

Abstract
Diamond–Blackfan anemia (DBA) is a rare genetic hypoplasia of erythroid progenitors characterized by mild to severe anemia and associated with congenital malformations. Clinical manifestations in DBA patients are quite variable and genetic testing has become a critical factor in establishing a diagnosis of DBA. The majority of DBA cases are due to heterozygous loss‐of‐function mutations in ribosomal protein (RP) genes. Causative mutations are fairly straightforward to identify in the case of large deletions and frameshift and nonsense mutations found early in a protein coding sequence, but diagnosis becomes more challenging in the case of missense mutations and small in‐frame indels. Our group recently characterized the phenotype of lymphoblastoid cell lines established from DBA patients with pathogenic lesions in RPS19 and observed that defective pre‐rRNA processing, a hallmark of the disease, was rescued by lentiviral vectors expressing wild‐type RPS19. Here, we use this complementation assay to determine whether RPS19 variants of unknown significance are capable of rescuing pre‐rRNA processing defects in these lymphoblastoid cells as a means of assessing the effects of these sequence changes on the function of the RPS19 protein. This approach will be useful in differentiating pathogenic mutations from benign polymorphisms in identifying causative genes in DBA patients.
Keywords: Diamond–Blackfan anemia, functional assay, ribosomal protein, RPS19, VUS
1. INTRODUCTION
Diamond–Blackfan anemia (DBA) is a congenital disorder of the bone marrow characterized by normochromic macrocytic anemia and associated with physical malformations and increased risk of malignancies (Lipton & Ellis, 2009; Vlachos et al., 2008). The penetrance is incomplete and a wide range of clinical manifestations may occur even among affected members of the same family. First‐line therapy is steroid treatment; options in steroid‐resistant patients are chronic transfusions or hematopoietic stem cell transplantation. DBA is usually caused by heterozygous mutations in ribosomal protein (RP) genes that lead to haploinsufficiency. To date, mutations in 19 RP genes (RPS19, RPS24, RPS17, RPL35A, RPL5, RPL11, RPS7, RPS10, RPS26, RPL26, RPL15, RPL31, RPS29, RPS28, RPL27, RPS27, RPS15A, RPL35, RPL18) have been identified in DBA patients (Cmejla, Cmejlova, Handrkova, Petrak, & Pospisilova, 2007; Doherty et al., 2010; Draptchinskaia et al., 1999; Farrar et al., 2008, 2014; Gazda et al., 2006, 2008, 2012; Gripp et al., 2014; Ikeda et al., 2017; Landowski et al., 2013; Mirabello et al., 2014, 2017; Wang et al., 2015). Rare mutations in GATA1, that abrogate the production of the full‐length protein (Parrella et al., 2014; Sankaran et al., 2012), and in TSR2, encoding a RPS26 interactor (Gripp et al., 2014), have also been described.
In DBA, the deficiency of a RP leads to the reduction in the number of ribosomes and this is particularly harmful for the red cell progenitors. Ribosome biogenesis is a complex process that requires the involvement of hundreds of different structural and accessory molecules. Mature 18S ribosomal RNA (rRNA), which forms the 40S subunit, together with mature 28S and 5.8S rRNAs, which are components of the 60S subunit, are all produced by sequential nucleolytic cleavages of a large polycistronic 45S precursor. Mutations in RPs of the small (RPS) or large (RPL) ribosomal subunit affect various steps of pre‐rRNA maturation, resulting in the impairment of ribosome biogenesis and function. Since the alterations of pre‐rRNA processing cause the accumulation of specific rRNA precursors depending on the mutated RP gene (Boria et al., 2010; Farrar et al., 2014; Flygare et al., 2007), pre‐rRNA analysis has been proposed as a potential aid for making a DBA diagnosis (Farrar et al., 2014; Quarello et al., 2016). RPS19 haploinsufficiency affects the maturation of 40S ribosomal subunits by specifically affecting the conversion of 21S pre‐rRNA into 18SE pre‐rRNA (Choesmel et al., 2007; Flygare et al., 2007), thus leading to accumulation of 21S pre‐rRNA and increase of 21S/18SE ratio.
While many of the mutations found in RP genes are nonsense, frameshift, and splice site mutations that can be easily interpreted as pathogenic based on their predicted effects on the expression of a protein, the pathogenicity of missense variants remains often controversial. Over 25% of all DBA patients’ mutations are found in RPS19 and more than 40 different missense mutations and small in‐frame indels have been described in this gene (Boria et al., 2010; Konno et al., 2010; Ozono et al., 2016; Smetanina et al., 2015; Wang et al., 2015). Their pathogenic significance is often difficult to evaluate and may present an obstacle to genetic testing of the proband as well as of family members who are silent carriers of the disease. This is particularly relevant when trying to identify a suitable donor for hematopoietic stem cell transplantation. To be able to counsel patients in these families, it is necessary to fully understand the role played by these variants of unknown significance (VUS) on protein expression and function.
In silico tools can aid in the interpretation of VUS and rely on the following criteria: (i) absence or very low frequency of the variant in the general population, (ii) change of an evolutionary conserved codon, (iii) non‐conservative amino acid substitution, (iv) cosegregation of the variant with the disease phenotype in the family under study (Richards et al., 2015). However, it is not recommended to use these predictions as the sole source of evidence to reach a diagnostic conclusion, and functional studies should support the in silico results. A VUS in RPS19 can be classified as benign when the analysis of rRNA maturation in patient cells points to defects in a different gene. On the contrary, the observation of pre‐rRNA processing alterations consistent with RPS19 loss of function, is not sufficient to interpret the VUS as pathogenic, since the patient could simultaneously carry a disease‐causing mutation, responsible for the defective rRNA processing, in another RP gene.
We recently characterized the phenotype of lymphoblastoid cell lines (LCLs) established from DBA patients with loss‐of‐function mutations in RPS19 (Aspesi et al., 2017). Aberrant pre‐rRNA processing and other pathological features were rescued by gene complementation, using an RPS19 transgene carried by a lentiviral vector (Aspesi et al., 2017). We reasoned that this complementation assay could be employed to investigate the effects of a wide range of VUS on RPS19 function (Figure 1). Here, we reviewed the literature regarding RPS19 mutations and selected a total of 12 RPS19 variants for functional analysis.
