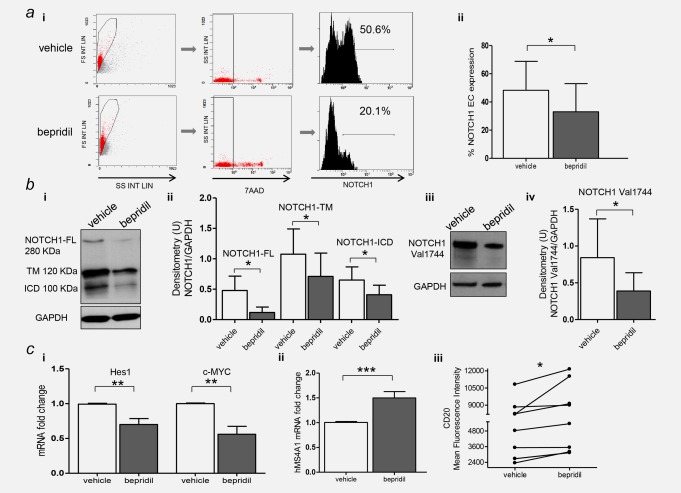
The antitumor efficacy of bepridil was associated with inhibition of NOTCH1 activity. (a) Representative flow cytometry histogram of the expression of the extracellular (EC) bound portion of NOTCH1 in CLL cells incubated with bepridil or vehicle control (i). Bar graphs show the means ± SD of NOTCH1‐EC fluorescence in 23 treated cases (ii). (b) Representative western blot (i) and quantitative densitometry analysis (ii) of the full‐length (FL), transmembrane (TM) and intracellular domain (ICD) forms of NOTCH1 protein levels in bepridil‐treated CLL compared to vehicle controls. Representative western blot (iii) and quantitative densitometry analysis (iv) of the anti‐NOTCH1(Val1744) antibody. Densitometry units (U) were calculated relative to GAPDH. (c) Analysis of HES1, c‐MYC (i) and hMS4A1 (ii) gene expression in CLL cells treated with bepridil (N=10). mRNA levels were normalized to GAPDH and represented as fold change using untreated cells as a reference. Panel (iii) shows a dot‐and‐line diagram of the CD20 Mean Fluorescence Intensity levels in primary CLL cells. Results are the mean ± SD of all the samples analyzed. The corresponding p‐values are reported. *p <0.05, **p < 0.01, ***p < 0.001 according to Student's test. [Color figure can be viewed at http://wileyonlinelibrary.com]
It has been previously demonstrated that NOTCH1 inhibition increased CD20 expression in CLL cells.37 To further demonstrate the anti‐NOTCH1 activity of bepridil, we measured transcript levels of MS4A1 and the expression of its encoded CD20 protein, in treated CLL cells. As shown in Figure 2 cii and iii, the expression of both MS4A1 and CD20 were significantly upregulated by bepridil treatment of both wild type and NOTCH1 mutated CLL cells.
It has been shown that various components of the microenvironment increase NOTCH1 activation in CLL cells.9, 38 Here, we examined whether bepridil was able to modulate the NOTCH1 expression in CLL cells co‐cultured on a stromal layer. Under co‐culture conditions, bepridil treatment led to a significant reduction of surface expression of NOTCH1 on CLL cells (45.8%±19.4 vs. 69.98%±9.9 of untreated cells, N = 6 p < 0.05) (Supporting Information Fig. S4), suggesting that bepridil not only affects constitutive NOTCH1 levels but even those induced by the microenvironment.
Several reports indicate that Ca2+ homeostasis is important in NOTCH1 maturation, trafficking and processing.39, 40 Bepridil has been previously reported to increase intracellular Ca2+ in human neutrophils by endoplasmic reticulum (ER) stores emptying.41 To find a mechanism for the anti‐NOTCH1 activity of bepridil, we determined its ability to alter intracellular Ca2+ in CLL using a Fluo‐4 calcium mobilization assay. After IgM stimulation of CLL cells, bepridil induced a large increase in Fluo‐4 fluorescence indicating high levels of intracellular Ca2+ fluxes (Supporting Information Fig. S5). Similar effects have been already described for thapsigargin, a Ca2+ channel inhibitor with anti‐NOTCH1 activity that is commonly used for emptying intracellular ER stores.24, 34
Altogether these data show that bepridil targets the NOTCH1 pathway perturbing the Ca2+ homeostasis in CLL cells.
Bepridil induces selective cytotoxicity in primary CLL cells via apoptosis
Sensitivity to bepridil in primary patients was evaluated by incubating CLL cells with different concentrations of bepridil ranging from 1 to 3 μM and viability was measured after 24 hr by Annexin V labeling (N = 6). Bepridil treatment significantly reduced the viability of primary CLL cells at a 2.5 μM concentration (Fig. 3 a). Specifically, viability significantly decreased to 34 ± 22.2% compared to 57 ± 21.6% of the vehicle control in 66 CLL samples tested (p <0.001) (Fig. 3 b; Supporting Information Table S1). Conversely, bepridil did not affect the viability of T cells from CLL patients nor the viability of B and T cells from healthy donors, demonstrating a selective activity against neoplastic B cells compared to normal hematopoietic cells (Figs. 3 b and 3 c).
