

Abstract
Aim
We performed acute and chronic studies in healthy and diet‐induced obese animals using mouse‐specific or monkey‐specific dual GLP‐1R/GCGR agonists to investigate their effects on food intake, body weight, blood glucose control and insulin secretion. The selective GLP‐1R agonist liraglutide was used as comparator.
Methods
The mouse‐specific dual agonist and liraglutide were tested in lean wild type, GLP‐1R knockout and diet‐induced obese mice at different doses. A chronic study was performed in DIO mice to investigate the effect on body weight, food consumption and total energy expenditure (TEE) in obese and diabetic monkeys with a focus on body weight and energy intake.
Results
The mouse‐specific dual agonist and liraglutide similarly affected glycaemic control. A higher loss in body weight was measured in dual agonist‐treated obese mice. The dual agonist significantly enhanced plasma glucose excursion in overnight fed GLP‐1R−/− mice, probably reflecting a potent GCGR agonist activity. It increased TEE and enhanced fat and carbohydrate oxidation, while liraglutide produced no effect on TEE. In obese and diabetic monkeys, treatment with the monkey‐specific dual agonist reduced total energy intake to 60%‐70% of baseline TEI during chronic treatment. A decrease in body weight and significant improvement in glucose tolerance was observed.
Conclusions
In DIO mice and non‐human primates, dual agonists elicited robust glycaemic control, similar to the marketed GLP‐1R agonist, while eliciting greater effects on body weight. Results from DIO mice suggest that the increase in TEE is caused not only by increased fat oxidation but also by an increase in carbohydrate oxidation.
Keywords: diabetes; diet‐induced obesity; dual agonism, energy expenditure; GLP‐1; glucagon; mouse; non‐human primates
Short abstract
This article includes a video abstract available at: https://vimeo.com/269278603
1. INTRODUCTION
Glucagon‐like peptide‐1 receptor (GLP‐1R) agonism is an established approach for the treatment of type 2 diabetes (T2DM). Activation of peripheral GLP‐1 receptors promotes glucose‐dependent insulin secretion and reduces the rate of gastric emptying while central GLP‐1R activation reduces food intake, leading to consequential weight loss. In the context of GLP‐1R agonist‐induced improvement of glycaemic control, the addition of glucagon receptor agonism (GCGR) appears contradictory, as the acute physiological response to glucagon is counter regulatory to the glucose‐lowering action of insulin. Glucagon exerts its hyperglycaemic effects through a transient increase of hepatic glucose production.1, 2, 3, 4, 5, 6 However, the description of oxyntomodulin as an endogenous and naturally occurring enteroendocrine peptidic hormone, activating both GLP‐1R and GCGR, inspired the development of dual GLP‐1R/GCGR agonists.7, 8 Oxyntomodulin combines the anorectic and glucose‐lowering effect of GLP‐1R agonists with the increased energy expenditure mediated by the GCGR. Chronic infusion of oxyntomodulin in GLP‐1R knock‐out mice (GLP‐1R−/−) produced a decrease in body weight, but to a lower extent as compared to wild‐type (WT) mice. This revealed that the anti‐obesity effect of oxyntomodulin requires activation of both GLP‐1R and GCGR.9 Stimulation of both receptors reverses diet‐induced obesity (DIO) in high‐fat fed mice10 and reduces body weight in overweight and obese human subjects.11 Recent research efforts have focused on developing dual acting GLP‐1R/GCGR agonists which combine GLP‐1‐related reduction of appetite and food intake with glucagon‐related increase in energy expenditure12. Pre‐clinical data from rodents indicate that dual GLP‐1R/GCGR agonists elicit greater weight loss when compared to selective GLP‐1R agonists.8 Similarly, Lao et al. reported that their dual GLP‐1R/GCGR agonist showed higher weight loss in diet‐induced obese rhesus monkeys and better glucose control in diabetic rhesus monkeys than GLP‐1R agonist gold standard liraglutide.13 A recent report also indicates that administration of MEDI0382, a dual GLP‐1R/GCGR agonist, at toxicological doses, to lean and healthy cynomolgus monkeys led to significant reduction in body weight.14 The impact on body weight results from a significant suppression of energy intake but may also arise from increased energy expenditure as observed in a sub‐chronic study in rats,15 in a multiple dosing study for 4 weeks in DIO mice8 and in humans.16 Co‐administration studies in humans confirmed that the concomitant activation of GLP‐1R mitigates the potential metabolic risk and hyperglycaemic impact of GCGR activation. Notably, while GCG infusion was accompanied by a rise in plasma glucose levels, co‐infusion of GLP‐1 and GCG led to a rapid decrease in glucose, as a result of a synergistic insulinotropic effect.17 While resting energy expenditure was not affected by GLP‐1 infusion, it rose significantly with GCG alone and to a similar extent in combination with GCG and GLP‐1. In a follow‐up study, GCG or GLP‐1, given individually at sub‐anorectic doses, did not reduce food intake in humans. But co‐infusion at the same doses led to a significant reduction in food intake and increased energy expenditure, and protected against glucagon‐induced hyperglycaemia.18 And several studies confirm that GCG elicits thermogenic activity and significantly increases energy expenditure.5, 19, 20, 21
We recently reported that dual GLP‐1R/GCGR agonists, which are structurally based on the exendin‐4 sequence, benefit from the favourable physicochemical properties of exendin‐4.22 These new dual agonists carry a fatty acid side chain on position 14 to improve half‐life. In this paper, we focus on such dual agonists, with a receptor activity ratio favouring the GLP‐1R to ensure proper glucose control and show the pharmacological profile in mice and obese diabetic NHP, which is, in our view, the most relevant translational model for the human situation (Table 1). Because of the distinct species‐dependent variation in GCGR agonist activity observed in peptides, we synthesized surrogates that were mouse‐ and monkey‐specific dual agonists with relative GLP‐1R/GCGR activity preferences similar to the human dual GLP‐1R/GCGR agonist currently in clinical development (see http://clinicaltrials.gov, NCT02411825). Because of the overlapping pharmacology observed following activation of GLP‐1 and GCG receptors, GCGR target engagement to guide compound optimization poses an experimental challenge. The current studies were therefore designed to demonstrate the acute effects of mouse‐specific dual agonists compared to the reference, that is, selective GLP‐1R agonist liraglutide in healthy WT and GLP‐1R−/− mice. With repetitive analysis of oxygen consumption and carbon dioxide production during chronic treatment, we investigated substrate utilization and were able to differentiate between fat and carbohydrate oxidation in DIO mice. In addition to the monitoring of change in body weight and food intake, total energy expenditure (TEE) and carbohydrate (CHO) and fat oxidation (FO) were determined in the chronic setting. We further performed chronic studies in DIO non‐human primates to evaluate efficacy of monkey‐specific dual agonist on glucose control and body weight as well as to interrogate biomarkers reflecting the distinct action of the dual approach.
Table 1.
Amino acid sequences of the mouse‐ and monkey‐specific dual GLP‐1R/GCGR agonists and the selective GLP‐1R agonist comparator liraglutide (top) with corresponding in vitro receptor agonist potencies (cAMP release) in HEK‐293 cell lines stably expressing human, mouse or monkey GLP‐1 or glucagon receptor (down)
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | C‐Term |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse dual agonist | H | D‐Ser | Q | G | T | F | T | S | D | L | S | K | Q | K[gGlu‐Palm] | D | S | R | R | A | Q | D | F | I | E | W | L | K | N | G | G | P | S | S | G | A | P | P | P | S | NH2 |
| Monkey dual agonist | H | D‐Ser | Q | G | T | F | T | S | D | L | S | K | Q | K[gGlu‐Stea] | D | E | Q | L | A | K | D | F | I | E | W | L | K | A | G | G | P | S | S | G | A | P | P | P | S | NH2 |
| Liraglutide | H | A | E | G | T | F | T | S | D | V | S | S | Y | L | E | G | Q | A | A | K[gGlu‐Palm] | E | F | I | A | W | L | V | R | G | R | G | OH |
| Compounds tested | EC50 human [pM] | EC50 mouse [pM] | EC50 monkey [pM] | |||
|---|---|---|---|---|---|---|
| GLP‐1R | GCGR | GLP‐1R | GCGR | GLP‐1R | GCGR | |
| Mouse dual agonist | 3.9 | 0.7 | 2.3 | 25 | ||
| Monkey dual agonist | 4.8 | 9.1 | 6.3 | 119.3 | ||
| Liraglutide | 6.4 | n.a. | 4.4 | n.a. | 4.1 | n.a. |
| GLP‐1(7‐36)amide | 0.9 | n.a. | 0.9 | 1.1 | ||
| Glucagon | 43.9 | 0.4 | 43.5 | 1.3 | 30.8 | 1.5 |
Abbreviation: n.a., not active.
2. MATERIALS AND METHODS
2.1. Animals and housing conditions – mouse studies
All mouse‐related experimental procedures were conducted in accordance with the German Animal Protection Law, as well as according to international animal welfare legislation and rules.
2.1.1. DIO and lean mice
Mouse studies were performed in male diet‐induced obese C57Bl/6N mice or in female lean C57Bl/6N (WT) and female GLP1R knock‐out (GLP‐1R−/−) mice. All mice were obtained from Jackson Laboratory (Bar Harbor, Maine). Homozygous GLP‐1R−/− (CD1‐GLPtm1Ddr) mice were bred and raised at Jackson Laboratory according to Scrocchi et al.23 Because our GLP‐1R−/− samples contain CD1, an outbred strain, we used the related inbred strains SJL, SWR and FVB to represent the alleles found in CD1. These strains were compared to C57BL/6J (B6) in a panel of polymorphic SNP markers (177 markers covering chromosomes 1‐19). The SNP analysis indicated that, in the GLP‐1R−/− colony, the percent of B6 is ~19.2% and the percent of CD1 is ~80.8%.
Male C57Bl/6N mice were housed and pre‐fed a high‐fat diet (ssniff adjusted to Teklad diet TD.97366 [E15797], EF HF purified diet, ssniff Spezialdiäten; GmbH, Soest, Germany) for 18 weeks, reaching a plateau in body weight of 40 to 50 g. All obese HFD‐fed mice were randomized and stratified for body weight prior to initiation of the chronic study and separated into n = 8‐16 mice per group and cage (makrolon type 4).