Figure 1.
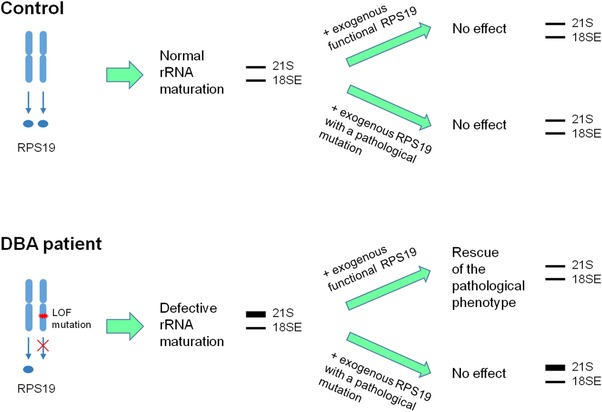
Scheme of the complementation assay. Cells from DBA patients have a loss‐of‐function mutation in RPS19 that cause the accumulation of 21S rRNA. Expression of either wild‐type RPS19 or RPS19 with a benign sequence variant allows the rescue of the rRNA processing defect. On the contrary, expression of a RPS19 transgene carrying a deleterious mutation does not recover the pathological phenotype
2. MATERIALS AND METHODS
2.1. Cells
LCLs were established as described previously from two DBA patients (P1 and P2) and one healthy subject (C), after approval of the local ethics committee (Aspesi et al., 2017). Both patients had a heterozygous loss‐of‐function mutation in RPS19 and their genotypes were: P1: c.283_284delG, p.Gly95Alafs*16; P2: c.36_37insAG, p.Glu13Argfs*17 (RefSeq NG_007080.3, NM_001022.3, NP_001013.1). Cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) and were incubated at 37°C in a humidified atmosphere with 5% CO2.
2.2. Selection of variants for the complementation assay
From the list of 165 RPS19 mutations described in the literature, we excluded protein‐truncating variants, that is, variants predicted to lead to nonsense‐mediated decay (NMD) and expected to have severe effects on gene function, as well as variants that disrupt a canonical splice site. For the present analysis, we considered only VUS: missense variants, small in‐frame indels, and truncating variants located in the last or penultimate exon. Stop codons located in the penultimate exon less than 50–55 bases from the final intron are not supposed to cause NMD (Le Hir, Izaurralde, Maquat, & Moore, 2000), thus the three nonsense variants in our list (c.376C>T p.Gln126*, c.382C>T p.Gln128*, c.406G>T p.Gly136*) could theoretically produce truncated proteins. We chose to select for the complementation assay only the variant located closest to the 3′ end of the transcript, c.406G>T p.Gly136*.
The novel variant c.338_340delTGG p.Val113del was submitted to the DBA database LOVD v.2.0 Build 36 (Boria et al., 2008, 2010).
To predict the functional consequences of variants, we used the following in silico prediction tools: SIFT v.1.03, Polyphen‐2 (Polymorphism Phenotyping v2), Provean v1.1.3, Condel 2.0, Mutation Assessor release 3, and MutationTaster 2. Mutation Assessor evaluates the probability that a mutation affects protein function, therefore the output “low” indicates a neutral variant. For Condel, the score ranges from 0 (neutral) to 1 (damaging); all variants we studied had an output between 0.5 and 0.9 and we considered them “probably damaging.” To address the potential effects on gene splicing, we used GeneSplicer, Human Splicing Finder version 3.1, MaxEntScan, NetGene2 version 2.42, NNSplice version 0.9, and FSPLICE. The presence and frequency of VUS selected for the complementation assay were evaluated in population databases: 1000 Genomes, Genome Aggregation Database (GnomAD), Exome Variant Server (EVS), and Exome Aggregation Consortium (ExAC).
2.3. Websites
DBA database, https://www.dbagenes.unito.it/home.php
SIFT, https://sift.jcvi.org
Polyphen‐2, https://genetics.bwh.harvard.edu/pph2
Provean, https://provean.jcvi.org/index.php
Mutation Assessor, https://mutationassessor.org
MutationTaster, https://www.mutationtaster.org
GeneSplicer, https://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml
Human Splicing Finder, https://www.umd.be/HSF
MaxEntScan, https://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
NetGene2, https://www.cbs.dtu.dk/services/NetGene2
FSPLICE, https://www.softberry.com/berry.phtml?topic=fsplice&group=programs&subgroup=gfind
1000 Genomes, https://www.internationalgenome.org/
2.4. Complementation assay
Site‐directed mutagenesis on RPS19 cDNA was carried out to introduce the selected variants using the QuikChange Site directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Primers are available upon request. The presence of each mutation was confirmed by Sanger sequencing. Lentiviral vectors were produced after transient transfection of 293T cells with the third generation packaging plasmids (pMDLg/pRRE, pRSV‐REV, and pMD2‐VSVG) and with the transfer construct for each RPS19 transgene (Aspesi et al., 2014; Follenzi, Ailles, Bakovic, Geuna, & Naldini, 2000). One control (C) and two RPS19‐haploinsufficient LCLs (P1 and P2) were transduced with 10 multiplicity of infection to express the mutant RPs. Integration and expression of at least one copy of the cassette carrying both RPS19 and green fluorescence protein (GFP) sequences resulted in the emission of green fluorescence. GFP+ cells were sorted, recultured for 2–3 weeks and analyzed. Total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), followed by on‐column DNase treatment and purification with miRNeasy Mini Kit (Qiagen, Milano, Italy). For Northern blot analysis, 5 μg of total RNA was fractionated on 1.5% formaldehyde agarose gels, transferred to a positively charged nylon membrane (Roche, Monza, Italy) and immobilized on the membrane by UV‐crosslinking performed with 120 milliJoules/cm2. The oligonucleotide probe (5′‐CCTCGCCCTCCGGGCTCCGTTAATGATC‐3′) was labeled with [γ‐32P]ATP using T4 polynucleotide kinase and hybridized overnight with the membrane at 37°C in ULTRAHyb‐Oligonucleotide hybridization buffer (Ambion, Thermo Fisher Scientific, Waltham, MA, USA). The membrane was washed at 37°C with 6XSSC and subjected to phosphorimaging analysis (Flygare et al., 2007). The overexpression of RPS19 did not cause adverse effects on pre‐rRNA processing in control cells (Aspesi et al., 2017), nor did the expression of RPS19 mutants (Figure 2A).
