

Abstract
Liver fibrosis is a reversible wound‐healing response that occurs after liver injury. NADPH oxidases (NOXs) and reactive oxygen species (ROS) which are expressed in hepatocytes (HCs), hepatic stellate cells (HSCs), and Kupffer cells (KCs) play an important role in the development of hepatic fibrosis. In in vitro studies, we had shown that ursolic acid (UA) could reverse liver fibrosis by inhibiting the activation of NOX‐mediated fibrotic signaling networks in HSCs. In this study, we verified that UA could alleviate CCl4‐induced liver fibrosis by reducing the expression of NOXs/ROS in HCs, HSCs, KCs. Meanwhile, the phagocytic index α and clearance index K which represent phagocytosis of KCs were unchanged. Together, all our data demonstrated that UA induced the proliferation of HCs, promoted apoptosis in HSCs, and prevented activation of KCs in vivo by reducing the expression of NOXs/ROS in HCs, HSCs, KCs. Besides, UA had no effect on the host defense function.
Keywords: hepatic stellate cells, hepatocytes, Kupffer cells, ursolic acid
Abbreviations
- ALB
albumin
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AP
apocynin
- Bax
BCL2‐Associated X protein
- Bcl‐2
B‐cell lymphoma‐2
- CCl4
carbon tetrachloride
- CRP
C‐reactionprotein
- Gadd45
growth arrest and DNA damage inducible protein 45
- HCs
hepatocytes
- HSCs
hepatic stellate cells
- KCs
Kupffer cells
- MMP‐1
matrix metalloproteinase‐1
- NOXs
NADPH oxidase
- PCT
procalcitonin
- ROS
reactive oxygen species
- RT‐qPCR
reverse transcription‐quantitative polymerase chain reaction
- TBIL
total bilirubin
- TGF‐β1
transforming growth factor‐β1
- TIMP‐1
tissue inhibitor of metalloproteinase‐1
- TUNEL
TdT‐mediated dUTP nick‐end labeling
- UA
ursolic acid
- WB
Western‐blotting
- XIAP
X‐linked inhibitor of apoptosis protein
- α‐SMA
smooth muscle α‐actin
1. INTRODUCTION
Liver fibrosis is a common consequence of chronic liver injury and is caused by a variety of pathogenic processes, including viral infection, alcohol consumption, nonalcoholic fatty liver disease, cholestasis, autoimmune liver disease, and schistosomiasis japonica. Liver fibrosis leads to the accumulation of extracellular matrix or the formation of a fibrous scar (Thompson, Conroy, & Henderson, 2015; Zhang & Friedman, 2012). The transdifferentiation of hepatic stellate cells (HSCs) from a quiescent vitamin A storage cell to a proliferative myofibroblast is recognized as the central event in the development of hepatic fibrosis (Thompson et al., 2015). Hepatocytes (HCs) and Kupffer cells (KCs) are involved in the regulation of HSC activation through the release of TGF‐β1 (Liang, Kisseleva, & Brenner, 2016; Paik et al., 2014). Therefore, these cells appear to be specific cellular targets for antifibrotic therapy in chronic liver diseases of any etiology.
Recent studies have shown that NADPH oxidases (NOXs) and reactive oxygen species (ROS) play an important role in the development of hepatic fibrosis. Of the seven NOX family isoforms, the expression of NOX1, NOX2, and NOX4 has been widely studied in liver fibrosis. These three NOXs are expressed in HSCs, HCs and KCs, and they play an important role in the pathogenesis of hepatic fibrosis. Studies on the expression of NOXs in those cells and explorations of the drugs that block NOX activation in those cells are expected to achieve the goal of treating hepatic fibrosis (Altenhofer, Radermacher, Kleikers, Wingler, & Schmidt, 2015; Crosas‐Molist & Fabregat, 2015).
Ursolic acid (UA), a natural pentacyclic triterpenoid that is derived from berries, leaves, flowers, and fruits of medicinal plants, has been reported to have anti‐oxidation, anti‐inflammatory, anti‐ulcer, antibacterial, anti‐virus, anti‐tumor, anti‐obesity, hypoglycemic, antihypertensive, lipid‐lowering, liver protection, and immune regulation activities, among other (Cargnin & Gnoatto, 2017; Kim, Kim, Han, & Kim, 2015; Li, Ren, Luo, & Yang, 2016; Ma, Ding, Zhang, & Liu, 2015; Wang et al., 2011). In vitro studies have shown that UA could reverse liver fibrosis by inhibiting the activation of NOX‐mediated fibrotic signaling networks in HSCs, including the NF‐kB, PI3 K/Akt, P38MAPK, ERK1/2, JAK2‐STAT3, Hedgehog, and other signaling pathways (He et al., 2015). However, no one has reported whether the mechanisms of UA anti‐liver fibrosis are related to a reduction in the expression of NOXs in HSCs, HCs, and KCs in vivo. In this study, therefore, we investigated the effects of UA on the activation of NOXs in HSCs, HCs, and KCs in SD rats.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
UA and apocynin were purchased from Sigma Chemical Co. (St. Louis, MO), and a TUNEL assay kit was purchased from Promega Chemical Co. (Madison, WI). CCl4 and olive oil were from Shan‐dong Xiya Reagent Bioengineering Institute (Shandong, China). Hydroxyproline, malondialdehyde and glycogen staining assay kit were from Nan‐jing Jiancheng Bioengineering Institute (Nanjing, China). Sirius red staining assay kit was from Beijing Solarbio Bioengineering Institute (Beijing, China). Lipopolysaccharides (LPS) ELISA kit wsa from Wuhan Elabscience Biotechnology Co. (Hubei, China). DCFH‐DA cells ROS assay kit was from Beyotime Biotechnology (Shanghai, China). Anti‐MMP1 antibody, anti‐TIMP1 antibody, anti‐TGFβ1 antibody, and anti‐F4/80 antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti‐caspase3 antibody was from Cell Signaling Technology, Inc. (Boston, MA). All the other antibodies were obtained from Sigma Chemical Co. (St. Louis, MO).
2.2. Animals and experimental design
All SD rats (160–200 g) were obtained from the department of animal science, Medical College of Nanchang University. All animals received humane care, and the experimental protocol was approved by the Committee of Laboratory Animals and was performed according to institutional guidelines. All animals were kept in an environmentally controlled room (23 ± 2 °C, 55 ± 10% humidity) with a 12 hr light/dark cycle, and they were allowed free access to food and water.
After a 1‐week housing period, seventy‐two SD rats were randomly divided into six groups: blank control group, liver fibrosis model group, apocynin prevention group, apocynin treatment group, UA prevention group, or UA treatment group. For the liver fibrosis model group, we gave rats an intragastric administration of carbon tetrachloride (CCl4) for 8 weeks at 2 ml/kg (20% olive oil dilution concentration) twice a week. For rats in the prevention groups, 2 weeks after gavaging with CCl4, we gavaged the rats with apocynin or UA (40 mg/kg/day) for the last 6 weeks at the same time. For rats in the treatment groups, we gavaged the rats with apocynin or UA for another four weeks alone, 8 weeks after gavaging with CCl4.