For acute functional testing of glucose tolerance in an intraperitoneal tolerance test (ipGTT) female lean WT and GLP‐1R−/− mice were used after fasting for 6 h, with n = 8 mice per group and cage and approximately 30 g of body weight. The lean mice were maintained on standard control diet (E15796, ssniff Spezialdiäten GmbH, Soest, Germany). Mice were housed in an environmentally controlled room at 23°C and 12 h:12 h light dark cycle, with light on at 06:00 am, and food and water offered ad libitum.
2.2. Experimental design
2.2.1. Intraperitoneal glucose tolerance test (ipGTT) and glucose profile under fasted or fed conditions
For acute studies in female lean WT C57Bl/6 N and GLP‐1R−/− mice, mice were used for determination of glucose tolerance (n = 8 per group). All mice were fasted for 6 hours. The mouse‐specific dual agonist or liraglutide was administered subcutaneously (s.c.) at doses of 10, 30 or 100 μg/kg. Compounds were administered according to kinetic profile 2 hours prior to the glucose bolus (2 g/kg body weight, i.p.). Plasma samples for glucose analysis were taken at t = 0, 15, 30, 60, 120 and 180 minutes after glucose challenge. Insulin was analysed 15 minutes after the glucose challenge. All mice were re‐fed after last blood sampling. In addition, a glucose profile was obtained under fed conditions with blood sampling at t = 0, 30, 60, 120, 240 and 360 minutes after compound administration.
2.2.2. Indirect calorimetry
For the DIO mouse study (C57Bl/6 N mice), after body weight plateaued in mice fed a high‐fat diet, mice were injected with the mouse‐specific dual agonist or liraglutide at 50 μg/kg twice daily (b.i.d.) for 32 days. A vehicle‐treated group was included as a control. Body weight and food intake were recorded daily. On days 2, 16 and 30 oxygen consumption and carbon dioxide production were determined via indirect calorimetry. For this measurement, mice were transferred to calorimetry cages with n = 2 per cage for acclimatization for 12 hours, followed by 24 hours of measurement with ad libitum access to food and water. At the end of chronic treatment, blood samples were collected from the tip of the tail for determination of glucose and insulin, as well as retro‐orbitally, for determination of possible serum parameters, which is recommended as an indicator of target engagement (ketone bodies, FGF21).
The setting consisted of 17 cages, of which 16 were used for the housing of mice during gas analysis. One cage served as a reference for corrections of O2 and CO2 measurements. All mice were accustomed to the special calorimetry cages at least 12 hours before the measurement. O2 consumption and CO2 production were measured every 16 minutes per cage for 1 minute (gas analyzers: Magnos 16 and Uras 14; ABB, Frankfurt/Main, Germany) and were recorded automatically by computer. All values were expressed as means of litre/d of VCO2 and VO2, respectively. For interpretation of substrate utilization, the respiratory quotient (RQ) was calculated as the quotient of VCO2/ VO2. 22
The Total Energy Expenditure was calculated according to the formula:
| (1) |
Nitrogen loss was set constantly to 0.2 g/d. As the treated mice differed in body mass and body composition, allometry of oxygen consumption was achieved by correcting TEE per metabolic body mass (kg0.75) per day.24, 25 Fat and carbohydrate oxidation (FO and CHO) were calculated according to Ferrannini26 and were modified27 by applying the following formulas:
| (2) |
and
| (3) |
and were expressed as g/day * kg‐0.75. The resting metabolic rate (RMR) or resting energy expenditure (REE) was determined by measuring the 3 lowest VO2 values during the inactive (lights on) phase of the mice, which reflects the resting situation of the animal. As the current studies were not performed within the thermoneutral zone (TNZ), the REE (RMR) differs clearly from the basal metabolic rate (BMR), which is usually determined in thermoneutrality and in the resting phase.
2.3. Animals and housing conditions – monkey study
The monkey study was performed at Kunming Biomed International (KBI, Yunnan Province, China). KBI adheres to the guidelines for the care and use of animals for scientific purposes established by the Chinese National Advisory Committee for Laboratory Animal Research (NACLAR) as well as the safety and quality assurance guidelines documented in the Guideline for Experiments Document of Kunming Biomed International (KBI‐01‐GE v2.0). These guidelines set out the responsibilities of all parties involved in the care and use of animals for scientific purposes, in accordance with widely accepted scientific, ethical and legal principles. The guidelines stipulating the proposed use of animals for scientific purposes must be evaluated by an Institutional Animal Care and Use Committee (IACUC) in compliance with the guidelines. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of KBI prior to the experimental phase.
2.3.1. DIO monkeys
The cynomolgus monkey (Macaca fascicularis) was selected as the test species of choice for more meaningful results that were translatable to humans. A total of 51 monkeys were trained in order to identify 30 monkeys [n = 8‐10 per group] that were used in the chronic treatment phase and fulfilled the following metabolic criteria: weight at least 8 to 16 kg, age 12 to 20 years, fasting glucose >110 mg/dL and fasting insulin >70 μU/mL. During the progression of the study, 2 individual monkeys in the vehicle‐treated group were excluded from data analysis because of aggressive progression of diabetes. All monkeys used in the study were KBI F2 generation and were born and raised in the KBI breeding facilities. Monkeys were individually housed in species‐ and size‐appropriate metabolic stainless steel caging, with ad libitum access to water and under controlled environmental conditions with room temperature of 18 to 29°C, relative humidity of 30%‐70% and a minimum of 10 air changes/hour. A time‐controlled lighting system was used (lights on 7:00 am to 7:00 pm) to provide a regular 12‐hour light/12‐hour dark diurnal cycle. Cages were cleaned at regular intervals for maintenance of hygiene during the entire study period. Monkeys were provided 3 meals per day, consisting of approximately 50 g of standard monkey formula feed (extruded pellets, 3.1 Kcal/g: protein 24%, fat 15%, carbohydrate 61%) in the morning (9:00 am‐10:00 am), one apple (150 g, ~80 Kcal) in the afternoon (2:00 pm‐3:00 pm), and 100 g of KBI proprietary high‐fat diet feed in the evening (extruded pellets, 3.47 Kcal/g: protein 14%, fat (porcine) 34%, carbohydrate 52%, sucrose 35%, 4:00 pm‐5:00 pm). The total offered amount of daily energy was ~680 Kcal/d. All food was withdrawn at 5:00 pm; hence, all monkeys were fasted overnight. After each feeding, all remaining food was withdrawn and intake was determined by weighing the left‐over food. Each batch of monkey chow was delivered with an accompanying certificate of analysis, detailing nutritional composition and levels of specified contaminants (eg, heavy metals, aflatoxin and insecticides). Ad libitum access to water was suspended on days when water intake was quantified (Days −6 to −4, Days 1 to 77). Animals were provided with ad libitum access to water via a 500‐mL water bottle with a drinking valve attachment and availability was monitored throughout the day. Water lost from the bottle was collected in a drip tray beneath the drinking valve. Monkeys were provided with up to 5000 mL of water per day, which was added as needed between 6:00 pm and 7:00 pm, prior to the dark period. Water samples were routinely analysed for specified microorganisms and environmental contaminants. There were no known contaminants present in the diet or water that could confound the data generated in this study. Monkeys were provided with enrichment toys or devices at all times. Enrichment items were changed weekly for all study animals according to KBI SOP. Enrichment items included: plastic chew toys, plastic balls, food puzzles and foraging trays. Animals were stratified by randomized block stratification into 3 homogenous groups according to body weight, fasted plasma glucose and %HbA1c.
2.4. Experimental design
For the DIO monkey study, animals were trained for 3 weeks to habituate them to the blood‐draw procedure and for presentation of the subscapular/cervical region for s.c. injection of compounds with minimal physical restraint. Behavioural signs were recorded continuously, including vocalizations, willingness to present arm for blood draw, necessity of restraint during blood draw and reactions toward technical staff. Body weight measurements were performed by transferring the monkeys into smaller cages to a calibrated weighing scale.
The run‐in period, from Day −49 to −1, was used for basal metabolic profiling (Day −25), ivGTT (Day −32), as well as for safety blood sampling (Day −44, −40, −3), while the training procedure was continued. Animal selection and stratification was done according to a stratified block randomization approach from Day −17 to −1, based on blood glucose and insulin.
Administration of a monkey‐specific dual GLP‐1R/GCGR agonist, liraglutide or vehicle was performed subcutaneously during the dosing period from Day 1 to 70, including a dose‐ramping period from Day 1 to Day 10. During the entire dosing period, food and water intake was monitored daily and body weight was measured every 3 to 4 days. Metabolic profiling for glucose and insulin was repeated during the dosing period on Day 43, while ivGTT was repeated on Day 46 of chronic treatment. On the day of ivGTT, compounds were administered 2.5 hours before the glucose bolus (0.5 g/kg body weight) was injected intravenously. For the procedure, animals were sedated with 10 mg/kg ketamine (i.m.). In total, 7 serial blood samples (1.0 mL each) were collected at 1, 3, 5, 10, 20, 40 and 60 minutes following the end of the i.v. glucose bolus.
2.4.1. Intravenous glucose tolerance test (ivGTT) in NHP
On the treatment ivGTT days, all dosing commenced at 2.5 hours before i.v. glucose bolus, that is, ivGTT time = 0 minutes. Dosing began at 6:30 am and continued in a staggered manner until all monkeys were dosed. Animals were sedated (10 mg/kg ketamine, i.m.) and removed from the home cage, weighed and transported to the procedure room. Vital signs were recorded and confirmed within normal parameters prior to initiating the ivGTT.
Animals were placed on their sides and carefully secured to the procedure table using tether cords. Animals were also placed on heating blankets and covered throughout the procedure. A 50% glucose solution was prepared according to body weight (0.5 g/kg body weight), that is, 1 mL of solution per kg body weight, and the final solution was filtered through a 22‐μm microfilter. The sampling site was cleaned using betadine (iodine) and 70% alcohol solutions and 1.0 mL of baseline blood was collected by venipuncture from the upper cephalic vein via an i.v. butterfly needle using a syringe. The 50% glucose solution bolus was administered over 30 seconds; t = 0 minutes began at the end of the bolus administration. A total of 7 serial blood samples (1.0 mL each) were collected at 1, 3, 5, 10, 20, 40 and 60 minutes from the end of the i.v. glucose bolus. All blood samples were collected into chilled K2EDTA tubes, and centrifuged within 30 minutes of collection at 4°C.