Figure 2.
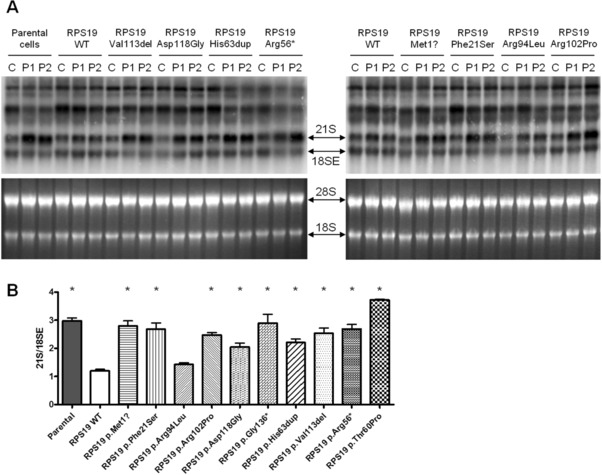
Complementation assay on VUS reported in DBA patients. A: Representative Northern blot experiments. Patient cells have an increased 21S/18SE rRNA ratio, that is corrected by the expression of a wild‐type RPS19 transgene but not by the expression of RPS19 carrying a pathogenic mutation. Upper panels show Northern blotting, lower panels show corresponding RNA gels stained by a fluorescent nucleic acid dye. C: control, P1: patient 1, P2: patient 2. B: Densitometry quantification of 21S/18SE ratio calculated on repeated Northern blot experiments. Asterisks represent statistically significant differences (P < 0.05) between samples with wild‐type and mutant exogenous RPS19. Error bars represent standard error of the mean
2.5. Quantitative RT‐PCR
RNA isolated from cells with wild‐type or mutant exogenous RPS19 was reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Real‐time PCR amplification of cDNA was performed in triplicate using Power SYBR® Green PCR Master Mix (Applied Biosystems) and specific primers for the target genes RPS19 and CDKN1A (p21). ACTB (β‐actin) was used as reference gene.
2.6. Statistical analysis
Northern blot bands were quantified using the ImageJ software. Results from P1 and P2 patient cells were considered as biological replicates. Differences in mean values between samples with either wild‐type or mutant exogenous RPS19 were analyzed with the Mann–Whitney test for two‐tailed data. Statistical significance was defined by a P value ≤0.05.
3. RESULTS
3.1. Selection of variants and in silico analyses
The DBA database (Boria et al., 2008, 2010) included 129 RPS19 mutations in 219 patients at its last update in 2010. We reviewed the literature to collect additional RPS19 mutations identified more recently and now have a total of 165 different RPS19 mutations reported in 313 DBA patients (Arbiv et al., 2017; Chae et al., 2010, 2014; Da Costa et al., 2013; Delaporta et al., 2014; Errichiello et al., 2017; Farrar et al., 2011; Gerrard et al., 2013; Ichimura et al., 2017; Konno et al., 2010; Kuramitsu et al., 2012; Landowski et al., 2013; Ozono et al., 2016; Pospisilova et al., 2012; Quarello et al., 2012; Smetanina et al., 2015; Solomon et al., 2014; Tsangaris et al., 2011; van Dooijeweert et al., 2017; Wang et al., 2015; Zhang et al., 2016). For our study, we selected only those variants for which there was no strong evidence of pathogenicity according to the genetic criteria outlined in Materials and Methods, and obtained 47 VUS reported in 122 patients (39% of RPS19‐mutated patients, approximately 10% of all DBA patients). We also included a new previously unpublished variant we identified in a DBA patient by next‐generation sequencing, c.338_340delTGG p.Val113del (Supp. Table S1). Thirteen of the VUS have already been found to be pathogenic by published functional studies (Angelini et al., 2007; Badhai et al., 2009; Chatr‐Aryamontri et al., 2004; Chae et al., 2014; Choesmel et al., 2007; Cmejlova et al., 2006; Da Costa et al., 2003; Gazda et al., 2004; Hamaguchi et al., 2002; Idol et al., 2007;) (Supp. Table S1) and so were not tested in our study.
The VUS were analyzed by multiple in silico tools to predict the impact of sequence variants on protein function. Five missense variants that were analyzed were considered tolerated/benign by at least one of the six in silico tools indicating some degree of ambiguity as to whether or not they could be the pathogenic lesion in these patients. These were all selected for functional analysis using the complementation assay outlined here (Table 1). We also chose to study a nonsense VUS late in the coding sequence predicted to escape nonsense‐mediated mRNA decay (Le Hir et al., 2000) as well as two small in‐frame indels, whose impact could not be determined by most prediction algorithms (Table 1). As negative controls for the effect of known loss‐of‐function mutations, we chose a nonsense mutation, c.166C>T p.Arg56*, previously reported in four DBA patients (Boria et al., 2010; Proust et al., 2003; Willig et al., 1999) and the missense c.178A>C p.Thr60Pro, reported in one patient and predicted as damaging by every in silico tool we used. None of the variants chosen for functional analysis here were reported in 1000 Genomes, GnomAD, EVS, and ExAC.
Table 1.
VUS in RPS19 identified in DBA patients selected for complementation assay
| DNA change | Protein change | Mutation taster | Mutation assessor | Polyphen‐2 | Provean | SIFT | Condel | Affected patients |
|---|---|---|---|---|---|---|---|---|
| c.1A>G | p.Met1? | Disease causing | na | Probably damaging | Neutral | Damaging | Probably damaging | 5 |
| c.62T>C | p.Phe21Ser | Disease causing | Medium | Probably damaging | Deleterious | Tolerated | Probably damaging | 1 |
| c.187_189insCAC | p.His63dup | Disease causing | na | na | Deleterious | na | na | 1 |
| c.281G>T | p.Arg94Leu | Polymorphism | Medium | Benign | Deleterious | Tolerated | Probably damaging | 1 |
| c.305G>C | p.Arg102Pro | Disease causing | Medium | Benign | Deleterious | Damaging | Probably damaging | 1 |
| c.353A>G | p.Asp118Gly | Disease causing | Low | Benign | Deleterious | Tolerated | Probably damaging | 1 |
| c.406G>T | p.Gly136* | Disease causing | na | na | na | na | na | 1 |
| c.338_340delTGG | p.Val113del | Disease causing | na | na | Deleterious | na | na | 1 |
na, not available. RefSeq: NM_001022.3, NP_001013.1.