At the end of treatment, eight rats in each group were used for the biochemical analysis and histological evaluations. Rats were sacrificed, and blood samples of approximately 5 ml were drawn through a postcava puncture with vacuum blood collection tubes. The plasma was collected after centrifugation at 5,000 rpm for 10 min and stored in a −70 °C freezer for further analysis. The liver tissues were rinsed in ice‐cold 0.9% NaCl solution and stored in a −70 °C freezer for later use.
Four rats in each group were used for extracting primary HSCs, HCs and KCs, in situ perfusion and gradient centrifugation. The rats were anesthetized by an intraperitoneal injection of 10% chloral hydrate. We perfused phosphate buffered saline (PBS) through the inferior vena cava, and cut open the portal vein at the same time. According to various densities in those three kinds of cells, we then separated HCs, HSCs, and KCs using Collagenase IV (Gibco, Grand Island, NY, UA), Pronase E (Sigma Immunochemicals, St. Louis, MO, UA) and Precoll (Sigma) successively. HCs and HSCs were cultured in Dulbecco's modified essential medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) in humidified air containing 5% CO2 at 37 °C; however, KCs were cultured in 1640 medium (Gibco) supplemented with 10% FBS (Gibco) in humidified air containing 5% CO2 at 37 °C.
2.3. Blood and liver tests
The alanine aminotransferase (ALT), total bilirubin (TBIL), albumin (ALB), white blood cell (WBC), C‐reactive protein (CRP), and procalcitonin (PCT) contents of serum were estimated using an automatic biochemical analyzer (Department of clinical laboratory, the First Affiliated Hospital of Nanchang University, China). The malondialdehyde (MDA) and hydroxyproline (HYP) contents of serum or liver tissue were tested spectrophotometrically using commercial diagnostic kits.
2.4. Histological evaluations
Liver samples fixed in 10% buffered formalin were embedded in paraffin, cut into 5 µm sections, stained with hematoxylin and eosin (H&E), Sirius red, TdT‐mediated dUTP Nick‐End Labeling (TUNEL, to detect the apoptosis of HCs in liver tissue), or for immunohistochemistry (IHC, to detect proliferating cell nuclear antigen, PCNA) analysis before being observed and photographed using light microscopy.
For the semi‐quantitative of TUNEL and PCNA expression, we used a scoring method as follows. The percentage of positive HCs in liver tissue was determined in at least five areas at ×400 magnification and assigned to one of the following categories: 0 = <5%; 1 = 6–49%; 2 = 50–75%; 3 > 75% (Table A below). The staining intensity of the nucleus were scored as 0 point (uncolored), 1 point (light yellow), 2 point (brown), 3 point (tan) (Table B below). Finally, according to the products of the positive cells percentage and the staining intensity score, the results were decided.
| A The percentage of positive cells | |||
| 0–5% | 6–49% | 50–75% | >75.0% |
| 0 | 1 | 2 | 3 |
| B The staining intensity score | |||
| Uncolored | Light yellow | Brown | Tan |
| 0 | 1 | 2 | 3 |
2.5. Western blot analysis
Liver tissue protein was obtained using protein lysate (BestBio, Shanghai, China) for Western blotting. Cells were harvested and re‐suspended in PBS. Cell protein extracts were obtained by centrifuging at 12,000 rpm for 10 min. Protein levels were determined using the BCA assay kit (Tiangen, Beijing, China). Protein extracts (10–20 ml) were separated using 6–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and electrophoretically transferred onto polyvinylidene fluoride membranes. After blocking with 5% nonfat dry milk in Tris‐buffered saline, PVDF membranes were incubated overnight at 4 °C with rabbit anti‐NOX1 (1:1000), anti‐NOX2 (1:1000), anti‐NOX4 (1:1000), anti‐TGFβ1 (1:2000), anti‐MMP1 (1:2000), anti‐TIMP1 (1:2000), anti‐p67phox (1:500), anti‐p22phox (1:500), anti‐p47phox (1:500), anti‐caspase‐3 (1:1000), anti‐caspase‐9 (1:1000), anti‐GADD45 (1:1000), anti‐Bcl2 (1:800), anti‐Bax(1:1000), anti‐CD95L (1:200), anti‐CD95 (1:3000), anti‐XIAP (1:2000), or mouse anti‐α‐SMA (1:200), anti‐rac1(1:750), anti‐ collagenase I (1:1000). Next, a secondary horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse IgG antibody (Zhongshan Golden Bridge Biotechnology Co., Beijing, China) was applied, and specific bands were visualized using an ECL detection kit (Thermo Fisher Scientific Inc).
2.6. RNA preparation
Total liver RNA was prepared from frozen tissue samples or cells using a RNA simple Total RNA Kit and a Fast Quant RT Kit (Tiangen, Beijing, China). The concentration and purity of isolated RNA were determined by measuring optical density at 260 and 280 nm. The integrity of the RNA was verified using agarose gel electrophoresis (AGE).
2.7. RT‐qPCR
For reverse transcription‐polymerase chain reaction (RT‐PCR), we used the experimental procedure from the Fast Quant RT Kit and the Super Real Pre‐Mix Plus (SYBR Green, tiangen, Beijing, China). The primer sets were as follows (Table 1). All primers were obtained from Beijing Genomics Institute (BGI, Shenzhen, China).
Table 1.
Primer sequences for RT‐qPCR
| PCR gene name | Forward primer (5′–3′) | Reverse primer (5′–3′) | Size (bp) |
|---|---|---|---|
| Rat β‐actin | CACGATGGAGGGGCCGGACTCATC | TAAAGACCTCTATGCCAACACAGT | 240 |
| Rat NOX1 | TCCTAAACTACCGACTCTTC | GTCCCACATTGGTCTCCC | 134 |
| Rat NOX2 | ACCATTCGGAGGTCTTAC | CTGGGCACTCCTTTATTT | 190 |
| Rat NOX4 | AGGTGTCTGCATGGTGGTG | GAGGGTGAGTGTCTAAATTGGT | 182 |
| Rat p47phox | TCCTAAACTACCGACTCTTC | GTCCCACATTGGTCTCCC | 107 |
| Rat p67phox | ACCATTCGGAGGTCTTAC | CTGGGCACTCCTTTATTT | 171 |
| Rat rac1 | AGGTGTCTGCATGGTGGTG | GAGGGTGAGTGTCTAAATTGGT | 120 |
| Rat p22phox | TGTTGCAGGAGTGCTCATCT | CACCACAGCGGTCAGGTAC | 107 |
| Rat TGFβ1 | CCATGACATGAACCGACCCT | CCGGGTTGTGTTGGTTGTAG | 296 |
| Rat MMP1 | GCTGATACTGACACTGGTACTG | CAATCTTTTCTGGGAGCTC | 216 |
| Rat TIMP1 | CCACAGATATCCGGTTCGGCTACA | GCACACCCCACAGCCAGCACTAT | 214 |
| Rat collagen α1 | GGGGCAAGACAGTCATCGAA | GGATGGAGGGAGTTTACACGAA | 144 |
2.8. Extraction of primary HSCs, HCs, and KCs
2.8.1. Liver perfusion
Four rats were taken from each group. Rats were anesthetized by intraperitoneal injection of 10% chloral hydrate. The abdominal cavity was cut open aseptically and the inferior vena cava was punctured. The PBS solution was uniformly perfused by the syringe, at the same time, the hepatic portal vein was cut open till the liver tissue turned into earthy yellow. Injected into the hepatic portal vein with perfusion solution (0.05% Collagenase IV) 50 ml at 37 ° C, and digested 10 min.