2.4.2. Treatments
Liraglutide, a selective GLP‐1R agonist, was chosen as the positive control compound. It was purchased directly from Novo Nordisk distributors in Beijing and Kunming and stored at 4°C in accordance with requirements of the manufacturer. Victoza pens contain liraglutide at a concentration of 6 mg/mL. For the monkey study, the solution was diluted to the desired concentrations of 100, 200 and 400 μg/mL using the vehicle solution. Liraglutide was dosed during the dose‐ramping period from 10 (Day 1‐3) to 20 (Day 4‐6) to 30 μg/kg (Day 7‐9) and was maintained at 40 μg/kg s.c. (in 0.1 mL/kg of formulation) from Day 10 to completion of the study.
2.4.3. Mouse‐ and monkey‐specific dual GLP‐1R/GCGR agonist peptides
In a recent publication we described the rational design of dual GLP‐1/glucagon receptor agonists.22 Because of its favourable physico‐chemical and metabolic stability properties, the marketed GLP‐1R agonist exendin‐4 was used as the starting point for the design. Structural elements of glucagon were engineered into exendin‐4, resulting in a hybrid peptide with potent dual GLP‐1/glucagon receptor activity. A d‐serine residue was introduced into position 2 to avoid DPP4‐mediated degradation.28 In addition, a palmitic acid had been attached at the ε‐amino group of a lysine residue using a γ‐glutamic acid spacer, a strategy that has been successfully proven to prolong half‐life,29 for example, in the case of liraglutide, via oligomerization and reversible albumin binding. As a result of these design activities, the “mouse dual agonist” was obtained. Subcutaneous administration of this peptide (1 mg/kg) in healthy mice showed a half‐life of 3.2 hours, similar to that of liraglutide (3.6 hours) but with a lower exposure.22
Further modifications of the peptide structure in positions 14 to 20 and 28 led to the “monkey dual agonist”. Both the mouse and monkey dual agonists have a receptor activity ratio favouring the (mouse and monkey) GLP‐1R. Liraglutide, developed by Novo Nordisk, is a marketed chemically modified GLP‐1 analog, which carries a fatty acid sidechain in position 20, leading to a half‐life of 13 hours in humans following subcutaneous injection.30 It was used as a comparator compound for our studies. Amino acid sequences of these peptides are shown in Table 1.
GLP‐1 and glucagon were purchased from Bachem (Bubendorf, Switzerland). Liraglutide was used from a prefilled pen (Victoza, Novo Nordisk). The mouse‐ and monkey‐specific dual GLP‐1R/GCGR agonists were prepared at Sanofi by Fmoc solid‐phase synthesis and purified by reverse‐phase high‐performance liquid chromatography using water/acetonitrile (0.1% trifluoroacetic acid) gradients. The purified peptides were characterized by electrospray MS. For the monkey in vivo study, the peptide was transformed into its acetate salt.
2.4.4. Determination of mouse and monkey GLP‐1R and GCGR potency
Agonist activity of compounds at the receptors was determined by functional assays measuring cAMP response of HEK‐293 cell lines stably expressing human, mouse or monkey GLP‐1R or GCGR (Table 1). Levels of cAMP produced were determined using a kit from Cisbio Corp. (cat. no. 62AM4PEJ, Codolet, France) based on HTRF (Homogenous Time Resolved Fluorescence) as per the manufacturer's instructions.
The monkey‐specific dual GLP‐1R/GCGR agonist was provided directly from Sanofi to KBI in pre‐prepared vials of 2 mg. The compound quantity in each vial was diluted with a PBS vehicle volume adjusted for the specific mass of peptide in each vial. Each formulation vial was prepared and used on the same day of dosing. The monkey‐specific dual agonist was dosed during the dose‐ramping period from 1 (Day 1‐3) to 2 (Day 46) to 3 μg/kg (Day 7‐9) and was maintained at 4 μg/kg s.c. (in 0.05 mg/kg of formulation) from Day 10 to completion of the study.
2.4.5. PK measurements in obese diabetic monkey study
Blood samples for PK analysis were collected on Day 43, directly before and 1, 2, 4, 8 and 22 hours post dosing. The samples were transferred directly into potassium ethylene diamine tetra‐acetic acid (EDTA‐2 K) tubes. Plasma was separated by centrifugation at 2500×g for 10 minutes at 4°C. Plasma samples were analysed after protein precipitation via liquid chromatography mass spectrometry (LC/MS) for the dual agonist and by ELISA for liraglutide.
2.5. Statistical analysis
All data are presented as means ± SEM. A 1‐way analysis of variance for factor treatment or 2‐way analysis including factor time were used, followed by Dunnett's test for multiple comparisons vs the vehicle or the high‐fat diet control group, and separately for comparison of the dual agonist and the reference liraglutide. Statistical significance was considered with P < .05. All analyses were performed using SAS (version 9.2) under HP‐UX via interface software EverStat V6.0‐alpha5.
3. RESULTS
3.1. Acute and multiple dose studies with mouse‐specific dual GLP‐1R/GCGR agonist in mice
The simultaneous activation of GLP‐1 and glucagon receptors (GCGR) was investigated in acute and chronic studies. In acute studies, the anti‐diabetic effect was tested by measuring the 6‐hour fed glucose profile and glucose tolerance in an intraperitoneal glucose tolerance test (ipGTT).
In the 6‐hour glucose profile obtained under continuous ad libitum feeding conditions, the mouse‐specific dual GLP‐1R/GCGR agonist and selective GLP‐1R agonist liraglutide were administered at 10, 30 or 100 μg/kg to healthy C57Bl6/N mice. Liraglutide significantly reduced blood glucose levels at 6 hours, but only at the 30 μg/kg (−13.7 ± 1.9%; P < .01) and 100 μg/kg (−26.0 ± 3.0%; P < .001) dose levels. In contrast, the dual agonist reduced glucose at all dose levels tested, reaching −25.5 ± 3.2% at 10 μg/kg, −31.0 ± 2.0% at 30 μg/kg and −36.9 ± 1.8% at 100 μg/kg after 6 hours (Figure 1).
Figure 1.
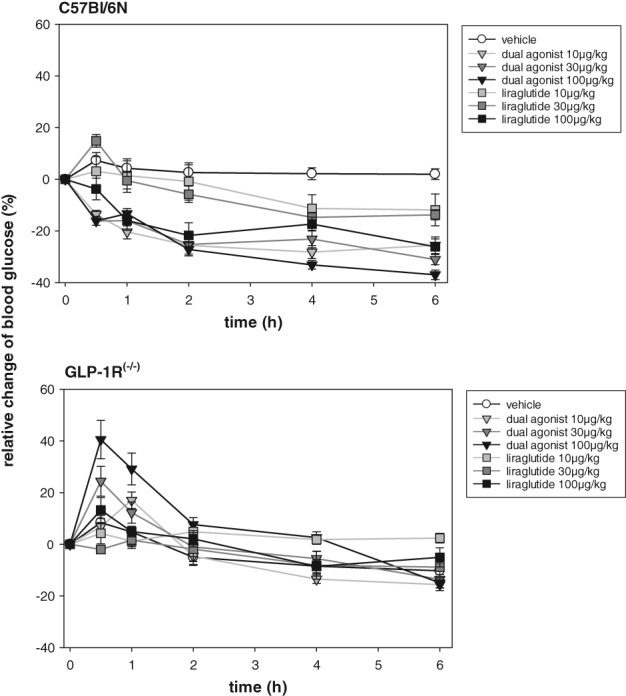
Acute studies with mouse‐specific dual GLP‐1R/GCGR agonist in healthy C57Bl/6N and GLP‐1 receptor knock out (GLP‐1R−/−) mice. Glucose profile under fed conditions injected s.c. with mouse‐specific dual GLP‐1R/GCGR agonist or liraglutide at 10, 30 or 100 μg/kg doses. Values are normalized to t = 0 minutes and shown as relative change from baseline. Values are given as mean ± SEM, n = 8/group
The same experimental design performed in GLP‐1R−/− mice revealed a transient dose‐dependent increase in glucose in dual agonist‐treated mice 30 minutes after compound administration, reaching statistical relevance at 100 μg/kg (P < .001) (Figure 1). No significant difference in blood glucose was observed 6 hours after treatment. As expected, liraglutide treatment did not affect blood glucose in GLP‐1R−/− mice.
In an ipGTT, a significant, dose‐dependent improvement in glucose tolerance was observed for both drug treatment groups in C57Bl6/N mice at the lowest dose injected (10 μg/kg). However, at selected doses, liraglutide was less effective than the dual agonist, as observed for the area under the curve calculations (Figure 2).
Figure 2.
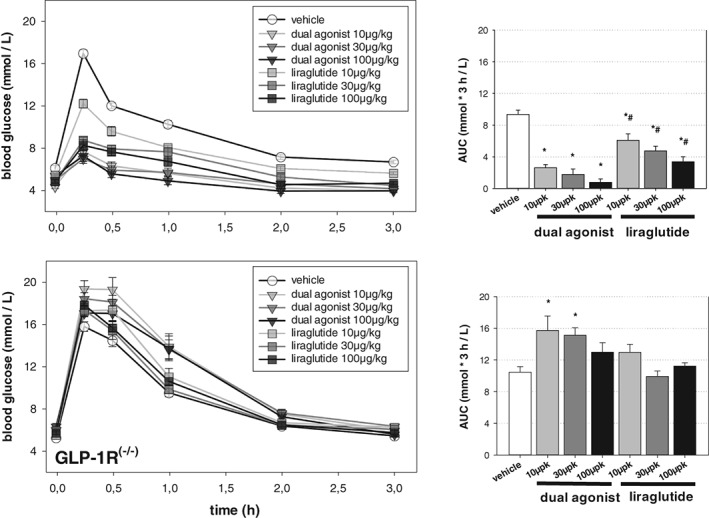
Acute studies with mouse‐specific dual GLP‐1R/GCGR agonist in healthy C57Bl/6N and GLP‐1 receptor knock out (GLP‐1R−/−) mice. Intraperitoneal glucose tolerance test (ipGTT) after 6 hours of fasting. Vehicle or compounds (10, 30 or 100 μg/kg, s.c.) were administered 2 hours prior to the glucose bolus (2 g/kg), which was administered i.p. at t = 0 minutes. Values are given as mean ± SEM, n = 8/group. *P < .05 vs vehicle control. #P < .05 vs dual agonist
In GLP‐1R−/− mice, injection of the dual agonist (10 or 30 μg/kg) significantly increased glucose excursion in the ipGTT (15 minutes after glucose bolus) (Figure 2) (P < .01). Increasing the dual agonist dose to 100 μg/kg increased glucose excursion compared to vehicle‐treated controls, but the increase did not reach statistical significance. Liraglutide did not affect the ipGTT in GLP‐1R−/− mice.