Figure 3.
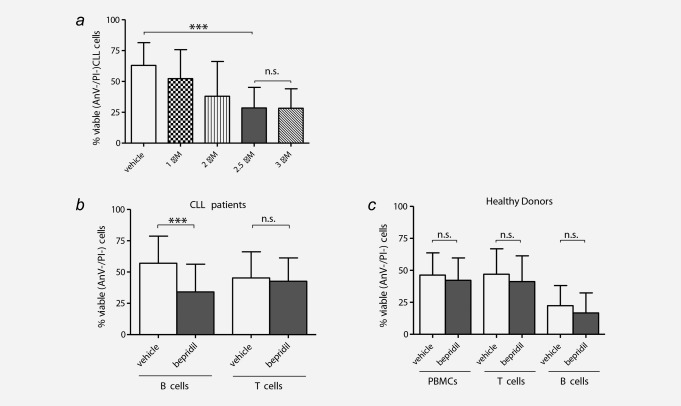
Selective cytotoxic effects of bepridil against primary CLL cells. (a) Primary CLL cells from six cases were incubated in the presence of vehicle control (DMSO) or increasing concentrations of bepridil (1–3 µM) for 24 hr. Viability was determined by Annexin‐V/PI flow cytometry. (b) Primary tumor B (N = 66) and normal T (N = 16) lymphocyte subpopulations from patients with CLL were incubated with 2.5 µM bepridil or vehicle control (DMSO) and viability was assessed at 24 hr culture. Data demonstrated a significant reduction of B CLL viability in bepridil treated compared to vehicle (34%±2.7 vs. 57%±2.6) while T cells were unchanged (42.6% ± 4.6 vs. 45.3% ± 5.2). (c) Peripheral blood mononuclear cells (PBMC) (N = 13), B (N = 10) and T (N = 16) lymphocytes from healthy donors were exposed to bepridil 2.5 µM or vehicle for 24 hr. Viability was evaluated by flow cytometric analysis of Annexin‐V/PI staining. PBMC, B and T cells mean viability values of bepridil treated vs. untreated cells were 41.2% ± 5.5 vs. 46.9% ± 5.5, 16.7 ± 4.9 vs. 22.3 ± 4.9 and 39.1% ± 5 vs. 43.4% ± 4.9, respectively. Results are presented as the percentage of viable (AnV‐/PI‐) cells, mean ± SD, ***p < 0.001 according to Student's test.
The cytotoxicity induced by bepridil was due to apoptosis, as indicated by a significantly increased percentage of Annexin V/propidium iodide positive apoptotic CLL cells compared to vehicle‐treated cells (40.2 ± 20.2% vs. 22.8 ± 13.6% respectively, p < 0.001) (Fig. 4 ai and ii). These apoptotic effects were supported by the detection of increased degradation of the PARP protein to its 89 kDa inactive fragment (N = 12 p < 0.05) (Fig. 4 bi and ii). Cleavage of PARP suggests that caspase activation contributed to bepridil‐induced apoptosis of CLL cells. Indeed, treatment of bepridil resulted in an increase in active cleaved products of caspase‐3 concurrent with a decrease in the pro‐form in selected CLL samples (Fig. 4 ci and ii). In addition, bepridil treatment was associated with a reduction of MCL‐1 protein expression (p < 0.01) (Fig. 4 di and ii) and a fivefold upregulation of NOXA transcript levels (Fig. 4 e).
Figure 4.
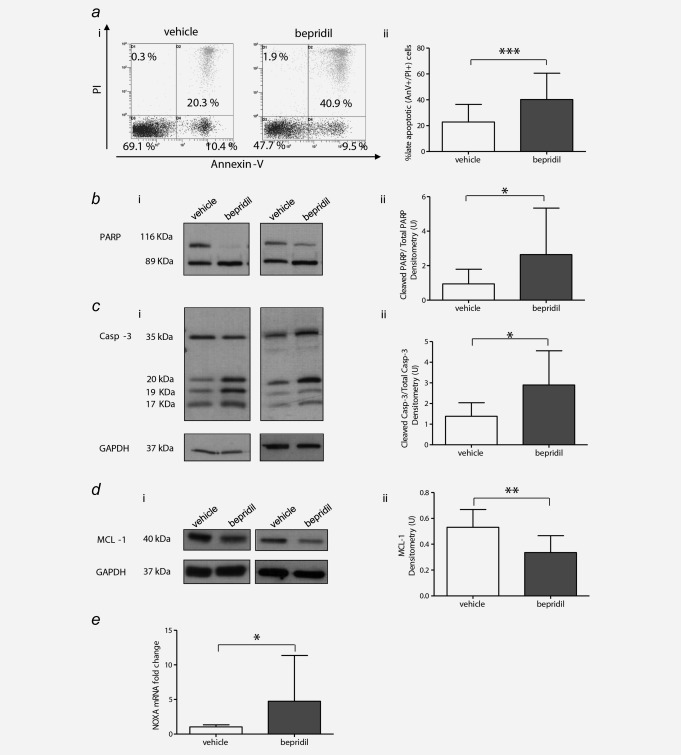
Bepridil‐induced apoptosis of CLL cells in vitro. (a) Representative dot plots (i) and bar graph (ii) of Annexin V‐FITC/PI staining results of bepridil‐induced apoptosis in B cells from CLL patients (N = 66). (b–d) Representative western blots (i) and quantitative densitometry analysis (ii) of cleaved PARP, caspase‐3 and MCL1 protein levels in bepridil‐treated CLL compared to vehicle controls (N = 12, N = 7, N = 11). (e) Analysis of NOXA gene expression in CLL cells treated with bepridil (N = 10). mRNA levels were normalized to GAPDH and represented as fold change using vehicle‐treated cells as a reference. Resulted are the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to Student's test.