2.8.2. Extraction of primary HCs
The liver tissue was completely cut off into in a plate.
Lift the liver with ophthalmic forceps, and remove the capsule, blood vessels, connective tissue.
Tear the liver tissue and obtaine suspension of liver cells. The suspension was filtered through a 150 mesh stainless steel mesh, and the filtrate was collected.
Centrifuge at 50g for 5 min, and the cell was collected.
The cells were resuspended in PBS, and centrifuged again (50g, 5 min).
The cells were suspended in DMEM conditioned medium and cultured overnight in a humidified air containing 5% CO2 at 37 °C.
2.8.3. Extraction of primary HSCs and KCs
The remaining liver tissue in Step 2 was cut into pieces and added with four times volume digestion solution (0.2% Collagenase IV + 0.2% Pronase E) for 30 min with water bath shaking at 37 °C.
After digestion, the tissue was mechanical separated softly until there was no obvious tissue mass.
Centrifuge at 50g for 3 min, and the supernatant was collected.
The supernatant was centrifuged at 500g for 5 min, and the sediment was collected.
The sediment was resuspended in PBS. We slowly added to the upper layer of well‐prepared separating medium with 70% and 30% Precoll non‐continuous gradient.
Centrifuge at 900g for 15 min. All layers of cells were aspirated slowly. The junction of 70% and 30% were KCs and liver sinusoidal endothelial cells, and upper 30% layer were HSCs.
For HSCs, resuspended in DMEM and centrifuge at 500g for 5 min, The sediment was resuspended in DMEM again and cultured overnight in a humidified air containing 5% CO2 at 37 °C.
KCs were resuspended in 1,640 complete medium and cultured into Petri dishes. Depending on the different speed of the adherent wall, we extracted the primary HSCs, and KCs. After cultured with 30 min, we discarded unadherent cells, replaced the fresh medium, and the adherent were KCs. KCs were cultured in a humidified air containing 5% CO2 at 37 °C.
2.9. Identification of primary HSCs, HCs, and KCs
2.9.1. Identification of primary HSCs (α‐SMA immunofluorescence identification) and KCs (F4/80 immunofluorescence identification)
The slides in the culture plate which had climbed with cells were immersed in PBS for three times, 3 min at a time.
Fix the slides with 4% paraformaldehyde for 15 min, and then wash the slides with PBS for three times, 3 min at a time.
Permeate the slides with 0.5% Triton X–100 (prepared with PBS) at room temperature for 20 min.
The slides were immersed in PBS for three times, 3 min at a time. Blot PBS with absorbent paper, and drop normal goat serum on the slides, blocking at room temperature for 30 min.
Blot serum with absorbent paper. Each slide was dropped with primary antibody dilution (1:100) and placed in wet box, incubating overnight at 4 °C.
Add fluorescent secondary antibody: PBST washed the slides three times, 3 min at a time. Blot PBST with absorbent paper, and add fluorescent secondary antibody dilution to the slides (1:200), incubating in wet box at 37 °C for 1 hr. PBST washed the slides three times, 3 min at a time. (from the step of adding fluorescent secondary antibody, the remained steps were operated as far as possible in the darkroom).
Counterstain nuclear: DAPI was added to the slides. After incubation for 5 min, PBST washed the slides four times, 5 min at a time.
Blot the liquid on the slides with absorbent paper, and then seal the slide with the antifluorescent quencher. Capture image under a fluorescence microscope.
2.9.2. Identification of primary HCs (by schiff periodic acid shiff, PAS)
The slides in the culture plate which had climbed with cells were immersed in PBS for three times, 3 min at a time.
Fix the slides with 4% paraformaldehyde for 15 min, and then wash the slides with PBS for three times, 3 min at a time.
Soak the slides with 1% periodic acid aqueous solution for 10 min. Wash the slides with PBS for 2 min.
Add Schiff's reagent on the slides, incubating the slides in the dark at room temperature for 30 min. Dip the slides with PBS for 10 min.
Counterstain with Mayer hematoxylin for 2 min, and wash the slides with water until the nucleus turned blue.
Dehydration with anhydrous ethanol, and transparention with dimethylbenzene.
Seal the slides with neutral balsam after dryness. Capture image under a microscope.
2.10. Determination of ROS
2.10.1. For ROS in HCs or HSCs
-
1.
After cell separation, HCs or HSCs were made into a single cell suspension, and the cells were uniformly inoculated into 6‐well plates at approximately 4 × 105 cells per well. The cells were cultured overnight in a humidified air containing 5% CO2 at 37 °C;
-
2.
Digest cells with 0.25% trypsin without EDTA. After termination of digestion, centrifuge at 1,500 rpm for 5 min. Supernatant was discarded and sediments were resuspended in PBS, and the above steps were repeated three times;
-
3.Detection using DCFH‐DA cells ROS assay kit:
-
a.Add 1 ml of PBS with diluted DCFH;
-
b.Incubate cells at 37 °C for 20 min. Mix every 3 min;
-
c.Wash the cells three times with serum‐free medium;
-
d.Add 500 ul of PBS resuspended cells;
-
a.
-
4.
Detection with flow cytometer.
2.10.2. For ROS in KCs
After cell separation, KCs were made into a single cell suspension, and the cells were uniformly inoculated into 6‐well plates at approximately 4 × 105 cells per well. The cells were cultured overnight in humidified air containing 5% CO2 at 37 °C;
Prepare 10−3 mmol/ml DCFH‐DA working solution by adding 10−2 mmol/ml DCFH‐DA: PBS = 1: 9;
Digest cells with 0.25% trypsin without EDTA. After termination of digestion, use part of the supernatant for protein determination. Add the protein concentration that calculated from the supernatant into the DCFH‐DA working solution, and mix. Set PBS for the control wells. Incubate solution at 37 °C for 30 min;
Detection of fluorescence intensity: the excitation wavelength was 485 nm and the emission wavelength was 525 nm. The results were expressed as fluorescence intensity/mg protein. ROS (fluorescence intensity/mg protein) = fluorescence intensity/(protein concentration × 0.19).
2.11. Test host defense function of KCs
2.11.1. Carbon particle clearance test detected the phagocytic function of KCs
Rats in each group were injected with ink (Carbon suspension, 10 ml/kg) in the tail vein 1 hr before the rats were sacrificed.
Then, 20 µl blood was collected from the eyeball of rats at 1, 5, 10 min, respectively. The blood was dissolved in 2 ml 0.1% of Na2CO3 solution.
The absorbance (A value) was measured at 650 nm with a spectrophotometer.
Weigh the wet weight of liver and spleen, calculate clearance index K and phagocytic index α based the formula:
2.12. Detection of bacterial infection
Peripheral blood (from eyeball) was taken weekly after drug treatment, and white blood cell (WBC), C‐reactive protein (CRP), and procalcitonin (PCT) in serum were estimated. Lipopolysaccharides (LPS) was tested by ELISA kit.