We used indirect calorimetry to assess whether chronic engagement of the GCGR was associated with an increase in energy expenditure. The mouse‐specific dual agonist was compared to either control or liraglutide (both peptides administered at 50 μg/kg b.i.d., s.c.). Throughout the initial 4 days of dosing, both liraglutide and the dual agonist decreased daily food intake compared to vehicle‐treated mice. The reduced energy intake returned to pre‐treatment levels on day 5 with both drugs. Thus, cumulative food intake for the remainder of the study period was similar among all groups (Figure 3 A). Both groups treated with drugs continuously lost body weight, although food consumption normalized on day 5 of treatment (Figure 3 B). However, in liraglutide‐treated mice, body weight loss was lower and reached a plateau earlier than observed in the dual agonist‐treated mice (Figure 3 C). At the end of the study, liraglutide treatment elicited −12.9% ± 1.4% change in body weight as compared to −21.1% ± 2.1% elicited by the dual agonist.
Figure 3.
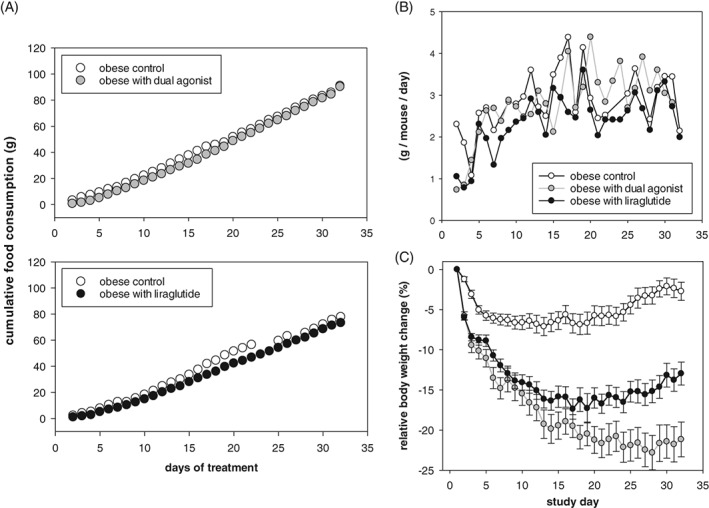
Food consumption during a 32‐day chronic study in DIO mice that received liraglutide or mouse‐specific dual agonist (50 μg/kg b.i.d. subcutaneously). A, cumulative food intake and B, expressed as daily food intake per mouse. Although food consumption normalized during the study, mice of both treatment groups decreased in body weight (C), with the dual agonist having a greater effect. Food intake values are shown as mean group values with n = 8 per cage. Relative change in body weight is given as mean ± SEM, n = 8‐16/group. *P < .05 vs vehicle control. #P < .05 vs dual agonist
This superior weight loss effect of the dual GLP‐1R/GCGR agonist vs selective GLP‐1R agonism was further investigated with concomitant measurement of O2 consumption and CO2 production on days 2, 16 and 30. Two single studies were performed with mouse‐specific dual agonist or liraglutide (administered at 50 μg/kg b.i.d.) and compared with vehicle‐treated control mice for each treatment group. We observed a metabolic switch from primarily fat oxidation during acute treatment to a more “mixed fuel” and substrate utilization during chronic treatment in the dual agonist treated mice. The initial appetite suppressing impact of both the dual agonist and liraglutide was reflected by a fasting‐induced decrease in TEE (Figure S1 in Appendix S1). Also, vehicle‐treated controls react with lowered CHO, mainly as a result of adaptation to the measurement environment. Energetic requirements are maintained by fat oxidation (FO) of endogenous fat depots. In contrast to liraglutide, the dual agonist significantly increased CHO during chronic treatment, which was accompanied by a significant increase in TEE on days 16 and 30 (Figure 4; day 16 not shown). Liraglutide increased TEE during prolonged treatment to a lesser extent than the dual agonist, but TEE was still significantly elevated compared to vehicle‐treated mice. We focused the analysis of substrate utilization on the nocturnal phase during which mice typically are active and consume energy. Compared to liraglutide and vehicle, the dual agonist‐treated mice displayed elevated CHO and FO throughout the chronic study, leading to a significant and persistent increase in TEE (Figure 5).
Figure 4.
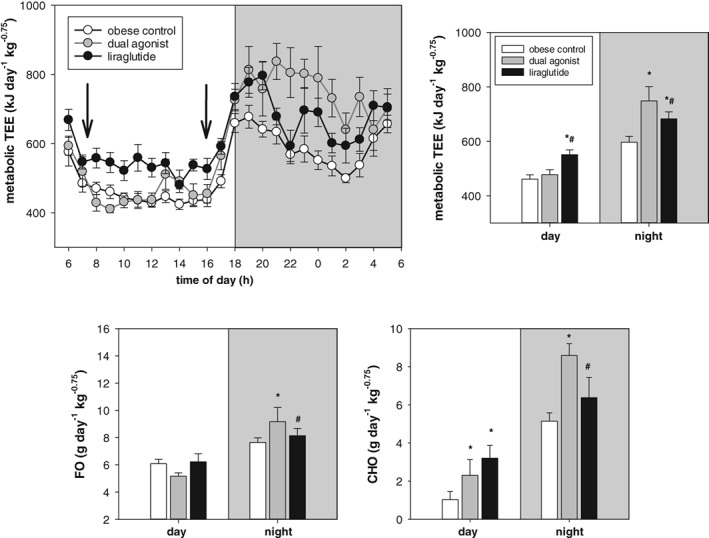
Energy expenditure related to metabolic body weight. Day 30 of chronic treatment with 50 μg/kg, s.c. of liraglutide or mouse‐specific dual agonist. Values are given as mean ± SEM, n = 8‐16/group. *P < .05 vs vehicle control. #P < .05 vs dual agonist
Figure 5.
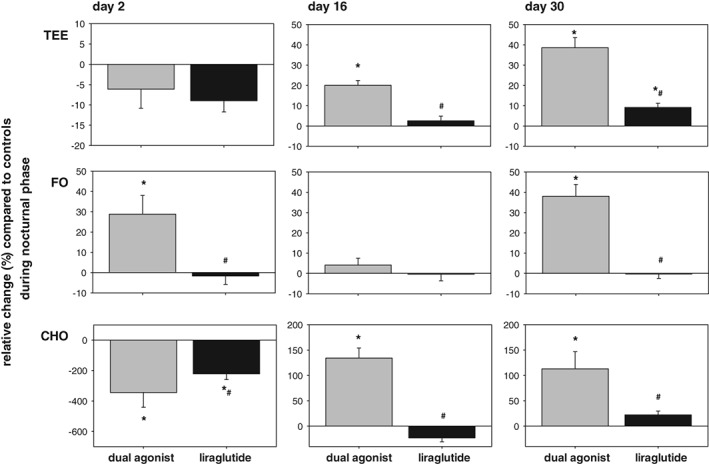
Metabolic switch as relative change of substrate utilization in drug‐treated (50 μg/kg, b.i.d.) subjects as compared to DIO vehicle‐treated controls in night/activity phase (grey bars, dual mouse‐specific GLP‐1R/GCGR agonist; black bars, liraglutide). Values are given as mean ± SEM, n = 8‐16/group. *P < .05 vs vehicle control. #P < .05 vs dual agonist
Liraglutide and the dual agonist increased REE during the chronic treatment period relative to their obese controls. After hypometabolic adaptation with lowered REE compared to controls on day 2 of treatment, dual agonism led to an increase by 25% on day 16 and by 9.8% on day 30 vs liraglutide, which increased REE by 8.0% on day 16 and by 12.4% on day 30.
In search of a GCGR agonist‐associated target engagement biomarker, liver glycogen content, total plasma ketone bodies and plasma FGF21 levels were measured. Seven days of treatment with the dual agonist (50 μg/kg, b.i.d.) led to a significant reduction in liver glycogen (Figure 6 A) (P < .001), whereas 7 days of treatment with liraglutide (50 μg/kg, b.i.d.) did not reduce, but rather slightly increased, liver glycogen content (P < .05). Serum ketone bodies increased significantly in dual agonist‐treated mice (Figure 6 B) (P < .01) without any observed change in liraglutide‐treated mice. Plasma FGF21 levels showed a trend toward an increase in dual‐agonist‐treated vs liraglutide‐ or vehicle‐treated mice (Figure 6 C).
Figure 6.