Bepridil induces cytotoxicity in CLL cells independent of NOTCH1 mutational status and other prognostic markers
The cytotoxic activity of bepridil treatment of CLL cells was heterogeneous displaying a variable effect on the induction of cell death. Because of this broad spectrum of results, we investigated whether the presence of CLL prognostic factors, including the IGHV mutational status, ZAP‐70 expression, or the most frequent cytogenetic abnormalities (17p, 11q and 13q deletions and trisomy 12), sensitize the cell to bepridil‐induced apoptosis. To this end, we analyzed samples from CLL cases that included different prognostic subtypes (Supporting Information Table S1). We found that there were no significant differences in the response to bepridil in CLL cells based on the analysis of traditional laboratory predictors of chemotherapy response (Figs. 5 a–5 e). Moreover, we did not find any association between bepridil sensitivity and the most frequent genomic alterations recently described in CLL cells. NOTCH1 activation in CLL relies on both receptor–ligand interaction and the presence of mutations. NOTCH1 status did not influence the bepridil response in the CLL samples studied (Fig. 5 d). There was also no influence of p53 deletion or SF3B1 status on the apoptotic response to bepridil in the CLL samples tested. In addition, we demonstrated that bepridil was toxic to both untreated and previously treated CLL patient's cells, the latter which are more frequently resistant to in vitro drug treatments (Supporting Information Fig. S6).
Figure 5.
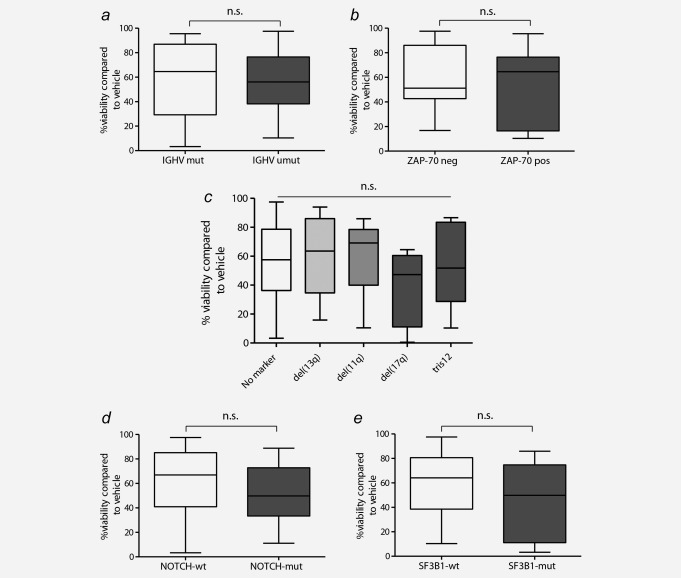
Cytotoxic effects of bepridil were independent of prognostic markers. The cytotoxic effects of bepridil were evaluated in the contest of common prognostic markers of CLL including (a) IGHV mutational status (mutated N=22, unmutated N = 42), (b) ZAP70 expression (ZAP70‐negative N = 27, ZAP70‐positive N = 24), (c) cytogenetic subgroups (No marker N = 17, del13q N = 23, del11q N = 6, del17q N = 7, tris12 N = 10), (d) NOTCH1 (wild type N = 45, mutated N = 21) and (e) SF3B1 (wild type N = 55, mutated N = 8) mutations. Viability was determined by Annexin‐V/PI flow cytometry and was calculated relative to untreated controls.
Bepridil overcomes the protective effects of mesenchymal stromal or nurse‐like cell‐based microenvironment and impairs CLL cells chemotaxis
Given that marrow stromal cells provide important survival and drug resistance signals to CLL cells, we tested whether bepridil could interfere with this protective stromal effect. Thus, we examined the cytotoxic properties of bepridil in the context of micro‐environmental stimuli by co‐culturing primary CLL cells with different stromal cells, including primary mesenchymal cells (MSCs), HS‐5 and OP9 stromal cell lines. As shown in Figure 6 a and Supporting Information S7a, bepridil at a 2.5 μM concentration still reduced viability and induced apoptosis in CLL cells under co‐culture conditions using MSCs derived from either healthy donors or CLL patients. In addition, co‐culture with HS‐5 or OP9 stromal cells demonstrated bepridil to overcome stromal protection of CLL cells against apoptosis (Fig. 6 a). As shown in Supporting Information Figure S7b, the presence of a stromal layer did not affect the apoptotic activity of bepridil at long‐term exposure. There was also no visible effect of bepridil on the structure or adhesion of different stromal cell layers (Supporting Information Fig. S7c). The cytotoxic effects of bepridil against CLL cells were also conserved in the contest of autologous nurse‐like cells micro‐environmental protection (Fig. 6 a).