2.13. Statistical analysis
All statistical analyses were performed using the SPSS software, version 17.0. Data were expressed as the means ± SEM. Statistical differences between means were determined using analysis of variance (ANOVA), Student's t test, Mann‐Whitney rank sum test, or Kruskal—Wallis H test. A value of p < 0.05 was selected prior to the experiments to indicate statistical significance.
3. RESULTS
3.1. UA alleviated CCl4‐induced liver fibrosis
First, the effect of UA on the regression of CCl4‐induced liver injury was observed. As shown in Figures 1a and 1b, CCl4 treatment caused inflammatory cellular infiltration, hepatocellular necrosis, the formation of fibrous septum, deposition of collagen fibers, and formation of a pseudo‐lobe. After UA or AP intervention, the listed visible liver histology injuries were alleviated. Disorder in the hepatic lobular structure, fibrous space and collagen deposition in liver tissue were also significantly lower after prevention or treatment compared to the CCl4 group. Indeed, quantitative analysis demonstrated a significant reduction in the fibrotic score and area (Figures 1c and 1d). The hydroxyproline contents of rat serum and hepatic tissue declined compared to the CCl4 group (Figures 1e and 1f). Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). Those results showed that UA could alleviate CCl4‐induced liver fibrosis.
Figure 1.
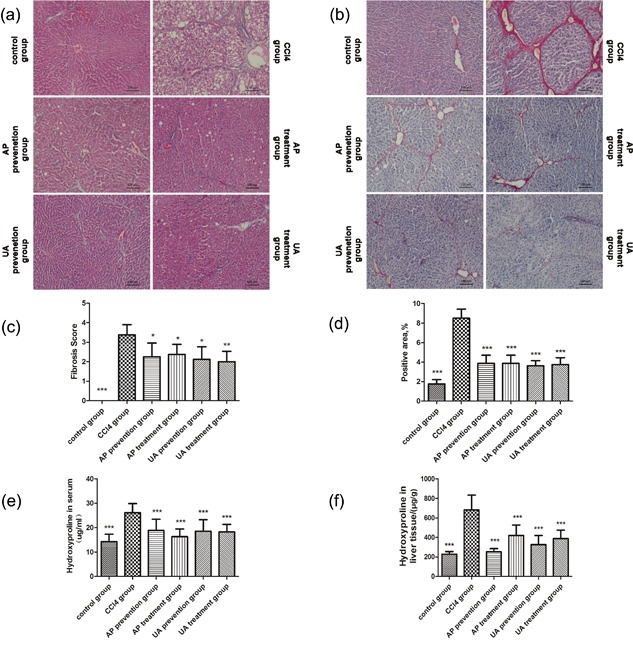
Effect of UA on regression of CCl4‐induced liver injury. (a) hematoxylin‐eosin (HE) staining. (b) Sirius red staining. Morphometrical analysis for fibrotic score (c) and area (d) were performed for five random fields in each preparation, and the average percentages of fibrotic area are plotted. Detection of hydroxyproline content in serum (e) and liver tissue (f) by colorimetry. All values are expressed as the means ± S.E.M. (n = 8). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001. Original magnification, ×100
3.2. The effect of UA intervention on liver function and oxidative stress
We measured the alanine aminotransferase (ALT), total bilirubin (TBIL), albumin(ALB), and malondialdehyde (MDA) contents of rats serum or liver tissue. CCl4 treatment resulted in high expression of ALT (Figure 2a), TBIL (Figure 2b), MDA (Figures 2d and 2f), and low expression of ALB (Figure 2c). After UA or AP intervention, the expression of the listed targets was reduced. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). We concluded that UA could alleviate CCl4‐induced hepatic dysfunction and oxidative stress.
Figure 2.
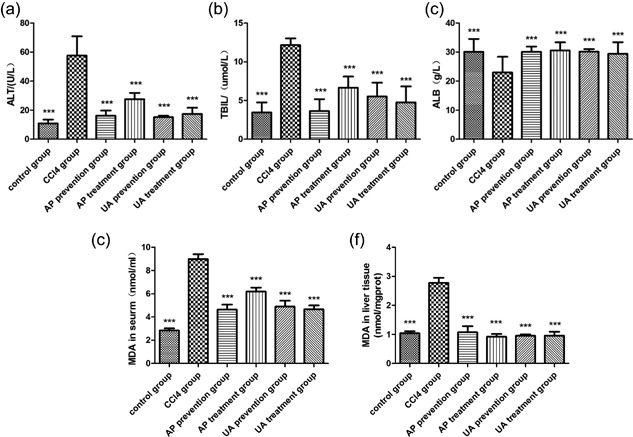
Effect of UA intervention on liver function and oxidative stress. (a) Automatic biochemical analyzer estimated ALT in rats’ serum. (b) TBIL in rats serum. (c) ALB in rats’ serum. MDA commercial diagnostic kits were used to estimate MDA in rats serum (d) and liver tissue (e). All values are expressed as the means ± S.E.M. (n = 8). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001
3.3. The effect of UA intervention on proliferation and apoptosis in HCs
Subsequently, the effect of UA intervention on proliferation and apoptosis of HCs was evaluated. We used immunohistochemistry (IHC) to detect proliferating cell nuclear antigen (PCNA) in liver tissue (Figure 3a). We utilized TdT‐mediated dUTP Nick‐End Labeling (TUNEL) staining to detect apoptosis in HCs (Figure 3b). CCl4 treatment led to increased apoptosis in HCs, and it led to decreased proliferation of HCs; After UA or AP intervention, the proliferation of HCs was increased, and apoptosis in HCs was decreased. The PCNA positive score and TUNEL positive score are shown in a table (Figure 3c). Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). These results showed that UA could reduce apoptosis in HCs, but increased the proliferation of HCs.
Figure 3.
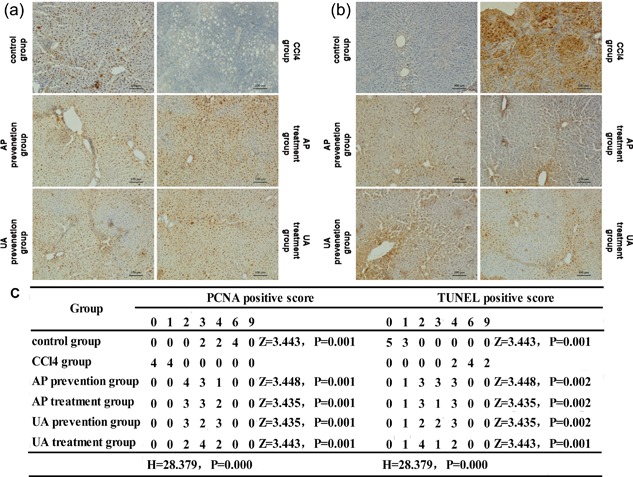
Effect of UA intervention on proliferation and apoptosis of HCs. (a) Immunohistochemistry (IHC) to detect proliferating cell nuclear antigen (PCNA) of HCs in liver tissue. (b) TdT‐mediated dUTP Nick‐End Labeling (TUNEL) staining to detect the apoptosis of HCs in liver tissue. (c) PCNA positive score and TUNEL positive score. Original magnification, ×100. If the data were class grouping data, Kruskal Wallis H test was used to compare between groups, and Mann‐Whitney U test was used to compare the groups
3.4. The effect of UA intervention on genes and proteins related to fibrosis
The effect of UA on the expression of type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 genes and proteins in liver tissue was tested. As shown in Figure 4a, CCl4 treatment induced high expression of type I collagen, TGF‐β1, and TIMP‐1 genes, but it induced low expression of the MMP‐1 gene. After UA or AP intervention, the expression of type I collagen, TGF‐β1, and TIMP‐1 genes were reduced, and the expression of the MMP‐1 gene was increased. This was the same as the effect of UA on type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 proteins. As shown in Figures 4b and 4c, CCl4 treatment induced high expression of type I collagen, TGF‐β1, and TIMP‐1 proteins, but a low expression of the MMP‐1 protein. After UA or AP intervention, the expression of type I collagen, TGF‐β1, and TIMP‐1 proteins were reduced, and the expression of the MMP‐1 protein was increased. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). These results demonstrated that UA could improve the expression of CCl4‐induced fibrosis‐related genes and proteins.