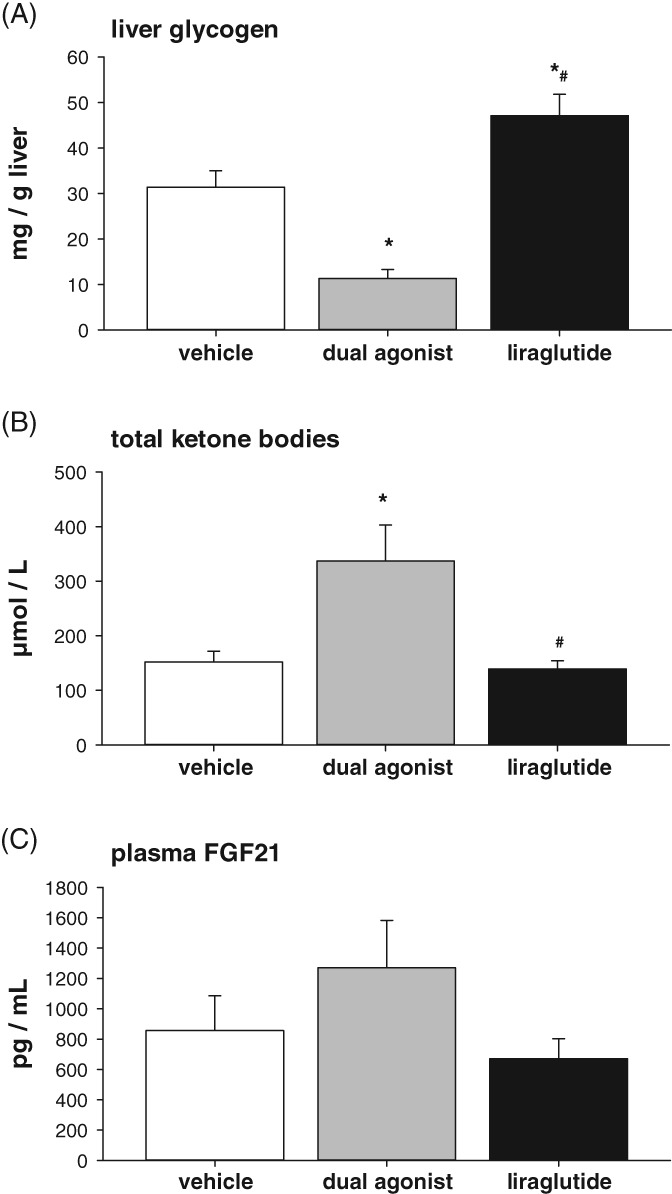
Potential blood biomarker reflecting target engagement of GCGR agonism in chronic‐treated DIO mice with 50 μg/kg (s.c., b.i.d.) liraglutide or mouse‐specific dual agonist. A, liver glycogen, B, ketone bodies and C, plasma FGF21. Values are given as mean ± SEM, n = 4‐8/group. *P < .05 vs vehicle control. #P < .05 vs dual agonist
3.1.1. Chronic and multiple‐dose study with monkey‐specific dual GLP‐1R/GCGR agonist in obese and diabetic non‐human‐primates (NHPs)
We used the impact on total energy intake and the accompanying body weight loss as the major indicators of tolerability during the multiple‐dose efficacy study in cynomolgus monkeys (Macaca fascicularis). To facilitate chronic treatment at the maximum tolerated dose, dose escalation was performed every third day. A monkey‐specific dual GLP‐1R/GCGR agonist, similar in its relative receptor‐activity preference to the mouse‐specific dual agonist, was compared to the selective GLP‐1R agonist liraglutide (Table 1). Mean body weights of the 3 groups varied between 10.8 and 11.7 kg. During the run‐in period (prior to initiation of treatment) the mean TEI of all groups was approximately 550 Kcal/d. Liraglutide induced a moderate but non‐significant decrease in food intake throughout the dose escalation period, from days 1 to 3 (10 μg/kg) until day 10 (40 μg/kg) onwards. The maximal reduction of TEI was approximately 25% during the initial dosing days for the highest dose, and this recovered slowly but continuously (Figure 7). NHPs treated with the monkey‐specific dual GLP‐1R/GCGR agonist showed stronger effects on TEI, following the escalation steps of 1 to 2 to 3 μg/kg and the maintenance dose of 4 μg/kg. Each dose step of the dual agonist produced a significant reduction in TEI compared to vehicle‐treated, but also to liraglutide‐treated, monkeys when escalating the dose to 1 and 3 μg/kg. From day 10 and onwards, the dual agonist induced a significant decrease in TEI when compared to vehicle (P < .01), whereas the drop in TEI was not statistically significantly different from that of liraglutide. The maximal reduction in TEI was approximately 50% and recovered only moderately until the end of the study. While vehicle‐treated monkeys lost 2.3% ± 0.8% of body weight during the study, liraglutide treatment led to a significant weight loss of 5.1% ± 1.1% (P < .001). The monkey‐specific dual GLP‐1R/GCGR had the highest impact on body weight and decreased body weight by 8.2% ± 1.2% (P < .001) (Figure 8).
Figure 7.
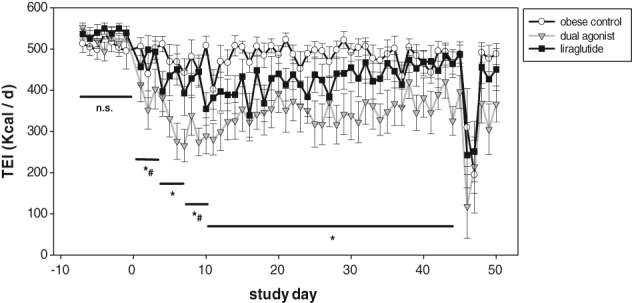
Chronic treatment of obese and diabetic monkeys (M. fascicularis) with monkey‐specific dual agonist and liraglutide. Total energy intake (TEI) per day consisting of: 46.5 g standard monkey formula feed as breakfast from 9:00 to 10:00 am; 150 g apple as lunch from 2:00 to 3:00 pm; 100 g KBI HFD as dinner from 4:00 to 5:00 pm. Drop in TEI on day 46 was the result of performance of the ivGTT. Values are given as mean ± SEM, n = 8‐10/group. *P < .01 dual agonist vs vehicle control. #P < .01 dual agonist vs liraglutide (days −7 to 0, days 1 to 3, days 4 to 6, days 7 to 9, days 10 to 45)
Figure 8.
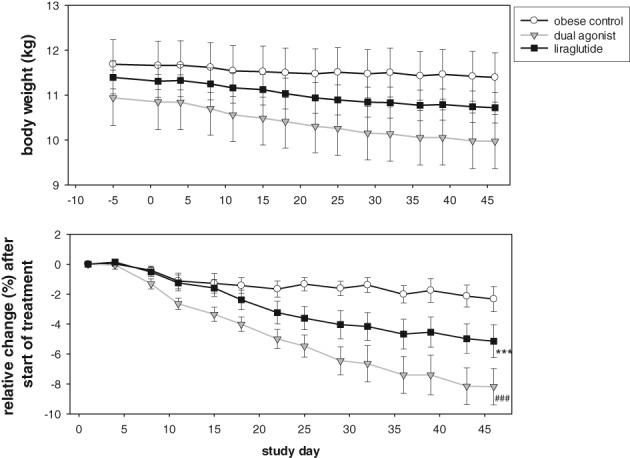
Absolute body weight (top) and relative body weight change (bottom) during study period. Values are mean ± SEM, n = 8‐10/group. *P < .001 liraglutide vs vehicle control. #P < .001 dual agonist vs vehicle control
During the entire study period, metabolic profiling was performed at different time points (Figures S2 and S3 in Appendix S1). Plasma levels of glucose, insulin, ketone bodies, and FGF21 were analysed on day −25, the first day on maintenance dose (day +10) and close to the end of chronic treatment (day +43). These values were all measured prior to performing the functional intravenous glucose tolerance test (ivGTT), which was done on days +46/+47.
Because of exclusion of 2 monkeys in the vehicle group, who became insulin‐dependent during the study, baseline mean glucose values (162.9 ± 36.9 mg/dL) for the vehicle‐treated monkeys were slightly lower compared to the treatment groups (213.9 ± 23.2 mg/dL for the monkey‐specific dual GLP‐1R/GCGR agonist and 233.6 ± 28.3 mg/dL for the liraglutide‐treated group). Nevertheless, plasma glucose and insulin levels of the vehicle‐treated monkeys included in the study remained unchanged throughout the study period. Treatment with escalating doses of liraglutide led to a drop in blood glucose from >230 mg/dL to levels between 150 and 170 mg/dL, which were maintained throughout the study period. The monkey‐specific dual agonist had a significantly greater effect on blood glucose lowering compared to liraglutide. After administration of the monkey‐specific dual GLP‐1R/GCGR agonist blood glucose initially decreased to below 100 mg/dL and then gradually returned to values of approximately 120 mg/dL (Figure S2 in Appendix S1). Monitoring fasting plasma glucose (FPG) on selected study days (days −3 and +1: both basal values; days +17, +24, +38 and +45: during chronic treatment) confirmed these results (Table 2). Fasted glucose values after initiation of treatment were obtained on day +17 and were compared to fasted glucose on day +1. FPG values were determined directly before breakfast. While the dual agonist significantly decreased FPG throughout the study period from 228.6 ± 22.5 mg/dL (day +1, determined as basal) to 127.1 ± 14.5 mg/dL (day +45), liraglutide showed a significant decrease only on days +17 and +38, not decreasing below 170.2 ± 21.1 mg/dL. In contrast, plasma glucose values for the vehicle‐treated monkeys remained at the same level of approximately 170 mg/dL throughout the study period.
Table 2.
Fasted plasma glucose (mg/dL) in obese and diabetic monkeys (M. fascicularis) on days −3 (basal), +1 (basal), +17, +24, +38 and +45 (all treatment). Values are given as mean ± SEM, n = 8‐10/group
| Compounds tested | Day −3 (basal) | Day +1 (basal) | Day +17 | Day +24 | Day +38 | Day +45 |
|---|---|---|---|---|---|---|
| Fasting plasma glucose [mg / dL] | ||||||
| Vehicle | 170.1 ± 26.3 | 173.9 ± 23.5 | 177.8 ± 27.0 | 175.0 ± 24.5 | 168.3 ± 25.1 | 176.5 ± 22.0 |
| Dual agonist | 211.3 ± 23.1 | 228.6 ± 22.5 | 138.1 ± 15.4* | 145.6 ± 14.9* | 124.3 ± 15.8* | 127.1 ± 14.5* |
| Liraglutide | 226.3 ± 14.6 | 233.4 ± 15.7 | 182.3 ± 18.0* | 194.4 ± 19.8 | 170.2 ± 21.1* | 200.0 ± 15.9# |
P < .05 vs baseline.
P < .05 vs dual agonist.