Figure 6.
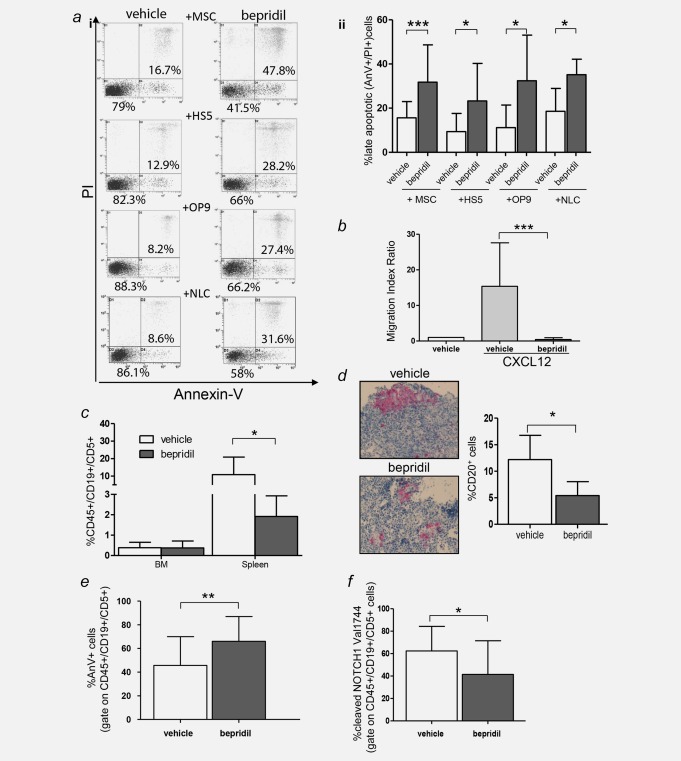
Bepridil overcomes CLL microenvironment protection, reduces cell migration and exerts antitumor activity in vivo. (a) Representative dot plots (i) of Annexin‐V/PI analysis performed on CLL cells treated with bepridil in the presence of primary mesenchymal cells (MSCs), HS‐5, OP9 cell lines and autologous nurse like cells (NLC). Statistical graphs of late apoptosis (ii) calculated relative to untreated controls (MSCs N = 34; HS‐5 N = 8; OP9 N = 14; NLC N = 6). Resulted are the mean ± SD of all the samples analyzed, *p < 0.05, ***p < 0.001 according to Student's test. (b) Statistical graphs of CLL cells chemotaxis toward CXCL12 (200 ng/mL) in presence or absence of bepridil. Data represent the mean ± SD of three‐independent experiments, each performed in triplicate. ***p < 0.001 according to Student's test. (c) Quantitative recovery of human CD45+/CD19+/CD5+ cells in the bone marrow and spleen of NSG mice evaluated by flow cytometry analysis. (d) Immunohistochemistry analysis and quantification of CD20+ human CLL cells engrafted in the spleen of vehicle or bepridil‐treated mice (original magnification 100×). (e) Percentage of apoptosis estimated by Annexin V staining in CD45+/CD19+/CD5+ cells recovered from the spleen of treated (N = 21) vs. untreated mice (N = 22). Data are mean ± SD. **p < 0.01. (f) Bar graphs show the means ± SD of cleaved‐NOTCH1‐Val1744 fluorescence on splenic CD45+/CD19+/CD5+ CLL cells of 16 bepridil‐treated vs. 17 untreated mice. *p < 0.05.
NOTCH1 activation has been recently associated with increased chemotaxis of CLL cells into tissue microenvironments.15 To evaluate the effects of bepridil treatment in the migratory potential of CLL primary cells we performed a CXCL12‐induced chemotaxis assay. As shown in Figure 6 b, bepridil treatment significantly decreased the migration index of CLL cell compared to vehicle (15.3 ± 12.2 vs. 0.4 ± 0.5; N = 3; p < 0.001)
Altogether these data suggest the potential efficacy of bepridil for the eradication of CLL cells in vivo.