Numeric output data are shown in Supp. Table S1. Websites and software versions are reported in the Materials and Methods section.
Finally, we assessed whether the VUS we selected caused the creation or loss of splice sites by in silico tools. These prediction tools have low specificity (∼60%–80%), but quite high sensitivity (∼90%–100%) in predicting splice site abnormalities, and therefore have a low false negative rate (Houdayer et al., 2012; Richards et al., 2015). None of the VUS included in this study were predicted to affect splicing, but the variant c.353A>G that, according to Human Splicing Finder, MaxEntScan, and FSPLICE, could activate a cryptic donor splice site (data not shown).
3.2. Analysis of VUS by complementation assay
Our previous findings showed that transfection of RPS19‐haploinsufficient LCLs with a vector expressing RPS19 cDNA corrected the abnormal accumulation of 21S pre‐rRNA, a well‐established defect in ribosome biogenesis caused by the loss of RPS19 function. Ribosome biogenesis defects such as these have been a hallmark of DBA pathogenesis in patients harboring mutations in RP genes. To assess the function of individual variants selected, we asked whether the variants could rescue the pre‐rRNA processing defect in the complementation assay outlined in Figure 1. Cells expressing the mutant transgenes showed an increased level of RPS19 transcript compared to parental cells, as measured by quantitative RT‐PCR (Supp. Figure S1A); Sanger sequencing performed on cDNA demonstrated the presence of the mutated transcripts (Supp. Figure S1B). Processing of pre‐rRNAs was evaluated by Northern blotting and results obtained by expressing wild‐type or mutant transgenes were compared. Representative experiments are shown in Figure 2A. Patient cells with no exogenous RPS19 (i.e., parental cells) or with the negative control p.Arg56* RPS19 had a mean 21S/18SE rRNA ratio of 2.97 ± 0.21 (standard deviation, SD) and 2.69 ± 0.31, respectively, whereas patient cells expressing the wild‐type transgene had a ratio of 1.20 ± 0.11, similar to the value of cells from healthy donors, that was 1.13 ± 0.13 (Figure 2A and B). Densitometry of Northern blots (Figure 2B) showed statistically significant differences (P < 0.05) between samples with wild‐type and mutant exogenous RPS19, demonstrating that all tested VUS were unable to recover the pathogenic phenotype of RPS19‐deficient cells. The only exception was c.281G>T p.Arg94Leu (mean 21S/18SE ratio 1.43 ± 0.12), which showed no statistical difference from wild‐type RPS19.
According to our data, we propose an arbitrary 21S/18SE cut‐off value ≥2 to define pathogenicity and ≤1.5 to indicate normal protein function. None of the mutants we analyzed showed a 21S/18SE ratio between these two values.
3.3. Analysis of variants found in population databases
Population databases such as 1000 Genomes, GnomAD, EVS, and ExAC were searched for polymorphisms in RPS19 that cause missense substitutions. Overall amino acid changes in RPS19 were extremely rare in population databases. No missense variant was present in 1000 Genomes, which includes data from more than 5,000 healthy subjects. The two most common variants in GnomAD, EVS, and ExAC, were c.68A>G p.Lys23Arg and c.164C>T p.Thr55Met (Table 2). These variants are presumed to be benign since it seems unlikely that individuals included in these databases would have a rare disease like DBA. Interestingly, variants c.68A>G p.Lys23Arg and c.164C>T p.Thr55Met were predicted to be benign only by one out of six and two out of six bioinformatic tools, respectively. Northern blot analysis showed that these variants failed to rescue the pre‐rRNA processing defects in patient lymphoblasts (Figure 3A). The mean 21S/18SE ratios for variants p.Lys23Arg and p.Thr55Met were 2.09 ± 0.13 and 2.01 ± 0.07, respectively, and were significantly different from data obtained by the wild‐type transgene, suggesting that these amino acid substitutions impair, at least partially, protein function (Figure 3A and B).
Table 2.
VUS in RPS19 selected from population databases
| Population database (MAF) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA change | Protein change | 1000 genomes | GnomAD | ExAC | EVS | Mutation taster | Mutation assessor | Polyphen‐2 | Provean | SIFT | Condel |
| c.68A>G | p.Lys23Arg | Not reported | 0.0001292 | 0.0001895 | 0.0002071 |
|
Medium score 2.29 |
|
Deleterious score −2.601 | Damaging score 0.03 | Probably damaging score 0.617 |
| c.164C>T | p.Thr55Met | Not reported | 0.0002261 | 0.0002734 | 0.0002478 |
|
Medium score 2.08 |
|
Neutral score −1.889 | Tolerated score 0.06 | Probably damaging score 0.647 |
MAF, minor allele frequency. RefSeq: NM_001022.3, NP_001013.1.
Mutation Taster: the score for amino acid substitutions reflects the physicochemical difference between the original and the mutated amino acid but does not influence the prediction. Mutation Assessor: the Functional Impact score is reported. PROVEAN: a score equal to or below the predefined threshold (−2.5) predicts a deleterious effect for the protein variant; a score above the threshold indicates that the variant is predicted to have a neutral effect. SIFT: the score predicts whether an amino acid substitution affects protein function, and ranges from 0.0 (deleterious) to 1.0 (tolerated).
Websites and software versions are shown in the Materials and Methods section.
Figure 3.