Circulating levels of insulin in obese, diabetic and insulin‐resistant monkeys were similar during the run‐in phase and increased during feeding times to approximately 600 μU/mL. No further increases were seen upon treatment with liraglutide or with the monkey‐specific dual GLP‐1R/GCGR agonist. Upon chronic treatment, insulin secretion strongly decreased in the liraglutide‐treated NHP's and a further decrease was apparent in the dual agonist‐treated NHP's, probably reflecting improved insulin sensitivity.
PK analysis on day 43 showed that the 40 μg/kg daily dose of liraglutide translated into ~6‐fold higher compound levels than the 4 μg/kg daily dose of the monkey‐specific dual agonist (Cmax of 279 ± 51 ng/mL vs 43 ± 7 ng/mL, respectively) (Table 3). Circulating levels of liraglutide in the obese monkeys are therefore somewhat higher than the levels reported for liraglutide 3 mg in human clinical studies (Liraglutide 3.0 mg for Weight Management, NDA 206‐321, FDA Briefing Document), underlining the relevance of the chosen doses for liraglutide.
Table 3.
Plasma drug concentration (ng/mL) on day 43/44 after administration of 4 μg/kg s.c. of monkey‐specific dual GLP‐1R/GCGR agonist and 40 μg/kg s.c. of liraglutide
| Hours post dosing | ||||||
|---|---|---|---|---|---|---|
| Compounds tested | 0 | 1 | 2 | 4 | 8 | 22 |
| Plasma drug concentration [ng / mL] | ||||||
| Monkey dual agonist | 10.6 ± 3.89 | 27.6 ± 5.48 | 37.5 ± 6.73 | 43.2 ± 6.95 | 37.7 ± 5.82 | 14.3 ± 3.89 |
| Liraglutide | 87.8 ± 17.2 | 145.0 ± 24.6 | 206.0 ± 36.7 | 279.0 ± 51.0 | 184.0 ± 31.4 | 93.7 ± 19.9 |
Values are are given as mean ± SEM, n = 8‐10/group.
For the investigation of surrogate biomarkers for GCGR activity in monkeys, ketone bodies and FGF21 plasma protein were measured (Figure S3 in Appendix S1). Individual total ketone bodies showed an increase in dual agonist‐treated monkeys from 37.2 to 85.2 μmol/L during baseline measurement to >200 μmol/L on day +10, which was the last escalation step of dose‐ramping. Values were significantly elevated compared to the vehicle group, varying from 32.9 to 84.9 μmol/L at day +10, or liraglutide‐treatment, varying from 64.1 to 124.1 μmol/L. This could be attributed to the reduced food intake of two monkeys, with less than 75 Kcal/day in the dual agonist group, which subsequently led to fasting‐induced elevation of total ketone bodies. However, after 43 days of daily treatment, no differences could be detected. Also, FGF21 levels showed a trend toward elevated levels on day +10, as well as at the end of chronic treatment. While values varied between 300 and 1100 pg/mL over 24 hours in the vehicle‐treated group, maximal levels of FGF21 for liraglutide‐treated monkeys were in the range of 1300 pg/mL and, for the monkey‐specific dual GLP‐1R/GCGR agonist‐treatment, approximately 2100 pg/mL.
The impact of treatment on β‐cell function and insulin secretion was tested by ivGTT after overnight fasting of the monkeys. Baseline values were measured on day −32/−31, the impact of chronic treatment was investigated on days +46/+47. Because of the diabetic conditions of the monkeys, the t = 0 minute glucose values were in the hyperglycaemic range, and insulin values were elevated above 100 μU/mL (Figure 9). The ivGTT data of vehicle‐treated monkeys were stable during the study, concerning both glucose and insulin. In the compound‐treated groups there were obvious differences between baseline and treatment tests for glucose, as well as for insulin measurements. In all compound‐treated groups, first‐phase insulin secretion was restored, and an early insulin peak was observed after i.v. glucose challenge. The most obvious results were obtained for the dual GLP‐1R/GCGR agonist (Figure 9). Glucose excursion was significantly decreased (AUC glucose, −26.6% ± 9.0% mean ± SEM; P < .05). Insulin secretion revealed a highly variable, but not statistically relevant, increase (individual AUC insulin varying from −39.6% to >26 000%). Glucose excursion of liraglutide‐treated monkeys showed a slight, but statistically non‐significant, decrease (AUC glucose, −10.3% ± 2.4%) and, again, a highly variable, but not significant, elevation in insulin secretion (individual AUC insulin varying from −58.0% to >560%) (Figure 9).
Figure 9.
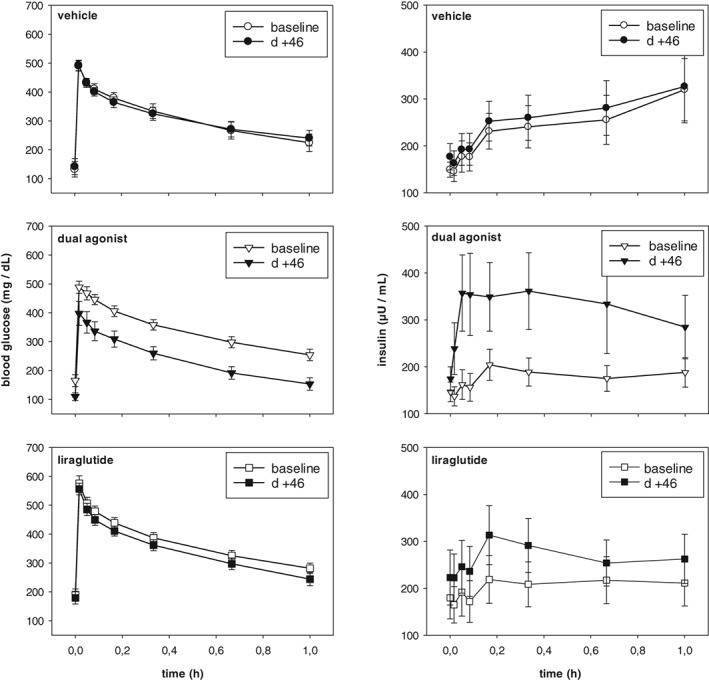
Impact of chronic treatment with monkey‐specific dual agonist on glucose control. Intravenous Glucose Tolerance Test (ivGTT) on day 46 of chronic treatment compared to baseline values in overnight fasted obese and diabetic monkeys. Top: vehicle‐treated obese controls; middle: monkey‐specific dual GLP‐1R/GCGR agonist; bottom: liraglutide. Compounds/vehicle were administered 2.5 hours prior to the glucose challenge. Values are given as mean ± SEM, n = 8‐10/group
On day −25%, HbA1c measurements were performed to obtain baseline values and also on day +43 following chronic treatment. While % HbA1c in the vehicle‐treated group slightly deteriorated during the study period (increase from 6.4% ± 0.5% to 6.9% ± 0.5%) and liraglutide treatment only moderately improved % HbA1c (8.3% ± 0.6% at baseline to 7.8% ± 0.6%), a significant decrease and, therefore, improvement in % HbA1c was observed in NHPs treated with the monkey‐specific dual GLP‐1R/GCGR agonist (Figure 10). The baseline value of 8.0% ± 0.7% decreased to 6.1% ± 0.4% on day +43. For estimating long‐term effects of treatment with dual agonists, we measured aspartate aminotransferase (AST) as a liver‐specific biomarker and alanine aminotransferase (ALT) as an indicator of liver damage on day 32 of chronic treatment in DIO mice and during the run‐out period on day 43 in the obese and diabetic monkey study (Table SA). No statistically relevant change, but rather a decrease, was monitored for AST, while ALT was significantly reduced in DIO mice treated with the mouse‐specific dual agonist. No impact of treatment was monitored in diabetic and obese monkeys.
Figure 10.
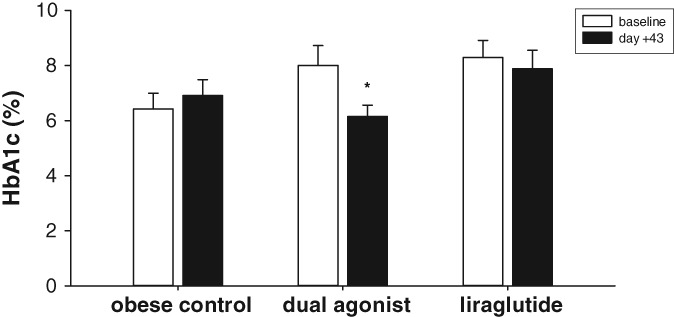
HbA1c determination after 43 days of chronic treatment compared to baseline. Values are given as mean ± SEM, n = 8‐10/group. *P < .05 vs baseline
4. DISCUSSION
Oxyntomodulin was originally characterized as a bioactive enteroglucagon,31 with activity at the glucagon receptor (GCGR), but was subsequently demonstrated to also have insulinotropic activity through activation of the GLP‐1 receptor (GLP‐1R).32 Oxyntomodulin is generated by intestinal L‐cells, as well as by brainstem neurons, from posttranslational processing of the preproglucagon peptide.33 Subsequent clinical studies of oxyntomodulin demonstrated significant efficacy concerning weight loss, resulting from the combined reduction of energy intake and increase of energy expenditure.11, 16 As natural gastrointestinal hormones such as GLP‐1, glucagon or oxyntomodulin are rapidly degraded by serum proteases and are renally cleared, a number of stabilized oxyntomodulin analogs,10, 28, 34 as well as GLP‐1R/GCGR dual agonists based on glucagon,8, 12, 14, 35, 36, 37 have been described as therapeutic agents for the management of obesity and associated metabolic disorders such as type 2 diabetes. However, the apparent challenge associated with translating the beneficial weight loss associated with a dual GLP‐1R/GCGR agonist is to curb the potential glucose‐elevating effects associated with excess glucagon pharmacology. The challenge for dual GLP‐1R/GCGR agonists is to establish the optimal ratio of GLP‐1R activation and GCGR agonism adequate to trigger increased energy expenditure without unwanted activation of counter‐regulatory responses that lead to continuous elevation of plasma glucose. This would place a greater burden on already stressed β‐cells and will probably lead to detrimental exhaustion of the β‐cells following chronic administration.38
Day and colleagues39 recommended that the relative ratio of GLP‐1R and GCGR dual agonism must be chosen carefully for each species. A rather GLP‐1R‐biased agonistic ratio leads to weight loss and glucose lowering. On the other hand, extensive enhancement of the relative GCGR agonistic potency yielded greater weight loss but at the expense of considerable glucose elevation.39
GCGR agonism is subject to considerable species variation. To assure optimal translational predictability we have designed several species‐specific dual GLP‐1R/GCGR agonists based on exendin‐4, which carry a fatty acid side chain for half‐life elongation. They are characterized by somewhat higher agonist activity on the GLP‐1R compared to the GCGR to ensure proper glucose control. The data presented here have been generated with mouse‐ and monkey‐specific dual GLP‐1R/GCGR agonists, which share pharmacological properties with SAR425899, the clinical GLP‐1R/GCGR candidate (manuscript in preparation).