Figure 4.
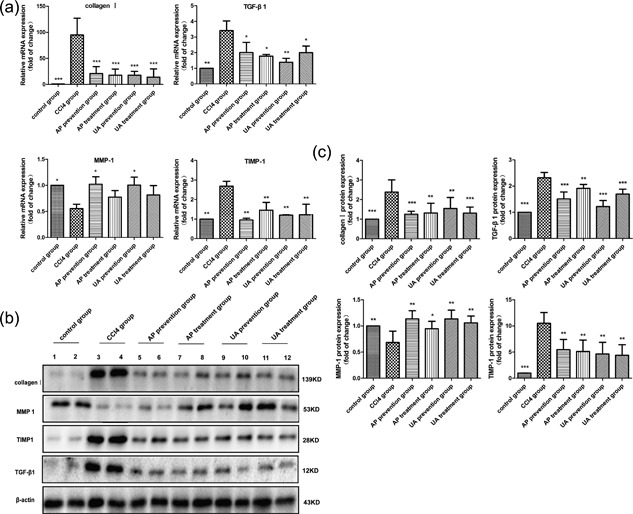
Effect of UA on the expression of type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 genes and proteins. (a) RT‐qPCR tested the expression of type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 genes. (b) WB tested the expression of type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 proteins. (c) Histogram analysis on the contents of type I collagen, TGF‐β1, MMP‐1, and TIMP‐1 proteins. All values are expressed as the means ± S.E.M. (n = 4). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001
3.5. The effect of UA intervention on NOX proteins in liver tissue
Recent studies have indicated that NADPH oxidases (NOXs) play an important role in the pathogenesis of liver fibrosis (Liang et al., 2016). We speculated that UA relieved liver fibrosis by reducing the expression of NOXs. The effect of UA on the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 proteins were detected in liver tissue. As shown in Figure 5a–h, CCl4 treatment induced a high expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 proteins in liver tissue. After UA or AP intervention, the listed proteins was alleviated. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). These results demonstrated that UA could ease CCl4‐induced liver fibrosis by reducing the expression of NOX proteins.
Figure 5.
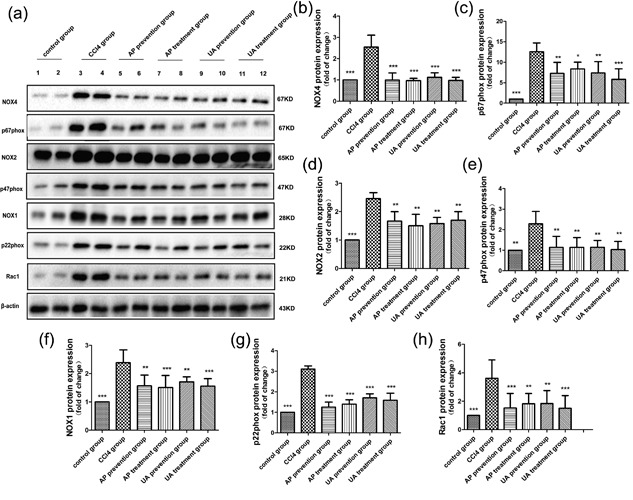
Effect of UA intervention on the expression of NOX proteins. (a) WB detected the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 proteins in liver tissue. Quantitative analysis of NOX4 (b), p67phox (c), NOX2 (d), p47phox (e), NOX1 (f), p22phox (g), and Rac1 (h) proteins in liver tissue. All values are expressed as the means ± S.E.M. (n = 4). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p <n0.001
3.6. The effect of UA intervention on the expression of NOX mRNAs in HSCs, HCs, and KCs
Research shows that NOXs expressed in hepatic stellate cells (HSCs), hepatic cells (HCs), and Kupffer cells (KCs) play a key role in liver fibrosis (Paik et al., 2014). We wanted to explore the effect of UA intervention on the expression of NOX mRNAs in HSCs, HCs, and KCs in vivo. We used in situ perfusion and gradient centrifugation to extract primary HSCs, HCs, and KCs from rats. We then utilized a glycogen staining assay kit to identify primary HCs, and we used Immunofluorescence (IF) to identify primary HSCs and KCs. As shown in Figure 6 (the first row), HCs contain abundant glycogen. After PAS dyeing, the cytoplasm became purple; α‐SMA, and DAPI dual‐positive cells were detected in primary HSCs (the second row), and F4/80 and DAPI dual‐positive cells were detected in primary KCs (the third row). Next, we inspected the expression of NOX mRNAs in HSCs, HCs, and KCs in vivo. As shown in Figure 6, CCl4 treatment resulted in high expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 genes in HCs (B), HSCs (C) and KCs (D). After UA or AP intervention, the expression of the listed genes declined to varying degrees. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). These results showed that UA could relieve CCl4‐induced liver fibrosis by reducing the expression of NOX mRNAs in HSCs, HCs, and KCs.
Figure 6.