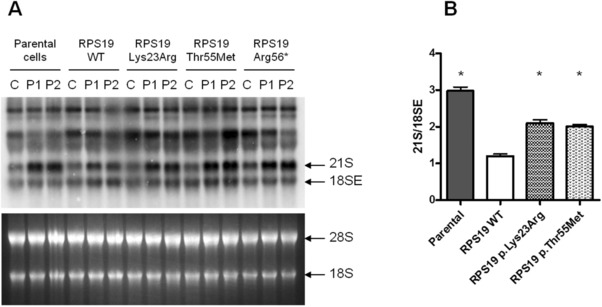
Complementation assay on VUS found in population databases. A: Representative Northern blot experiment. Upper panel shows Northern blot, lower panel shows the corresponding RNA gel stained by a fluorescent nucleic acid dye. C: control, P1: patient 1, P2: patient 2. B: Densitometry quantification of 21S/18SE ratio shows that neither variant could rescue the defective rRNA processing in patients cells. Asterisks represent statistically significant differences (P < 0.05) between samples with wild‐type and mutant exogenous RPS19. Error bars represent standard error of the mean
3.4. Evaluation of p21 transcript level
RPS19 deficiency induces stabilization of p53 and increased level of its target p21; such alterations are recovered by expression of the RPS19 transgene (Aspesi et al., 2017). We performed quantitative RT‐PCR to measure the level of p21 transcript in patient cells expressing the mutant transgenes. The results validated the data obtained by Northern blot analysis, since the expression of RPS19 mutants could not normalize the level of p21, but the expression of mutant p.Arg94Leu led to a clear, though not significant, decrease of p21 (Figure 4). Interestingly, the high p21 levels measured for the VUS p.Lys23Arg and p.Thr55Met corroborate their interpretation as deleterious variants (Figure 4).
Figure 4.
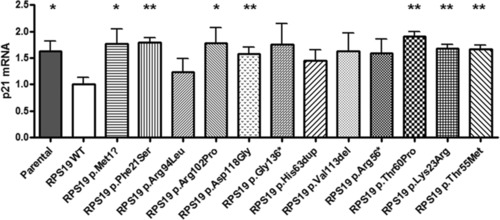
Level of p21 transcript in patient cells expressing wild‐type or mutant RPS19. The increased level of p21 typical of DBA patient cells is corrected by the expression of a wild‐type RPS19 transgene but not by the expression of RPS19 carrying a pathogenic mutation. Results obtained by quantitative RT‐PCR performed on P1 and P2 patient cells were considered biological replicates. Asterisks represent statistically significant differences between samples with wild‐type and mutant exogenous RPS19: *P < 0.05; **P < 0.01. Error bars represent standard error of the mean
4. DISCUSSION
Our work was aimed at creating a complementation assay to assess the function of RPS19 VUS and clarify their involvement in the pathogenesis of DBA. Novel sequence variants in genes known to be involved in DBA have been increasingly detected, but our understanding of the clinical significance of these novel variants is often limited. Many of the most challenging variants change only one amino acid, making it difficult to discern benign variants from pathogenic mutations that disrupt protein function. In silico prediction tools can be of aid in the interpretation of sequence variants but their output should be validated by functional studies.
It is widely accepted that the underlying pathophysiology in the majority of DBA patients is haploinsufficiency for a RPs and an ensuing disruption of ribosome biogenesis. RPS19 is the most frequently mutated gene in DBA and structure/function relationships of known pathogenic mutations have been characterized previously. The crystal structure of RPS19 from the archeon Pyroccocus abyssi was used to group RPS19 missense mutations in two classes: class I included residues essential for the folding and stability of the protein, whereas class II included mutations that affected surface residues and presumably impaired the capacity of RPS19 to engage intermolecular interactions (Gregory et al., 2007). In another study, eleven missense mutations and one trinucleotide insertion were expressed in human HEK293 cells (Angelini et al., 2007). Some mutants, corresponding to class I mutants (p.Val15Phe, p.Leu18Pro, p.Ala57Pro, p.Ala61Glu, p.Gly127Glu, and p.Leu18_Ala19insGlu) were expressed at a barely detectable level, whereas the others (all class II mutants: p.Pro47Leu, p.Trp52Arg, p.Arg56Gln, p.Arg62Gln, p.Arg62Trp, p.Arg101His), although more stable, were not assembled into mature ribosomes.
Additional studies have shown that missense mutations can influence the localization of RPS19 and disrupt function by mislocalization. For example, mutant proteins such as p.Val15Phe, p.Gly127Gln, and p.Leu131Pro could not reach the nucleolus and their expression was dramatically decreased compared with the wild‐type protein (Cretien et al., 2008; Da Costa et al., 2003).
Because of the limitations associated with assessing a protein's function based on sequence analysis alone, there is a significant need to be able to functionally annotate novel sequence variants for their potential involvement in DBA pathogenesis. We have developed a new complementation assay to assess a variant's function based on its ability to rescue a pre‐rRNA processing defect in lymphoblasts derived from patients haploinsufficient for RPS19. This assay is not restricted to RPS19 and could potentially be developed for other known DBA genes.
We functionally tested 12 variants: nine were already described in the literature as mutations responsible for DBA, one was a new variant we identified in a DBA patient and two variants were selected from population databases. Among the variants we analyzed, only the missense c.281G>T p.Arg94Leu successfully rescued the rRNA processing defect in patient cells. This variant was reported as pathogenic after the identification in a female DBA patient whose parents were not tested (Boria et al., 2010). Our results suggest that this variant may actually be benign and further studies should be carried out in this patient to establish the causative gene. The remaining variants c.1A>G p.Met1?, c.62T>C p.Phe21Ser, c.187_189insCAC p.His63dup, c.305G>C p.Arg102Pro, c.353A>G p.Asp118Gly and c.406G>T p.Gly136* were not able to complement RPS19 deficiency. Among the variant failing to complement was the new variant c.338_340delTGG p.Val113del, whose impact was difficult to predict even by bioinformatic algorithms.
The most surprising result from our analysis was that two rare variants selected from population databases, c.68A>G p.Lys23Arg, and c.164C>T p.Thr55Met failed to complement the pre‐rRNA processing defect in patient lymphoblasts. The latter variant was previously observed in a DBA patient who also carried a second variant, p.Val15Phe, on the same allele. In this case, p.Val15Phe was considered pathogenic, whereas p.Thr55Met was interpreted as benign because only protein localization was being studied (Boria et al., 2008; Da Costa et al., 2003).