As a selective GLP‐1R agonist comparator, we chose the current market leader in the class liraglutide. It has a profile similar to the studied dual agonists, in terms of GLP‐1R activity as well as half‐life and exposure in the studied species.22 The current studies were thus designed to investigate key metabolic end points, as well as an array of biomarkers of potential specificity for receptor activation following acute treatment or over 30 days of multiple dosing with potent dual GLP‐1R/GCGR agonists. The investigations performed with GLP‐1R(−/−) mice clearly showed that the mouse‐specific agonist is a potent dual acting agonist, elevating blood glucose in case of invalid GLP‐1 receptor response. Therefore, the results are in line with data published by Lynch and colleagues,35 showing that the glucose homeostasis and insulin‐releasing action of a dual GLP‐1R/GCGR agonist is dependent on GLP‐1R activation.
4.1. Contribution of energy expenditure to beneficial body weight control
An increase in energy expenditure leading to a beneficial impact on reduction in body weight in humans remains controversial. We performed indirect calorimetry studies, illustrating a switch of substrate preference during chronic treatment with dual GLP‐1R/GCGR agonists, starting with preferred fat oxidation and gradually increasing carbohydrate oxidation. The incidence of increased oxygen consumption and energy expenditure during chronic treatment of DIO mice with dual GLP‐1R/GCGR agonists is not novel and has been shown in previous studies.8, 35 While Day and colleagues8 observed a tendency toward a decreased respiratory exchange ratio (RER or RQ, respiratory quotient), Lynch et al. claimed that RER remained unchanged.35 Our results confirmed the latter observations during chronic treatment. While vehicle‐treated mice showed an RER at 0.78 ± 0.003, which reflected the high‐fat diet and preferred fat oxidation, the dual agonist‐treated mice did not differ from that, with an RER at 0.79 ± 0.002. However, as the elevated fat oxidation should decrease RER further, the increased carbohydrate oxidation apparently compensates for the expected decrease in the case of a dual mouse‐specific agonist. The increased energy expenditure could be linked, only in part, to increased locomotor activity,35, 40 but others did not report an elevation in spontaneous physical activity.8 However, our results can provide further clarification by matching the increased energy expenditure to the additional carbohydrate oxidation obtained from GCGR‐stimulated carbohydrate release from hepatic storage.
Co‐infusion of native glucagon and GLP‐1 revealed that beneficial effects on food consumption, body weight and glucose control could be achieved, compared to treatment with mono‐agonists.17, 18 It was further shown that resting energy expenditure was not affected by GLP‐1 infusion alone, but rose significantly when human volunteers were treated with glucagon alone and, to a similar extent, in combination with GLP‐1. The increase of REE in both liraglutide‐ and dual GLP‐1R/GCGR agonist‐treated mice in the current study presumably implies a higher relative metabolic contribution of lean mass and metabolically active organs as well as increased heat loss through a relatively greater surface area. Whether this is specific to mouse metabolism or could be translated into human metabolism will be a future topic for investigation.
Dakin and colleagues15 reported that repeated twice‐daily intracerebroventricular (ICV) administration of oxyntomodulin for 7 days caused an inhibition of food intake in rats. Treated animals gained less weight than controls, but exhibited increased core body temperature, suggesting enhanced energy expenditure. This hyperthermic reaction was maintained throughout the entire study period. A concomitant pair‐fed group exhibited a significant hypothermic response compared to saline‐injected ad libitum‐fed controls. Our measurements, covering the acute impact of treatment up to chronic effects with switching metabolism, confirm these data. As a result of continued GCGR stimulation, the mouse‐specific acute adaptation of metabolism for energy saving and homeostasis changes to hypermetabolism, leading to a negative energy balance and greater body weight loss. Baggio and colleagues41 underlined the notion that anorectic effects by acute oxyntomodulin ICV infusion were preserved in GCGR−/− mice but abolished in GLP‐1R−/− mice, while oxygen consumption was reduced acutely by exendin‐4 but not by oxyntomodulin in wild‐type mice. In humans, a four‐hour continuous infusion of potent GLP‐1R‐ or GCGR‐activating peptides with short half‐lives, mimicking an acute treatment, could not confirm additional effects on resting energy expenditure.42 Thus far, no published reports indicate the potential of measuring energy expenditure in non‐human primates, and it therefore remains hypothetical whether a dual agonism of GLP‐1R and GCGR also elevates REE or TEE in NHPs. Nevertheless, it has been unequivocally shown that oxyntomodulin, the endogenous dual GLP‐1R/GCGR agonist, reduces energy intake and body weight in overweight and obese human subjects. A pre‐prandial 3‐times daily injection of oxyntomodulin over a four‐day period increased energy expenditure while reducing energy intake.15 Also, co‐administration of a GLP‐1 agonist with a glucagon infusion resulted in an increase of energy expenditure in humans.16 But these human studies infused GCG for only 45 to 210 minutes. A recently published study investigated whether sustained hyperglucagonaemia affects energy expenditure and hepatic glucose production in humans.43 While no difference in RER, carbohydrate or fat oxidation could be measured, GCG‐induced stimulation of energy expenditure was sustained for a minimum of 12 hours. However, the extent of increase in response to the dose of GCG used was of only marginal significance, and was of longer duration when insulin secretion was inhibited by somatostatin. Serum FGF21 levels were suppressed even during GCG infusion, indicating that circulating FGF21 was not involved in the increased energy expenditure. Our findings in diabetic and obese monkeys support this result and are in conflict with previous studies,44, 45 indicating that GCG administration significantly increased plasma FGF21 concentration and that glucagon is a required factor for FGF21 synthesis.46 However, whether the physiological switch of endogenous nutrient utilization observed in mice translates to humans, and whether this correlates with the circulating FGF21 level, remains an open question and the possibility cannot be excluded.
Daily administration of the monkey‐specific dual GLP‐1R/GCGR agonist at a 10‐fold lower dose compared to the selective GLP‐1R agonist liraglutide (4 vs 40 μg/kg, s.c.) in the present obese NHP study caused greater body weight loss. Body weight reduction was greater despite a 6‐fold lower exposure of the dual agonist at the highest dose tested. The findings demonstrate that liraglutide must be administered at a much higher dose to be equi‐effective to the monkey‐specific dual GLP‐1/GCGR agonist.
Henderson and colleagues14 have previously reported body weight loss in cynomolgus monkeys with the dual GLP‐1/GCGR agonist MEDI0382. However, the studies were conducted in lean and healthy monkeys as part of toxicological testing with MEDI0382 administered s.c. at doses well above the pharmacological range. In this manuscript we report for the first time the translatability of dual GLP‐1R/GCGR pharmacology from DIO mice to obese, diabetic and insulin resistant DIO monkeys following acute and chronic dosing. Our results are in accordance with a preliminary meeting report from Lao and colleagues13 that showed superior pharmacological properties of a dual GLP‐1R/GCGR agonist as compared to GLP‐1R monoagonist liraglutide. We observed a diminishing impact of a dual GLP‐1R/GCGR agonist on TEI reduction in monkeys similar to that observed in mice, while body weight loss continued. Glycaemic control was also assessed in the monkeys. We observed a decrease in fed plasma glucose values that translated into a 1.86% reduction in HbA1c compared to baseline at the end of the study (liraglutide, −0.41%). Nevertheless, a reliable biomarker demonstrating GCGR activity could not be identified; gluconeogenetic liver genes as PCK1 or G6Pase did not change in comparison to liraglutide treatment (data not shown). Also, hepatic FGF21 gene expression was not different during chronic treatment with the monkey surrogate. As elevated total ketones, most likely reflecting the acute impact of the last dose‐escalation step on day 10, disappeared completely on day 43 of chronic treatment with a high maintenance dose, it does not seem reasonable to use it as a potential predictor of GCGR stimulation (Figures S2 and S3 in Appendix S1). Rather, plasma levels of FGF21 and ketone bodies indicate a change in fuel utilization. To what extent these variables reflect specific GCGR target engagement or are, rather, a consequence of an acute negative energy balance is at present unclear. Therefore, in the absence of available data concerning energy expenditure, the search for a specific GCGR‐induced biomarker in mice and non‐human primates continues.
Our results from the DIO mice studies on body weight with a mouse‐specific dual GLP‐1R/GCGR agonist do not result exclusively from reduced food intake. We propose that our observations regarding a mechanism‐related switch of substrate oxidation can be of further therapeutic benefit, and might explain the superior effect of dual GLP‐1R/GCGR agonists for body weight and glucose control in obese and diabetic patients.
Supporting information
Appendix S1. Supporting information files.
ACKNOWLEDGMENTS
We are grateful to Ferenc Levai and Katrin Schröter for providing the kinetic profiles of compounds used from monkey plasma samples. We further appreciate the great support and performance of the monkey study at KBI, China, with special thanks to Tony Wang, Rosario M. Perez, Paul Higgins and Shaohui (Jason) Ji. We thank Anish A. Konkar for reading the manuscript and providing valuable comments.
Conflict of interest
All authors were employed by Sanofi at the time the study was performed. Our employer manufactures and markets, among others, pharmaceuticals related to the treatment of diabetes. Some authors hold stock of Sanofi.
Author contributions
RE, AH, TW, MB and BZ contributed substantially to the concept and design of the non‐human primate study. RE, PW, JW, UB and SK contributed to the design and data evaluation of the mouse study. RE, MB, AK, AE and PJL drafted the manuscript. All authors approved the version to be published.