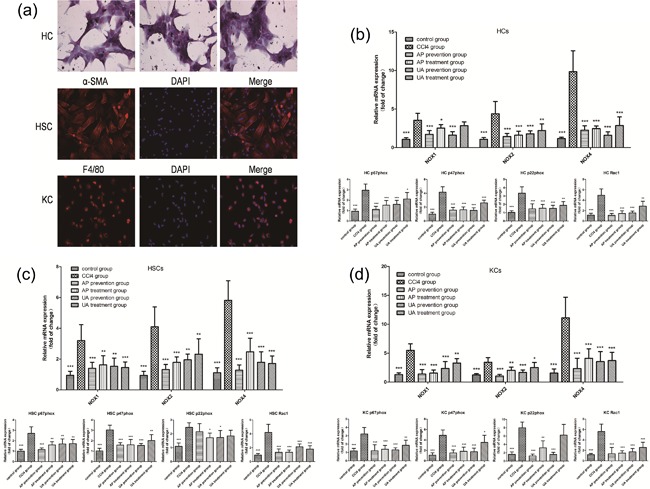
Effect of UA intervention on the expression of NOX mRNAs in HSCs, HCs, and KCs. (a) Isolation of primary HSCs, HCs, and KCs. First row: glycogen staining assay kit to identify primary HCs. Second row: IF to identify primary HSCs. Representative serial images for α‐SMA (red, left panel), DAPI (blue, middle panel), and merged image (right panel). Third row: IF to identify primary KCs. Representative serial images for F4/80 (red, left panel), DAPI (blue, middle panel), and merged image (right panel). RT‐qPCR to check the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 genes in HCs (b), HSCs (c), and KCs (d). All values are expressed as the means ± S.E.M. (n = 4). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001. Original magnification, ×200
3.7. The effect of UA intervention on ROS expressed in HCs, HSCs, and KCs
NOXs produce and . This occurs because NOXs deliver electrons to O2 molecules, and generate reactive oxygen species (ROS). The agglutination of intracellular ROS can lead to cell structure damage, oxidative stress, and inflammatory injury (Nisimoto, Diebold, Cosentino‐Gomes, & Lambeth, 2014; Weiskirchen, 2015). We observed the effect of UA intervention on ROS in HCs, HSCs, and KCs. As shown in Figure 7a, after HCs or HSCs were mixed with DCF‐DA working solution, the expression of ROS in primary cells was observed using confocal fluorescence microscopy. We next used flow cytometry to detect the ROS content. Histograms showed that CCl4 treatment caused high expression of ROS in HCs (Figure 7b), HSCs (Figure 7c), and KCs (Figure 7d). However, the expression of ROS in those three cell types partly declined after UA or AP intervention. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). Those results showed that UA could lighten CCl4‐induced liver fibrosis by reducing the expression of ROS in HSCs, HCs, and KCs.
Figure 7.
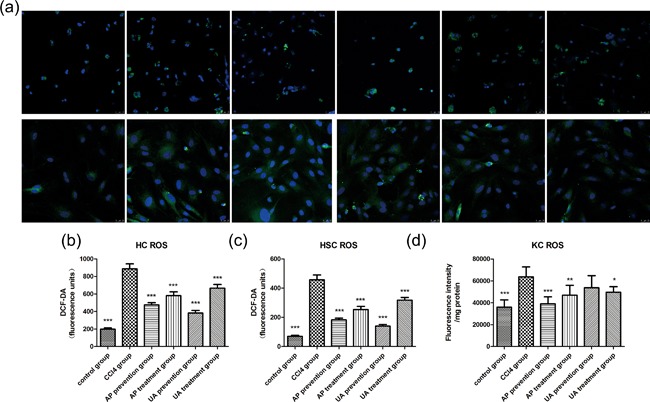
Effect of UA intervention on ROS expressed in HCs, HSCs, and KCs. (a) Confocal fluorescence microscopy observed ROS in primary HCs (upper row) or HSCs (down row) that were loaded with DCF‐DA working solution. Flow cytometry combined quantitative analysis tested ROS in HCs (b), HSCs (c), and KCs (d). All values are expressed as the means ± S.E.M. (n = 4). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001. Original magnification, ×200
3.8. The effect of UA intervention on anti‐ or pro‐apoptosis protein expression on HCs and HSCs
The apoptosis of HCs and the proliferation of HSCs play a key role in liver fibrosis. We tested the effect of UA on the expression of anti‐apoptotic proteins, including Bcl2 and XIAP, and pro‐apoptotic proteins, like Gadd45β, Bax, CD95L, CD95, Caspase3 and Caspase9 in HCs and HSCs. As shown in Figure 8a, CCl4 treatment led to a reduction in anti‐apoptotic proteins like Bcl2 and XIAP, but it led to an increase in pro‐apoptotic proteins like Gadd45β, Bax, CD95L, CD95, Caspase3, and Caspase9 in HCs. After UA or AP intervention, the listed effects were reversed, with anti‐apoptotic proteins up‐regulated and pro‐apoptotic proteins down‐regulated. As shown in Figure 8b, CCl4 treatment led to increased anti‐apoptotic proteins like Bcl2 and XIAP, but decreased pro‐apoptotic proteins like Gadd45β, Bax, CD95L, CD95, Caspase3, and Caspase9 in HSCs. After UA or AP intervention, anti‐apoptotic proteins decreased, and pro‐apoptotic proteins increased. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). Those results showed that UA could alleviate CCl4‐induced liver fibrosis by reducing apoptosis in HCs, or increasing apoptosis in HSCs.
Figure 8.

WB tested the effect of UA intervention on anti‐ or pro‐apoptosis protein expression in HCs and HSCs. (a) anti‐apoptotic proteins (Bcl2, XIAP), and pro‐apoptotic proteins (Gadd45β, Bax, CD95L, CD95, Caspase3, Caspase9) in HCs. (b) anti‐apoptotic proteins and pro‐apoptotic proteins in HSCs. All values are expressed as the means ± S.E.M. (n = 4). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001
3.9. Safety research about UA to reverse liver fibrosis
We wanted to know the effect of UA on host defense function. We used the carbon particle clearance test to assess the effect of UA on phagocytosis in KCs. In contrast to rats in the CCl4 group, the phagocytic index α (Figure 9b) and clearance index K (Figure 9c), which represent phagocytosis in KCs, were unchanged in the UA or AP prevention group. However, phagocytosis of KCs in the UA or AP treatment group had an advantage over the CCl4 group. There was no significant change in WBC (Figure 9a), LPS (Figure 9d), CRP (Figure 9e) between the CCl4 group and the other groups. CCl4 treatment generated over‐expression of PCT (Figure 9f). After UA or AP intervention, PCT was reduced. Nevertheless, there was no significant difference between the UA treatment group and the AP treatment group (p > 0.05), nor was there a significant difference between the UA prevention group and AP prevention group (p > 0.05). These results showed that UA could not weaken phagocytosis of KCs, but could not impair host defense function or increase the chance of bacterial infection.
Figure 9.

Safety assessment of UA on regression of CCl4‐induced liver fibrosis. White blood cells (WBC, weekly, a), C‐reactive protein (CRP, e), and procalcitonin (PCT, f) in serum were estimated by automatic biochemical analyzer. Lipopolysaccharides (LPS) was tested by a ELISA kit (d). Phagocytosis of KCs was evaluated by inspecting phagocytic index α (b) and clearance index K (c). α = (body weight/[liver weight + spleen weight]) × 1/3 K. K = (logA1—logA2)/(t2—t1)]. All values are expressed as the means ± S.E.M. (n = 8). CCl4 group compared to all other groups, respectively; *p < 0.05, **p < 0.01, ***p < 0.001
4. DISCUSSION
Hepatic fibrosis is caused by various pathogeneses, and it lead to extracellular matrix deposition or scar formation. The sustainable development of liver fibrosis can eventually develop into liver cirrhosis, or even liver cancer, which can threaten human health (Thompson et al., 2015; Zhang & Friedman, 2012). Although, early antiviral therapy can reverse liver fibrosis in patients with HBV and HCV infection (D'Ambrosio et al., 2012; Marcellin et al., 2013), for other causes of liver fibrosis and cirrhosis, there are still no effective anti‐fibrosis drugs applied to clinical treatment (Sun & Kisseleva, 2015), Therefore, developing anti‐liver fibrosis drugs is highly significant (Lee, Wallace, & Friedman, 2015). We used fibrotic rats as research subjects, and apocynin (AP) (Rahman et al., 2017) as a positive control, and we observe the effect of UA intervention (UA prevention and treatment) on reversing liver fibrosis. Compared to the liver fibrosis group, the fibrous septum and collagen deposition were reduced in the liver tissue of the UA prevention and treatment group (Figure 1a–d). There was an obvisous reduction in hydroxyproline in rats serum and liver tissue (Figures 1e and 1d). The expression of type I collagen, TGF beta 1, TIMP‐1 mRNA, and protein in liver tissue was reduced; However, the expression of the MMP‐1 mRNA and protein was increased (Figure 4a–d). These results showed that UA could alleviate CCl4‐induced liver fibrosis.