Bepridil exerts antitumor activity in a xenotransplant model of CLL
Since bepridil was able to effectively kill CLL cells in the presence of bone marrow stromal cells, we postulated that this compound would be effective against CLL cells in vivo. To test this possibility, we treated NSG mice with bepridil to determine its in vivo effects on xenografted CLL cells (Supporting Information Table S3: B‐CLL and T cells purity, clinical and molecular features). Bepridil 5 mg/kg was intraperitoneal (i.p.) administered daily over a 20‐day period starting 3 days after CLL injection. Mice were killed 4 weeks after transplant. Flow cytometry analysis revealed a statistically significant decrease in human CD45 + CD19 + CD5+ percentage in the spleen of bepridil‐treated mice compared to vehicle (1.9 ± 1% vs. 10.8 ± 10%, N = 13, p < 0.05) (Fig. 6 c). Furthermore, immunohistochemical analyses showed significantly reduced splenic engraftment of human CD20 positive cells in treated mice (Fig. 6 d). Bepridil significantly increased the expression of Annexin V on human CD45 + CD19 + CD5+ B cells compared to vehicle (Fig. 6 e; 66%±20.8 vs. 45.7%±24.3, p < 0.01), consistent with a high rate of apoptosis in vivo. The anti‐leukemic effects of bepridil correlated with a reduced NOTCH1 activity in CLL cells recovered from treated compared to untreated mice (Fig. 6 f; 41.4%±29.9 vs. 62.3%±21.9, p < 0.05). Bepridil treatment did not have a major effect in the neoplastic bone marrow cell population of treated animals. However, CLL recovery from bone marrow was limited and significantly lower than spleen as expected in this xenograft model.32
Mice in both control and bepridil‐treated groups did not show any overt signs of toxicity. There was no statistically significant difference in the body weight of the two groups at the end of the study (Supporting Information Fig. S8a). At necropsy, there was no evidence of gastrointestinal toxicity, and visual inspection of the organs did not show any overt toxicity. Histological analysis of the intestine showed normal goblet cell number with preservation of the architecture and proliferation of the intestinal epithelium (Fig. 5 c) (Supporting Information Fig. S8b) suggesting a lack of combined NOTCH1 and NOTCH2 inhibition in this tissue. This result further supports the notion that bepridil preferentially target NOTCH1 over NOTCH2 and suggest that this drug might be potentially effective for clinical treatment of CLL without the on‐target NOTCH toxicity found with GSI compounds.
Discussion
Growing evidence supported by recent studies shows that the NOTCH signaling network is frequently deregulated in CLL3 thus having tremendous potential as a new therapeutic target in this disease. Here, we challenged primary CLL cells with the ion flux modulator bepridil that has been validated to induce a NOTCH1 off signature in T‐ALL. Available in France and Japan, bepridil is an FDA‐approved non‐selective ion channels blocker. It is classified as a class IV antiarrhythmic drug, prescribed for the treatment of angina pectoris and supraventricular and ventricular arrhythmia.42, 43 Our results show that the NOTCH1 pathway was inhibited at concentrations of bepridil that induced apoptosis in CLL cells. Furthermore, despite the risk of a QTc‐prolongation (and more rarely torsades de pointes), at NOTCH1 inhibitory concentrations, bepridil can be safely administered in a hospital setting to people.44
GSIs represent the pioneering class of NOTCH inhibitors that have been extensively used in experimental works and administered in clinical trials.22 GSI treatment blocks gamma‐secretase‐mediated proteolytic activation of all four NOTCH receptors resulting in reduced levels of the NOTCH active intracellular fragment. Similarly, bepridil treatment results in decreased levels of activated NOTCH1 in CLL cells. Distinct from GSI, bepridil reduces the expression of the full‐length precursor, extracellular and transmembrane forms of NOTCH1 in CLL cells. These results, along with the evidence that bepridil does not affect NOTCH1 mRNA expression, suggest that bepridil effect on NOTCH1 occurs at a translational/post‐translational level, before the cleavage by the gamma‐secretase complex.
Several reports indicate that Ca2+ homeostasis is important in NOTCH1 maturation, trafficking and processing.24, 39, 40 Bepridil has been previously reported to increase intracellular Ca2+ in human neutrophils by ER stores emptying.41 Our data indicated similar alteration in Ca2+ fluxes, suggesting that bepridil might alter NOTCH1 translation, trafficking and/or maturation by reducing ER Ca2+ concentration in CLL. In line with the hypothesis that the ER exhaust might impede protein trafficking, multiple SERCA‐type Ca2+ channel inhibitors (i.e., thapsigargin, cyclopiazonic acid and clerodane diterpene casearin J (CJ),24, 34, 45 demonstrated a profound anti NOTCH1 activity in different disease models.
Small‐molecule inhibitors are thought to be less specific than therapeutic monoclonal antibodies. In CLL cells, bepridil demonstrated a specific down‐modulation of activated NOTCH1 whereas the expression of NOTCH2 remains unaffected. This selective reduction of the NOTCH1 protein represents a potential advantage of bepridil that is similar to antibody inhibitors. Indeed, NOTCH isoform‐specific antibodies work by binding‐specific regions of the NOTCH receptor and allow targeting of individual NOTCH family members. Recently, a NOTCH1 antibody against the negative regulatory region of the protein showed selective inhibition with good antitumor effects and limited toxicity.46 However, antibodies are large molecules, and delivery/access to leukemic cells in bulky lymph nodes could be problematic.47 We demonstrated that bepridil displays antileukemic effects against human CD19 + CD5+ cells engrafted in the spleen of xenotransplanted mice, suggesting a relevant activity against nodal disease.