There is growing evidence that RP mutations can be found in patients with very mild or absent hematologic manifestations, as previously described, for instance, in a family with no sign of DBA where a truncating germline mutation in RPS20 cosegregated with colon cancer (Nieminen et al., 2014). This is also supported by the recent report of two unrelated patients with congenital heart disease and mutations in RPS24 who were not anemic (Vlachos et al., 2018). Our observation that VUS reported in population databases could be involved in DBA pathogenesis highlights the need to deepen our knowledge about the possible presence in the general population of silent carriers of RP mutations and atypical cases of DBA.
Our results also emphasize the limited reliability of in silico tools for pathogenicity prediction. According to the data obtained by our complementation assay, Mutation Taster was the only tool with sensitivity and specificity equal to 1, whereas the other tools resulted in false negative and/or false positive predictions.
Several VUS have been reported also in other RP genes mutated in DBA patients (Arbiv et al., 2017; Doherty et al., 2010; Gerrard et al., 2013; Konno et al., 2010; Pospisilova et al., 2012; Smetanina et al., 2015; Tsangaris et al., 2011; van Dooijeweert et al., 2017). In the future, appropriate complementation assays could be implemented to extend the study of pathogenicity to other DBA genes, similarly to the approach we developed for RPS19.
In conclusion, we provided a strategy to distinguish disease‐causing mutations in RPS19 from benign polymorphisms and clarify their clinical significance. This information should assist clinicians in the counseling and management of DBA patients and their families.
Supporting information
SUPPLEMENTARY MATERIAL
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
Aspesi A, Betti M, Sculco M, et al. A functional assay for the clinical annotation of genetic variants of uncertain significance in Diamond–Blackfan anemia. Human Mutation. 2018;39:1102–1111. 10.1002/humu.23551
Contract grant sponsors: Diamond Blackfan Anemia Foundation; Fondazione Europea per la DBA; Gruppo di Sostegno DBA Italia; Banca del Piemonte; E‐RARE EuroDBA Consortium (BMBF #01GM1301, 01GM1609); ERC (261178).
Communicated by Maurizio Genuardi
REFERENCES
- Angelini, M. , Cannata, S. , Mercaldo, V. , Gibello, L. , Santoro, C. , Dianzani, I. , & Loreni, F. (2007). Missense mutations associated with Diamond‐Blackfan anemia affect the assembly of ribosomal protein S19 into the ribosome. Human Molecular Genetics, 16, 1720–1727. [DOI] [PubMed] [Google Scholar]
- Arbiv, O. A. , Cuvelier, G. , Klaassen, R. J. , Fernandez, C. V. , Robitaille, N. , Steele, M. G. , … Dror, Y. (2017). Molecular analysis and genotype‐phenotype correlation of Diamond‐Blackfan anemia. Clinical Genetics, 93, 320–328. [DOI] [PubMed] [Google Scholar]
- Aspesi, A. , Monteleone, V. , Betti, M. , Actis, C. , Morleo, G. , Sculco, M. , … Dianzani, I. (2017). Lymphoblastoid cell lines from Diamond Blackfan anaemia patients exhibit a full ribosomal stress phenotype that is rescued by gene therapy. Scientific Reports, 7, 12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspesi, A. , Pavesi, E. , Robotti, E. , Crescitelli, R. , Boria, I. , Avondo, F. , … Dianzani, I. (2014). Dissecting the transcriptional phenotype of ribosomal protein deficiency: Implications for Diamond‐Blackfan Anemia. Gene, 545, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badhai, J. , Fröjmark, A‐S. , Razzaghian, H. R. , Davey, E. , Schuster, J. , & Dahl, N. (2009). Posttranscriptional down‐regulation of small ribosomal subunit proteins correlates with reduction of 18S rRNA in RPS19 deficiency. FEBS Letters, 583, 2049–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boria, I. , Garelli, E. , Gazda, H. T. , Aspesi, A. , Quarello, P. , Pavesi, E. , … Dianzani, I. (2010). The ribosomal basis of Diamond‐Blackfan Anemia: Mutation and database update. Human Mutation, 31, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boria, I. , Quarello, P. , Avondo, F. , Garelli, E. , Aspesi, A. , Carando, A. , … Ramenghi, U. (2008). A new database for ribosomal protein genes which are mutated in Diamond‐Blackfan Anemia. Human Mutation, 29, E263–E270. [DOI] [PubMed] [Google Scholar]
- Chae, H. , Park, J. , Kim, M. , Lim, J. , Kim, Y. , Han, K. , … Kim, H. K. (2010). [Diamond‐Blackfan anemia confirmed by RPS19 gene mutation analysis: A case study and literature review of Korean patients]. Korean Journal of Laboratory Medicine, 30, 249–254. [DOI] [PubMed] [Google Scholar]
- Chae, H. , Park, J. , Lee, S. , Kim, M. , Kim, Y. , Lee, J‐W. , … Park, G. (2014). Ribosomal protein mutations in Korean patients with Diamond‐Blackfan anemia. Experimental & Molecular Medicine, 46, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr‐Aryamontri, A. , Angelini, M. , Garelli, E. , Tchernia, G. , Ramenghi, U. , Dianzani, I. , & Loreni, F. (2004). Nonsense‐mediated and nonstop decay of ribosomal protein S19 mRNA in Diamond‐Blackfan anemia. Human Mutation, 24, 526–533. [DOI] [PubMed] [Google Scholar]
- Choesmel, V. , Bacqueville, D. , Rouquette, J. , Noaillac‐Depeyre, J. , Fribourg, S. , Crétien, A. , … Gleizes, P‐E. (2007). Impaired ribosome biogenesis in Diamond‐Blackfan anemia. Blood, 109, 1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmejla, R. , Cmejlova, J. , Handrkova, H. , Petrak, J. , & Pospisilova, D. (2007). Ribosomal protein S17 gene (RPS17) is mutated in Diamond‐Blackfan anemia. Human Mutation, 28, 1178–1182. [DOI] [PubMed] [Google Scholar]