NADPH oxidases (NOXs) are members of a multi‐subunit transmembrane enzyme complex that is composed of seven members: NOX1, NOX2, NOX3, NOX4, NOX5, and two double oxidase Duox1, Doux2. The activation of phagocytic NOX2 requires two membrane proteins, NOX2 (gp91phox), and p22phox; three subcellular proteins, p40phox, p47phox, and p67phox; and two guanosine binding. NOX1, NOXA1, p22phox, and Rac GTPase are needed for the activation of NOX1; and NOX4, p22phox, and Poldip2 are co‐assembled into activated NOX4. NOXs are the main sources of ROS, and a large number of ROS in the cells may mediate oxidative stress and promote the development of hepatic fibrosis (Liang et al., 2016). Paik et al. (2011) found that NOX1 or NOX2 gene knockout in mice decreased ROS production, and alleviated CCl4 or bile duct ligation‐induced liver fibrosis. Lan, Kisseleva, and Brenner (2015) showed that the NOX1/4 double inhibitor GKT137831, or the NOX1/4 gene knockout could reduce the formation of ROS and the expression of inflammation‐related genes and block liver fibrosis. Qiu et al. (2016) showed that AP, the non‐specific blocking agent of NOXs, could stop atrial remodeling, and prevent myocardial fibrosis, (Jiang et al., 2010) showed that inhibiting the activation of NOX2 could inhibit the activation of HSCs. In this study, the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 proteins in liver tissue were decreased after UA or AP intervention (Figure 5a–h) compared to the liver fibrosis group. Malondialdehyde (MDA) in rats serum or liver tissue was down‐regulated (Figures 2d and 2f), and the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, Rac1 genes (Figure 6b–d) and ROS (Figure 7a–d) in HCs, HSCs, KCs were also reduced. These results demonstrated that UA could alleviate CCl4‐induced liver fibrosis by reducing the expression of NOXs/ROS.
The transformation of HSCs into proliferative myofibroblasts (MFBs) is the central event of hepatic fibrosis. Activated HSCs lead to liver metabolism reprogramming, increased autophagy and serious damage to parenchymal cells. This results in loss of the retinoids and enhanced contractility in HSCs, which releases growth factors and inflammatory signal factors in the liver microenvironment, liberates large amounts of extracellular matrix (ECM), and lead to an imbalance of matrix metalloproteinases (MMPs) and matrix inhibitor of metalloproteinase (TIMPs) (Lee et al., 2015; Robert, Gicquel, Bodin, Lagente, & Boichot, 2016). HCs and KCs also play an important role in liver fibrosis. Damaged HCs or activated KCs can secrete TGF‐β1, one of the strongest pro‐fibrosis factors, and accelerate the activation of HSCs (Jiang et al., 2012; Sun et al., 2017). It was shown that NOX1, NOX2, NOX4 were expressed in HCs and HSCs, whereas only NOX2 was expressed in KCs. Decreasing the expression of NOXs in HCs, HSCs, and KCs could inhibit apoptosis in HCs, suppress the activation of KCs and HSCs, and block the development of liver fibrosis (Paik et al., 2011). In this study, the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, Rac1 genes (Figure 6b–d) in HCs, HSCs, KCs was also reduced compared to the liver fibrosis group. These data showed that UA could alleviate CCl4‐induced liver fibrosis by reducing the expression of NOXs/ROS. The ALT and TBIL contents decreased, and ALB content increased (Figure 2a–c). The proliferation of HCs increased, while the apoptosis of HCs decreased (Figure 3a–c). Anti‐apoptotic proteins such as Bcl2 and XIAP were up‐regulated, but pro‐apoptotic proteins such as Gadd45β, Bax, CD95L,CD95, Caspase3, Caspase9 were down‐regulated in HCs (Figure 8a). After UA or AP intervention, anti‐apoptotic proteins down‐regulated, but pro‐apoptotic proteins were up‐regulated in HSCs (Figure 8b). These data indicated that UA could reduce the expression of NOXs/ROS, increase proliferation of HCs, decrease apoptosis in HCs, increase apoptosis in activated HSCs, decrease the activation of HSCs, and alleviate CCl4‐induced liver fibrosis.
Present studies show that KCs have a dual role in the development and progression of hepatic fibrosis. On the one hand, early KCs promote liver fibrosis by recruiting proinflammatory cells and secreting proinflammatory factors, chemokines, ROS and so on. On the other hand, advanced KCs promote the degradation of fibrosis by producing MMPs (especially MMP9, MMP12, and MMP13). In addition, KCs can swallow cell debris to reduce inflammation and liver fibrosis. Therefore, inhibiting the activation of KCs or increasing the phagocytosis of KCs can relieve liver fibrosis (Possamai, Thursz, Wendon, & Antoniades, 2014; Tacke, 2017; Tacke & Zimmermann, 2014). In this study, the expression of NOX4, p67phox, NOX2, p47phox, NOX1, p22phox, and Rac1 genes (Figure 6d) in KCs was also reduced compared to the liver fibrosis group. This was accompanied by a reduction in activated KCs; The phagocytic index α and clearance index K which represent phagocytosis of KCs were unchanged in the UA or AP prevention group (Figures 9b and 9c); but phagocytosis of KCs in UA or AP treatment group had an advantage over the CCl4 group. These results indicated that UA could lighten liver fibrosis by reducing the activation of KCs, and partially improve phagocytosis of KCs.
In the end, we discussed the effects of UA on bacterial infection and systemic inflammation during anti‐hepatic fibrosis. We found that there was no significant change in WBC, LPS, or CRP between the CCl4 group and the other groups. CCl4 treatment induced the over‐expression of PCT; After UA or AP intervention, PCT was reduced (Figures 9a and 9d–f). This showed that UA could be applied to safely alleviate CCl4‐induced rats liver fibrosis.
In conclusion, UA induces the proliferation of HCs, promotes apoptosis in HSCs, and prevents activation of KCs in vivo by reducing the expression of NOXs/ROS in HCs, HSCs, KCs. UA also partially improves phagocytosis of KCs, but it has no effect on the host defense function. The molecular mechanisms that undergird the effect of UA on liver fibrosis require further investigations, and future approaches toward clinical applications of UA need to be done too.
CONFLICTS OF INTEREST
All the authors who have taken part in this study declared that they have no conflicts of interest to this manuscript.
ACKNOWLEDGMENT
This work is supported by National Natural Science Foundation of China (approval number of project: 81260082).