The cytotoxic effect of bepridil was selective for tumor CLL cells, given that autologous T lymphocytes remain almost unaffected. In addition, this compound had little cytotoxic impact on normal human PBMCs, T and B cells in vitro suggesting that hematological side effects in a clinical setting could be minimized.
In our study, we found that the level of apoptosis induced by bepridil was independent of the strongest laboratory predictors of CLL response to chemotherapy,48, 49 suggesting other potential molecular targets beside NOTCH1. Indeed, our preclinical findings are similar to those of new PI3K and BTK inhibitors50, 51 that are currently used for high‐risk patients. Exploring additional bepridil combinations with kinase inhibitors targeting multiple branches of signaling pathways may identify further therapeutic opportunities for CLL patients.
Notably, our data demonstrated that bepridil inhibits signaling through wild‐type and mutated NOTCH1 receptors, exerting cytotoxic effects regardless the NOTCH1 mutational status. This is consistent with the constitutive NOTCH signaling activation that was implicated in CLL apoptosis resistance and indicated that mutations could be only one of the mechanisms explaining altered NOTCH signaling in this disease. Therefore, bepridil represents a repositioned drug with potential benefits for all CLL patients.
In addition, the anti‐leukemic effect of bepridil is able to substantially increase CD20 expression in CLL cells. Similar results have been demonstrated with the anti‐NOTCH1 activity of GSIs and specific siRNA against NOTCH1.38 These data further support the evidence that the cytotoxic effects of bepridil depend on NOTCH1 inhibition.
It has been reported that deregulated NOTCH signaling not only affects CLL cell apoptosis but also influences the interaction of leukemic cells with the BM microenvironment.37 In this work, bepridil effectively killed CLL cells in the presence of BM stromal cells, which usually confer resistance to many anticancer agents. It is well established that the stromal niche exerts a protective effect on CLL cells, thereby also affecting their drug sensitivity. Recent evidence implicates the contribution of the NOTCH signaling to the stromal cell‐mediated effects on CLL cells.37 Indeed, BM stromal cells express some NOTCH molecules52 that activate the NOTCH pathway and induce pro‐survival signals on CLL cells.53 Therefore, the inactivation of NOTCH exerted by bepridil disrupts the reciprocal interaction of BM stroma with CLL cells, thus representing an additional tool to overcome drug resistance and improve the therapeutic strategies for CLL.
Our in vivo experiments showed that bepridil was able to modulate the NOTCH1 signaling and control the growth of CD19 + CD5+ tumor cells infiltrating the spleen of immunodeficient mice. These results provide significant evidence that bepridil may circumvent the protective effects of the CLL cell microenvironment not only in vitro but also in the in vivo setting, showing the potential for CLL treatment. Additional studies aimed at improving the controlled delivery of this therapeutic agent for high efficacy and limited side effects are needed.
The clinical success of GSIs for anti‐NOTCH cancer therapy was delayed by the development of high‐grade gastrointestinal toxicity.54 This is likely an on‐target toxicity of NOTCH inhibition that induces gastrointestinal precursor cells to become secretory goblet cells.55, 56 To date, this GSI‐induced gut toxicity has been overcome by intermitting dosing schedules and attenuated by the concomitant use of glucocorticoids.57 Bepridil treatment was not associated with any significant gastrointestinal injury despite the inhibitory effect against leukemic B cells. This might be related to the antagonistic activity of bepridil that preferentially targets NOTCH1 vs. NOTCH2. This selective effect might be beneficial for a future application for a targeted therapy in CLL.
In conclusion, bepridil displays cytotoxic activity against CLL cells both in vitro and in vivo while sparing normal B and T lymphocytes. This anti‐neoplastic effect is associated with specific inhibition of the NOTCH1 signaling regardless the presence of different adverse prognostic markers, including NOTCH1 mutations. Given its preclinical activity and specificity, bepridil is an attractive compound for clinical development.
Author Contributions
S.B. and B.D.P. performed experiments and analyzed data, P.A., E.D and F.D.F. performed experiments and analyzed data, E.V. and C.R. assisted with in vivo experiments, D.S. and C.D. performed flow cytometry, T.Z. contributed to samples collection, A.D.T. performed molecular diagnostics, E.R. contributed to interpretation of data and edited the manuscript, G.A. analyzed data, G.R. and K.S. analyzed and interpreted data and edited the manuscript, M.D.I. and F.F. provided patient samples, clinical data and critical suggestions, P.S. led the project, designed experiments and wrote the manuscript. All authors critically reviewed the final manuscript.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Figure 8
Supporting Information Tbale 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information
Acknowledgements
We thank M.G. Cantelmi for her assistance in conducting the flow cytometry analysis, R. Iacucci for her help in cell separation, R. Ciurnelli for excellent technical help and G. Ricci for histopathologic advice.
Conflict of interest: The authors declare no potential conflict of interest.