Elvert R, Herling AW, Bossart M, et al. Running on mixed fuel‐dual agonistic approach of GLP‐1 and GCG receptors leads to beneficial impact on body weight and blood glucose control: A comparative study between mice and non‐human primates. Diabetes Obes Metab. 2018;20:1836–1851. 10.1111/dom.13212
Funding information There was no external funding source for this study.
This article includes a video abstract available at: https://vimeo.com/269278603
REFERENCES
- 1. Ritter S, Weatherford SC, Stone SL. Glucagon‐induced inhibition of feeding is impaired by hepatic portal alloxan injection. Am J Physiol. 1986;250:R682‐R690. [DOI] [PubMed] [Google Scholar]
- 2. Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573‐576. [DOI] [PubMed] [Google Scholar]
- 3. Quesada I, Tuduri E, Ripoll C, Nadal A. Physiology of the pancreatic α‐cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199:5‐19. [DOI] [PubMed] [Google Scholar]
- 4. Christensen M, Bagger JI, Vilsboll T, Knop FK. The alpha‐cell as target for type 2 diabetes therapy. Rev Diabet Stud. 2011;8:369‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones BJ, Tan T, Bloom SR. Minireview: glucagon in stress and energy homeostasis. Endocrinology. 2012;153:1049‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mighiu PI, Yue JTY, Filippi BM, et al. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med. 2013;19:766‐772. [DOI] [PubMed] [Google Scholar]
- 7. Pocai A. Unraveling Oxyntomodulin, GLP1’s enigmatic brother. J Endocrinol. 2012;15:335‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Day JW, Ottaway N, Patterson JT, et al. A new glucagon and GLP‐1 co‐agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749‐757. [DOI] [PubMed] [Google Scholar]
- 9. Kosinski JR, Huber J, Carrington PE, et al. The glucagon receptor is involved in mediating the body weight‐lowering effects of oxyntomodulin. Obesity. 2012;20:1566‐1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pocai A, Carrington PE, Adams JR, et al. Glucagon‐like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wynne K, Park AJ, Small CJ, et al. Subcutaneous Oxyntomodulin reduces body weight in overweight and obese subjects. Diabetes. 2005;54:2390‐2395. [DOI] [PubMed] [Google Scholar]
- 12. Tschöp M, Finan B, Clemmensen C, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24:51‐62. [DOI] [PubMed] [Google Scholar]
- 13. Lao J, Hansen BC, DiMarchi R, et al. Effect of GLP1R/GCGR dual agonist in monkeys. Diabetes. 2013;62(suppl 1):A257. [Google Scholar]
- 14. Henderson SJ, Konkar A, Hornigold DC, et al. Robust anti‐obesity and metabolic effect of a dual GLP‐1/glucagon receptor peptide agonist in rodents and non‐human primates. Diabetes Obes Metab. 2016;18:1176‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dakin CL, Small CJ, Park AJ, et al. Repeated ICV administration of oxyntomodulin causes a greater reduction in body weight gain than in pair‐fed rats. Am J Physiol Endocrinol Metab. 2002;283:E1173‐E1177. [DOI] [PubMed] [Google Scholar]
- 16. Wynne K, Park AJ, Small CJ, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes. 2006;30:1729‐1736. [DOI] [PubMed] [Google Scholar]
- 17. Tan TM, Tield BCT, McCullough A, et al. Coadministration of glucagon‐like Peptide‐1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62:1131‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cegla J, Troke RC, Jones B, et al. Coinfusion of low‐dose GLP‐1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63:3711‐3720. [DOI] [PubMed] [Google Scholar]
- 19. Nair KS. Hyperglcagonemia increases resting metabolic rate in man during insulin deficiency. J Clin Endocrinol Metab. 1987;64:896‐901. [DOI] [PubMed] [Google Scholar]
- 20. Heppner KM, Habegger KM, Day J, et al. Glucagon regulation of energy metabolism. Physiol Behav. 2010;100:545‐548. [DOI] [PubMed] [Google Scholar]
- 21. Salem V, Izzi‐Engbeaya C, Coello C, et al. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes Metab. 2016;18:72‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evers A, Haack T, Lorenz M, et al. Design of novel exendin‐based dual GLP‐1 / glucagon receptor agonists. J Med Chem. 2017;60:4293‐4303. [DOI] [PubMed] [Google Scholar]
- 23. Scrocchi LA, Brown TJ, MacLusky N, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon‐like peptide 1 receptor gene. Nat Med. 1996;2:1254‐1258. [DOI] [PubMed] [Google Scholar]
- 24. Herling A, Kilp S, Elvert R, et al. Increased energy expenditure contributes more to the body weight‐reducing effect of rimonabant than reduced food intake in candy‐fed wistar rats. Endocrinology. 2008;149:2557‐2566. [DOI] [PubMed] [Google Scholar]
- 25. Meyer CWE, Klingenspor M, Rozman J, Heldmaier G. Gene or size: metabolic rate and body temperature in obese growth hormone‐deficient dwarf mice. Obes Res. 2004;12:1509‐1518. [DOI] [PubMed] [Google Scholar]
- 26. Ferrannini E. The theoretical bases of indirect calorimetry: a review. Metabolism. 1988;37:287‐301. [DOI] [PubMed] [Google Scholar]
- 27. Boschmann M, Frenz U, Noack R, Aust L, Murphy CM. Energy metabolism and metabolite patterns of rats after application of dexfenfluramine. Int J Obes Relat Metab Disord. 1994;18:235‐242. [PubMed] [Google Scholar]
- 28. Santoprete A, Capitò E, Carrington PE, et al. DPP‐IV‐resistant, long‐acting oxyntomodulin derivatives. J Pept Sci. 2011;17(4):270‐280. [DOI] [PubMed] [Google Scholar]
- 29. Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon‐like peptide‐1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664‐1669. [DOI] [PubMed] [Google Scholar]
- 30. Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010;9:267‐268. [DOI] [PubMed] [Google Scholar]
- 31. Bataille D, Tatemoto K, Gespach C, Jörnvall H, Rosselin G, Mutt V. Isolation of glucagon‐37 (bioactive enteroglucagon/oxyntomodulin) from porcine jejuno‐ileum. Characterization of the peptide. FEBS Lett. 1982;146:79‐86. [DOI] [PubMed] [Google Scholar]
- 32. Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV. Oxyntomodulin (glicentin‐(33‐69)): pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul Pept. 1988;21:151‐166. [DOI] [PubMed] [Google Scholar]
- 33. Larsen PJ, Tang‐Christensen M, Holst JJ, Orskov C. Distribution of glucagon‐like peptide‐1 and other preproglucagon‐derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77:257‐270. [DOI] [PubMed] [Google Scholar]
- 34. Kerr BD, Flatt PR, Gault VA. A novel chemically modified analogue of Oxyntomodulin with Antihyperglycaemic, Insulinotropic and Anorexigenic actions. Biochem Pharmacol. 2010;80:1727‐1735. [DOI] [PubMed] [Google Scholar]
- 35. Lynch AM, Pathak N, Pathak V, et al. Novel DPP IV‐resistant C‐terminally extended glucagon analogue exhibits weight‐lowering and diabetes‐protective effects in high‐fat‐fed mice mediated through glucagon and GLP‐1 receptor activation. Diabetologia. 2014;57:1927‐1936. [DOI] [PubMed] [Google Scholar]
- 36. O'Harte FPM, Ng MT, Lynch AM, Conlon JM, Flatt PR. Novel dual agonist peptide analogues derived from dogfish glucagon show promising in vitro insulin releasing actions and antihyperglycaemic activity in mice. Mol Cell Endocrinol. 2016;431:133‐144. [DOI] [PubMed] [Google Scholar]
- 37. O'Harte FPM, Ng MT, Lynch AM, Conlon JM, Flatt PR. Dogfish glucagon analogues conter hyperglycaemia and enhance both insulin secretion and action in diet‐induced obese diabetic mice. Diabetes Obes Metab. 2016;18:1013‐1024. [DOI] [PubMed] [Google Scholar]
- 38. Meier JJ, Bonadonna RC. Role of reduced β‐cell mass versus impaired β‐cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36(suppl 2):S113‐S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Day JW, Gelfanov V, Smiley D, et al. Optimization of co‐agonism at GLP‐1 and glucagon receptors to safely maximize weight reduction in DIO‐rodents. Pept Sci. 2012;98:443‐450. [DOI] [PubMed] [Google Scholar]
- 40. Gault VA, Bhat VK, Irwin N, Flatt PR. A novel glucagon‐like peptide‐1 (GLP‐1)/glucagon hybrid peptide with triple‐acting agonist activity at glucose‐dependent insulinotropic polypeptide, GLP‐1, and glucagon receptors and therapeutic potential in high fat‐fed mice. J Biol Chem. 2013;288:35581‐35591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baggio LL, Huang Q, Brown TJ, Drucker DJ. Oxyntomodulin and glucagon‐like peptide‐1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127:546‐558. [DOI] [PubMed] [Google Scholar]
- 42. Bagger JI, Holst JJ, Harmann B, Andersen B, Knop FK, Vilsbøll T. Effect of Oxyntomodulin, glucagon, GLP‐1 and combined glucagon + GLP‐1 infusion on food intake, appetite and resting energy expenditure. J Clin Endocrinol Metab. 2015;100:4541‐4552. [DOI] [PubMed] [Google Scholar]
- 43. Chakravarthy M, Parsons S, Lassman ME, et al. Effects of 13‐hour hyperglucagonemia on energy expenditure and hepatic glucose production in humans. Diabetes. 2017;66:36‐44. [DOI] [PubMed] [Google Scholar]
- 44. Arafat AM, Kaczmarek P, Skrzypski M, et al. Glucagon increases circulating fibroblast growth factor 21 independently of endogenous insulin levels: a novel mechanism of glucagon‐stimulated lipolysis? Diabetologia. 2013;56:588‐597. [DOI] [PubMed] [Google Scholar]
- 45. Habegger KM, Stemmer K, Cheng C, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62:1453‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Müller TD, Finan B, Clemmensen C, RD DM, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev. 2017;97:721‐766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information files.