- Cmejlova, J. , Dolezalova, L. , Pospisilova, D. , Petrtylova, K. , Petrak, J. , & Cmejla, R. (2006). Translational efficiency in patients with Diamond‐Blackfan anemia. Haematologica, 91, 1456–1464. [PubMed] [Google Scholar]
- Da Costa, L. , Chanoz‐Poulard, G. , Simansour, M. , French, M. , Bouvier, R. , Prieur, F. , … Touraine, R. (2013). First de novo mutation in RPS19 gene as the cause of hydrops fetalis in Diamond–Blackfan anemia. American Journal of Hematology, 88, 340–341. [DOI] [PubMed] [Google Scholar]
- Da Costa, L. , Tchernia, G. , Gascard, P. , Lo, A. , Meerpohl, J. , Niemeyer, C. , … Mohandas, N. (2003). Nucleolar localization of RPS19 protein in normal cells and mislocalization due to mutations in the nucleolar localization signals in 2 Diamond‐Blackfan anemia patients: Potential insights into pathophysiology. Blood, 101, 5039–5045. [DOI] [PubMed] [Google Scholar]
- Cretien, A. , Hurtaud, C. , Moniz, H. , Proust, A. , Marie, I. , Wagner‐Ballon, O. , … Da Costa, L. (2008). Study of the effects of proteasome inhibitors on ribosomal protein S19 (RPS19) mutants, identified in patients with Diamond‐Blackfan anemia. Haematologica, 93, 1627–1634. [DOI] [PubMed] [Google Scholar]
- Delaporta, P. , Sofocleous, C. , Stiakaki, E. , Polychronopoulou, S. , Economou, M. , Kossiva, L. , … Kattamis, A. (2014). Clinical phenotype and genetic analysis of RPS19, RPL5, and RPL11 genes in Greek patients with Diamond Blackfan Anemia. Pediatric Blood & Cancer, 61, 2249–2255. [DOI] [PubMed] [Google Scholar]
- Doherty, L. , Sheen, M. R. , Vlachos, A. , Choesmel, V. , O'Donohue, M‐F. , Clinton, C. , … Gazda, H. T. (2010). Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond‐Blackfan anemia. American Journal of Human Genetics, 86, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dooijeweert, B. , van Ommen, C. H. , Smiers, F. J. , Tamminga, R. Y. J. , Te Loo, M. W. , Donker, A. E. , … Bartels, M. (2017). Pediatric Diamond‐Blackfan anemia in the Netherlands: An overview of clinical characteristics and underlying molecular defects. European Journal of Haematology, 100, 163–170. [DOI] [PubMed] [Google Scholar]
- Draptchinskaia, N. , Gustavsson, P. , Andersson, B. , Pettersson, M. , Willig, T. N. , Dianzani, I. , … Dahl, N. (1999). The gene encoding ribosomal protein S19 is mutated in Diamond‐Blackfan anaemia. Nature Genetics, 21, 169–175. [DOI] [PubMed] [Google Scholar]
- Errichiello, E. , Vetro, A. , Mina, T. , Wischmeijer, A. , Berrino, E. , Carella, M. , … Zuffardi, O. (2017). Whole exome sequencing in the differential diagnosis of Diamond‐Blackfan anemia: Clinical and molecular study of three patients with novel RPL5 and mosaic RPS19 mutations. Blood Cells, Molecules & Diseases, 64, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar, J. E. , Nater, M. , Caywood, E. , McDevitt, M. A. , Kowalski, J. , Takemoto, C. M. , … Arceci, R. J. (2008). Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond‐Blackfan anemia. Blood, 112, 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar, J. E. , Quarello, P. , Fisher, R. , O'Brien, K. A. , Aspesi, A. , Parrella, S. , … Ellis, S. R. (2014). Exploiting pre‐rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis. American Journal of Hematology, 89, 985–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar, J. E. , Vlachos, A. , Atsidaftos, E. , Carlson‐Donohoe, H. , Markello, T. C. , Arceci, R. J. , … Bodine, D. M. (2011). Ribosomal protein gene deletions in Diamond‐Blackfan anemia. Blood, 118, 6943–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flygare, J. , Aspesi, A. , Bailey, J. C. , Miyake, K. , Caffrey, J. M. , Karlsson, S. , & Ellis, S. R. (2007). Human RPS19, the gene mutated in Diamond‐Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood, 109, 980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi, A. , Ailles, L. E. , Bakovic, S. , Geuna, M. , & Naldini, L. (2000). Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV‐1 pol sequences. Nature Genetics, 25, 217–222. [DOI] [PubMed] [Google Scholar]
- Gazda, H. T. , Grabowska, A. , Merida‐Long, L. B. , Latawiec, E. , Schneider, H. E. , Lipton, J. M. , … Sieff, C. A. (2006). Ribosomal protein S24 gene is mutated in Diamond‐Blackfan Anemia. American Journal of Human Genetics, 79, 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda, H. T. , Preti, M. , Sheen, M. R. , O'Donohue, M‐F. , Vlachos, A. , Davies, S. M. , … Beggs, A. H. (2012). Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre‐ribosomal RNA processing defect in diamond‐blackfan anemia. Human Mutation, 33, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda, H. T. , Sheen, M. R. , Vlachos, A. , Choesmel, V. , O'Donohue, M‐F. , Schneider, H. , … Beggs, A. H. (2008). Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond‐Blackfan anemia patients. American Journal of Human Genetics, 83, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda, H. T. , Zhong, R. , Long, L. , Niewiadomska, E. , Lipton, J. M. , Ploszynska, A. , … Sieff, C. A. (2004). RNA and protein evidence for haplo‐insufficiency in Diamond‐Blackfan anaemia patients with RPS19 mutations. British Journal of Haematology, 127, 105–113. [DOI] [PubMed] [Google Scholar]
- Gerrard, G. , Valgañón, M. , Foong, H. E. , Kasperaviciute, D. , Iskander, D. , Game, L. , … Karadimitris, A. (2013). Target enrichment and high‐throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond‐Blackfan anaemia. British Journal of Haematology, 162, 530–536. [DOI] [PubMed] [Google Scholar]