References
- 1. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 2011;11:338–51. [DOI] [PubMed] [Google Scholar]
- 2. Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006;107:2223–33. [DOI] [PubMed] [Google Scholar]
- 3. Rosati E, Sabatini R, Rampino G. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B‐CLL cells. Blood 2009;113:856–65. [DOI] [PubMed] [Google Scholar]
- 4. Fabbri G, Holmes AB, Viganotti M, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2017;114:E2911–E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Falco F, Sabatini R, Del Papa B, et al. Notch signaling sustains the expression of Mcl‐1 and the activity of eIF4E to promote cell survival in CLL. Oncotarget 2015;6:16559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosati E, Sabatini R, De Falco F, et al. γ‐Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and notch down‐regulation. Int J Cancer 2013;132:1940–53. [DOI] [PubMed] [Google Scholar]
- 7. Baldoni S, Sportoletti P, Del Papa B, et al. NOTCH and NF‐κB interplay in chronic lymphocytic leukemia is independent of genetic lesion. Int J Hematol 2013;98:153–7. [DOI] [PubMed] [Google Scholar]
- 8. De Falco F, Sabatini R, Falzetti F, et al. Constitutive phosphorylation of the active Notch1 intracellular domain in chronic lymphocytic leukemia cells with NOTCH1 mutation. Leukemia 2015;29:994–8. [DOI] [PubMed] [Google Scholar]
- 9. Arruga F, Gizdic B, Serra S, et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia 2014;28:1060–70. [DOI] [PubMed] [Google Scholar]
- 10. Gaidano G, Foà R, Dalla‐Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest 2012;122:3432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Ianni M, Baldoni S, Rosati E, et al. A new genetic lesion in B‐CLL: a NOTCH1 PEST domain mutation. Br J Haematol 2009;146:689–91. [DOI] [PubMed] [Google Scholar]
- 12. Puente XS, Pinyol M, Quesada V, et al. Whole‐genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med 2011;208:1389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sportoletti P, Baldoni S, Del Papa B, et al. A novel NOTCH1 PEST domain mutation in a case of chronic lymphocytic leukemia. Leuk Lymphoma 2013;54:1780–2. [DOI] [PubMed] [Google Scholar]
- 15. Arruga F, Gizdic B, Bologna C, et al. Mutations in NOTCH1 PEST domain orchestrate CCL19‐driven homing of chronic lymphocytic leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia 2017;31:1882–93. [DOI] [PubMed] [Google Scholar]
- 16. Sportoletti P, Baldoni S, Cavalli L, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B‐CLL. Br J Haematol 2010;151:404–6. [DOI] [PubMed] [Google Scholar]
- 17. Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012;119:521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Villamor N, Conde L, Martinez‐Trillos A, et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia 2013;27:1100–6. [DOI] [PubMed] [Google Scholar]
- 19. Sportoletti P, Baldoni S, Del Papa B, et al. A revised NOTCH1 mutation frequency still impacts survival while the allele burden predicts early progression in chronic lymphocytic leukemia. Leukemia 2014;28:436–9. [DOI] [PubMed] [Google Scholar]
- 20. Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med 2013;210:2273–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Albi E, Baldoni S, Aureli P, et al. Ibrutinib treatment of a patient with relapsing chronic lymphocytic leukemia and sustained remission of Richter syndrome. Tumori 2017;103:e37–e40. doi: 10.5301/tj.5000667. [DOI] [PubMed] [Google Scholar]
- 22. Deangelo DJ, Stone RM, Silverman LB, et al. A phase I clinical trial of the notch inhibitor MK‐0752 in patients with T‐cell acute lymphoblastic leukemia/lymphoma (T‐ALL) and other leukemias. J Clin Oncol 2006;24(18_suppl):6585–6585. [Google Scholar]
- 23. Roti G, Stegmaier K. Targeting NOTCH1 in hematopoietic malignancy. Crit Rev Oncog 2011;16:103–15. [DOI] [PubMed] [Google Scholar]
- 24. Roti G, Carlton A, Ross KN, et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013;23:390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roti G, Ross KN, Ferrando AA, et al. Expression‐based screen identifies the calcium channel antagonist bepridil as a Notch1 modulator in T‐ALL. Blood 2009;114. no. 22 366. [Google Scholar]
- 26. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC‐1α‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- 27. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dohda T, Maljukova A, Liu L, et al. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T‐cell acute lymphoblastic leukemia cell lines. Exp Cell Res 2007;313:3141–52. [DOI] [PubMed] [Google Scholar]
- 29. Dovey HF, John V, Anderson JP, et al. Functional gamma‐secretase inhibitors reduce beta‐amyloid peptide levels in brain. J Neurochem 2009;76:173–81. [DOI] [PubMed] [Google Scholar]
- 30. Sportoletti P, Del Papa B, De Ioanni M, et al. Interleukin‐7‐engineered mesenchymal cells: in vitro effects on naive T‐cell population. Biol Blood Marrow Transplant 2006;12:1250–60. [DOI] [PubMed] [Google Scholar]