Gan D, Zhang W, Huang C, et al. Ursolic acid ameliorates CCl4‐induced liver fibrosis through the NOXs/ROS pathway. J Cell Physiol. 2018;233:6799–6813. 10.1002/jcp.26541
REFERENCES
- Altenhofer, S. , Radermacher, K. A. , Kleikers, P. W. , Wingler, K. , & Schmidt, H. H. (2015). Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxidants & Redox Signaling, 23(5), 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnin, S. T. , & Gnoatto, S. B. (2017). Ursolic acid from apple pomace and traditional plants: A valuable triterpenoid with functional properties. Food Chemistry, 220, 477–489. [DOI] [PubMed] [Google Scholar]
- Crosas‐Molist, E. , & Fabregat, I. (2015). Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biology, 6, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio, R. , Aghemo, A. , Rumi, M. G. , Ronchi, G. , Donato, M. F. , Paradis, V. , … Bedossa, P. (2012). A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology, 56(2), 532–543. [DOI] [PubMed] [Google Scholar]
- He, W. , Shi, F. , Zhou, Z. W. , Li, B. , Zhang, K. , Zhang, X. , … Zhu, X. (2015). A bioinformatic and mechanistic study elicits the antifibrotic effect of ursolic acid through the attenuation of oxidative stress with the involvement of ERK, PI3 K/Akt, and p38 MAPK signaling pathways in human hepatic stellate cells and rat liver. Drug Design, Development and Therapy, 9, 3989–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. X. , Chen, X. , Serizawa, N. , Szyndralewiez, C. , Page, P. , Schroder, K. , … Torok, N. J. (2012). Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radical Biology & Medicine, 53(2), 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J. X. , Venugopal, S. , Serizawa, N. , Chen, X. , Scott, F. , Li, Y. , … Torok, N. J. (2010). Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology, 139(4), 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. H. , Kim, J. N. , Han, S. N. , & Kim, H. K. (2015). Ursolic acid isolated from guava leaves inhibits inflammatory mediators and reactive oxygen species in LPS‐stimulated macrophages. Immunopharmacology and Immunotoxicology, 37(3), 228–235. [DOI] [PubMed] [Google Scholar]
- Lan, T. , Kisseleva, T. , & Brenner, D. A. (2015). Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS ONE, 10(7), e0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. A. , Wallace, M. C. , & Friedman, S. L. (2015). Pathobiology of liver fibrosis: A translational success story. Gut, 64(5), 830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Ren, D. , Luo, Y. , & Yang, X. (2016). Protective effects of ursolic acid against hepatotoxicity and endothelial dysfunction in mice with chronic high choline diet consumption. Chemico‐Biological Interactions, 258, 102–107. [DOI] [PubMed] [Google Scholar]
- Liang, S. , Kisseleva, T. , & Brenner, D. A. (2016). The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Frontier in Physiology, 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. Q. , Ding, J. , Zhang, L. , & Liu, C. M. (2015). Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clinics and Research in Hepatology and Gastroenterology, 39(2), 188–197. [DOI] [PubMed] [Google Scholar]
- Marcellin, P. , Gane, E. , Buti, M. , Afdhal, N. , Sievert, W. , Jacobson, I. M. , … Heathcote, E. J. (2013). Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5‐year open‐label follow‐up study. Lancet (London, England), 381(9865), 468–475. [DOI] [PubMed] [Google Scholar]
- Nisimoto, Y. , Diebold, B. A. , Cosentino‐Gomes, D. , & Lambeth, J. D. (2014). Nox4: A hydrogen peroxide‐generating oxygen sensor. Biochemistry, 53(31), 5111–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik, Y. H. , Iwaisako, K. , Seki, E. , Inokuchi, S. , Schnabl, B. , Osterreicher, C. H. , … Brenner, D. A. (2011). The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91 (phox) mediate hepatic fibrosis in mice. Hepatology, 53(5), 1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik, Y. H. , Kim, J. , Aoyama, T. , De Minicis, S. , Bataller, R. , & Brenner, D. A. (2014). Role of NADPH oxidases in liver fibrosis. Antioxidants & Redox Signaling, 20(17), 2854–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possamai, L. A. , Thursz, M. R. , Wendon, J. A. , & Antoniades, C. G. (2014). Modulation of monocyte/macrophage function: A therapeutic strategy in the treatment of acute liver failure. Journal of Hepatology, 61(2), 439–445. [DOI] [PubMed] [Google Scholar]
- Qiu, J. , Zhao, J. , Li, J. , Liang, X. , Yang, Y. , Zhang, Z. , … Li, G. (2016). NADPH oxidase inhibitor apocynin prevents atrial remodeling in alloxan‐induced diabetic rabbits. International Journal of Cardiology, 221, 812–819. [DOI] [PubMed] [Google Scholar]
- Rahman, M. M. , Muse, A. Y. , Khan, D. , Ahmed, I. H. , Subhan, N. , Reza, H. M. , … Sarker, S. D. (2017). Apocynin prevented inflammation and oxidative stress in carbon tetra chloride induced hepatic dysfunction in rats. Biomedicine & Pharmacotherapy=Biomedecine & Pharmacotherapie, 92, 421–428. [DOI] [PubMed] [Google Scholar]
- Robert, S. , Gicquel, T. , Bodin, A. , Lagente, V. , & Boichot, E. (2016). Characterization of the MMP/TIMP imbalance and collagen production induced by IL‐1beta or TNF‐alpha release from human hepatic stellate cells. PLoS ONE, 11(4), e0153118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, M. , & Kisseleva, T. (2015). Reversibility of liver fibrosis. Clinics and Research in Hepatology and Gastroenterology, 39(Suppl 1), S60–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. Y. , Li, X. F. , Meng, X. M. , Huang, C. , Zhang, L. , & Li, J. (2017). Macrophage phenotype in liver injury and repair. Scandinavian Journal of Immunology, 85(3), 166–174. [DOI] [PubMed] [Google Scholar]
- Tacke, F. (2017). Targeting hepatic macrophages to treat liver diseases. Journal of Hepatology, 66(6), 1300–1312. [DOI] [PubMed] [Google Scholar]
- Tacke, F. , & Zimmermann, H. W. (2014). Macrophage heterogeneity in liver injury and fibrosis. Journal of Hepatology, 60(5), 1090–1096. [DOI] [PubMed] [Google Scholar]
- Thompson, A. I. , Conroy, K. P. , & Henderson, N. C. (2015). Hepatic stellate cells: Central modulators of hepatic carcinogenesis. BMC Gastroenterology, 15, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Ikejima, K. , Kon, K. , Arai, K. , Aoyama, T. , Okumura, K. , Abe, W. , Sato, N. , & Watanabe, S. (2011). Ursolic acid ameliorates hepatic fibrosis in the rat by specific induction of apoptosis in hepatic stellate cells. Journal of Hepatology, 55(2), 379–387. [DOI] [PubMed] [Google Scholar]
- Weiskirchen, R. (2015). Hepatoprotective and anti‐fibrotic agents: It's time to take the next step. Frontiers in Pharmacology, 6, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. Y. , & Friedman, S. L. (2012). Fibrosis‐dependent mechanisms of hepatocarcinogenesis. Hepatology, 56(2), 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]