- 31. Gerhardt DM, Pajcini KV, D'altri T, et al. The Notch1 transcriptional activation domain is required for development and reveals a novel role for Notch1 signaling in fetal hematopoietic stem cells. Genes Dev 2014;28:576–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dürig J, Ebeling P, Grabellus F, et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res 2007;67:8653–61. [DOI] [PubMed] [Google Scholar]
- 33. Lopez‐Guerra M, Xargay‐Torrent S, Rosich L, et al. The γ‐secretase inhibitor PF‐03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1‐mutated CLL cells. Leukemia 2015;29:96–106. [DOI] [PubMed] [Google Scholar]
- 34. Roti G, Qi J, Kitara S, et al. Leukemia‐specific delivery of mutant NOTCH1 targeted therapy. J Exp Med 2018;215:197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aste‐Amézaga M, Zhang N, Lineberger JE, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One 2010;5:e9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Kalken CK, van der Hoeven JJ, de Jong J, et al. Bepridil in combination with anthracyclines to reverse anthracycline resistance in cancer patients. Eur J Cancer 1991;27:739–44. [DOI] [PubMed] [Google Scholar]
- 37. Pozzo F, Bittolo T, Arruga F, et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation‐driven epigenetic dysregulation. Leukemia 2016;30:182–9. [DOI] [PubMed] [Google Scholar]
- 38. Nwabo Kamdje AH, Bassi G, Pacelli L, et al. Role of stromal cell‐mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J 2012;2:e73. doi: 10.1038/bcj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aster JC, Simms WB, Zavala‐Ruiz Z, et al. The folding and structural integrity of the first LIN‐12 module of human Notch1 are calcium‐dependent. Biochemistry 1999;38:4736–42. [DOI] [PubMed] [Google Scholar]
- 40. Rand MD, Grimm LM, Artavanis‐Tsakonas S, et al. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol 2000;20:1825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen LW, Jan CR. Effect of the antianginal drug bepridil on intracellular Ca2+ release and extracellular Ca2+ influx in human neutrophils. Int Immunopharmacol 2001;1:945–53. [DOI] [PubMed] [Google Scholar]
- 42. Hollingshead LM, Faulds D, Fitton A. Bepridil. A review of its pharmacological properties and therapeutic use in stable angina pectoris. Drugs 1992;44:835–57. [DOI] [PubMed] [Google Scholar]
- 43. Nakazato Y. The resurfacing of bepridil hydrochloride on the world stage as an antiarrhythmic drug for atrial fibrillation. J Arrhythmia 2009;25:4–9. [Google Scholar]
- 44. Linn SC, van Kalken CK, van Tellingen O, et al. Clinical and pharmacologic study of multidrug resistance reversal with vinblastine and bepridil. JCO 1994;12:812–9. [DOI] [PubMed] [Google Scholar]
- 45. De Ford C, Heidersdorf B, Haun F, et al. The clerodane diterpene casearin J induces apoptosis of T‐ALL cells through SERCA inhibition, oxidative stress, and interference with Notch1 signaling. Cell Death Dis 2016;7:e2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu Y, Cain‐Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010;464:1052–7. [DOI] [PubMed] [Google Scholar]
- 47. Chames P, Van Regenmortel M, Weiss E, et al. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 2009;157:220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Byrd JC, Gribben JG, Peterson BL, et al. Select high‐risk genetic features predict earlier progression following chemoimmunotherapy with fludara‐bine and rituximab in chronic lymphocytic leukemia: justification for risk‐adapted therapy. JCO 2006;24:437–43. [DOI] [PubMed] [Google Scholar]
- 49. Montillo M, Hamblin T, Hallek M, et al. Chronic lymphocytic leukemia: novel prognostic factors and their relevance for risk‐adapted therapeutic strategies. Haematologica 2005;90:391–9. [PubMed] [Google Scholar]
- 50. Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI‐32765. Blood 2011;117:6287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3‐kinase‐ä inhibitor CAL‐101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010;116:2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liotta F, Angeli R, Cosmi L, et al. TLR3 and TLR4 are expressed by human bone marrowderived mesenchymal stem cells and can inhibit their T‐cell modulatory activity by impairing Notch signalling. Stem Cells 2008;26:279–89. [DOI] [PubMed] [Google Scholar]
- 53. Hajdu M, Sebestyén A, Barna G, et al. Activity of the notch‐signalling pathway in circulating human chronic lymphocytic leukaemia cells. Scand J Immunol 2007;65:271–5. [DOI] [PubMed] [Google Scholar]
- 54. Garber K. Notch emerges as new cancer drug target. J Natl Cancer Inst 2007;99:1284–5. [DOI] [PubMed] [Google Scholar]
- 55. Zecchini V, Domaschenz R, Winton D, et al. Notch signaling regulates the differentiation of postmitotic intestinal epithelial cells. Genes Dev 2005;19:1686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Milano J, McKay J, Dagenais C, et al. Modulation of notch processing by γ‐secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci 2004;82:341–58. [DOI] [PubMed] [Google Scholar]
- 57. Tosello V, Ferrando AA. The NOTCH signaling pathway: role in the pathogenesis of T‐cell acute lymphoblastic leukemia and implication for therapy. Ther Adv Hematol 2013;4:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Figure 8
Supporting Information Tbale 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information