- Gregory, L. A. , Aguissa‐Touré, A‐H. , Pinaud, N. , Legrand, P. , Gleizes, P‐E. , & Fribourg, S. (2007). Molecular basis of Diamond‐Blackfan anemia: Structure and function analysis of RPS19. Nucleic Acids Research, 35, 5913–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp, K. W. , Curry, C. , Olney, A. H. , Sandoval, C. , Fisher, J. , Chong, J. X.‐L. , … Sol‐Church, K. (2014). Diamond‐Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. American Journal of Medical Genetics Part A, 164A, 2240–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi, I. , Ooka, A. , Brun, A. , Richter, J. , Dahl, N. , & Karlsson, S. (2002). Gene transfer improves erythroid development in ribosomal protein S19‐deficient Diamond‐Blackfan anemia. Blood, 100, 2724–2731. [DOI] [PubMed] [Google Scholar]
- Le Hir, H. , Izaurralde, E. , Maquat, L. E. , & Moore, M. J. (2000). The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon‐exon junctions. The Embo Journal, 19, 6860–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer, C. , Caux‐Moncoutier, V. , Krieger, S. , Barrois, M. , Bonnet, F. , Bourdon, V. , … Stoppa‐Lyonnet, D. (2012). Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Human Mutation, 33, 1228–1238. [DOI] [PubMed] [Google Scholar]
- Ichimura, T. , Yoshida, K. , Okuno, Y. , Yujiri, T. , Nagai, K. , Nishi, M. , … Ohga, S. (2017). Diagnostic challenge of Diamond‐Blackfan anemia in mothers and children by whole‐exome sequencing. International Journal of Hematology, 105, 515–520. [DOI] [PubMed] [Google Scholar]
- Idol, R. A. , Robledo, S. , Du, H‐Y. , Crimmins, D. L. , Wilson, D. B. , Ladenson, J. H. , … Mason, P. J. (2007). Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells, Molecules & Diseases, 39, 35–43. [DOI] [PubMed] [Google Scholar]
- Ikeda, F. , Yoshida, K. , Toki, T. , Uechi, T. , Ishida, S. , Nakajima, Y. , … Ito, E. (2017). Exome sequencing identified RPS15A as a novel causative gene for Diamond‐Blackfan anemia. Haematologica, 102, e93–e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno, Y. , Toki, T. , Tandai, S. , Xu, G. , Wang, R. , Terui, K. , … Ito, E. (2010). Mutations in the ribosomal protein genes in Japanese patients with Diamond‐Blackfan anemia. Haematologica, 95, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramitsu, M. , Sato‐Otsubo, A. , Morio, T. , Takagi, M. , Toki, T. , Terui, K. , … Hamaguchi, I. (2012). Extensive gene deletions in Japanese patients with Diamond‐Blackfan anemia. Blood, 119, 2376–2384. [DOI] [PubMed] [Google Scholar]
- Landowski, M. , O'Donohue, M‐F. , Buros, C. , Ghazvinian, R. , Montel‐Lehry, N. , Vlachos, A. , … Gazda, H. T. (2013). Novel deletion of RPL15 identified by array‐comparative genomic hybridization in Diamond‐Blackfan anemia. Human Genetics, 132, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton, J. M. , & Ellis, S. R. (2009). Diamond‐Blackfan Anemia: Diagnosis, treatment, and molecular pathogenesis. Hematology/Oncology Clinics of North America, 23, 261–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello, L. , Khincha, P. P. , Ellis, S. R. , Giri, N. , Brodie, S. , Chandrasekharappa, S. C. , … Savage, S. A. (2017). Novel and known ribosomal causes of Diamond‐Blackfan anaemia identified through comprehensive genomic characterisation. Journal of Medical Genetics, 54, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello, L. , Macari, E. R. , Jessop, L. , Ellis, S. R. , Myers, T. , Giri, N. , … Savage, S. A. (2014). Whole‐exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond‐Blackfan anemia families. Blood, 124, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen, T. T. , O'Donohue, M‐F. , Wu, Y. , Lohi, H. , Scherer, S. W. , Paterson, A. D. , … Peltomäki, P. (2014). Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology, 147, 595–598.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozono, S. , Mitsuo, M. , Noguchi, M. , Nakagawa, S‐I. , Ueda, K. , Inada, H. , … Ito, E. (2016). Critical Diamond‐Blackfan anemia due to ribosomal protein S19 missense mutation. Pediatrics International, 58, 930–933. [DOI] [PubMed] [Google Scholar]
- Parrella, S. , Aspesi, A. , Quarello, P. , Garelli, E. , Pavesi, E. , Carando, A. , … Dianzani, I. (2014). Loss of GATA‐1 full length as a cause of Diamond‐Blackfan anemia phenotype. Pediatric Blood & Cancer, 61, 1319–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilova, D. , Cmejlova, J. , Ludikova, B. , Stary, J. , Cerna, Z. , Hak, J. , … Cmejla, R. (2012). The Czech National Diamond‐Blackfan Anemia Registry: Clinical data and ribosomal protein mutations update. Blood Cells, Molecules & Diseases, 48, 209–218. [DOI] [PubMed] [Google Scholar]
- Proust, A. , Da Costa, L. , Rince, P. , Landois, A. , Tamary, H. , Zaizov, R. , & SHIP Working Group on DBA (2003). Ten novel Diamond–Blackfan anemia mutations and three polymorphisms within the rps19 gene. Journal of Hematology, 4, 132–136. [DOI] [PubMed] [Google Scholar]
- Quarello, P. , Garelli, E. , Brusco, A. , Carando, A. , Mancini, C. , Pappi, P. , … Ramenghi, U. (2012). High frequency of ribosomal protein gene deletions in Italian Diamond‐Blackfan anemia patients detected by multiplex ligation‐dependent probe amplification assay. Haematologica, 97, 1813–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarello, P. , Garelli, E. , Carando, A. , Mancini, C. , Foglia, L. , Botto, C. , … Ramenghi, U. (2016). Ribosomal RNA analysis in the diagnosis of Diamond‐Blackfan Anaemia. British Journal of Haematology, 172, 782–785. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , & Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran, V. G. , Ghazvinian, R. , Do, R. , Thiru, P. , Vergilio, J. A. , Beggs, A. H. , … Gazda, H. T. (2012). Exome sequencing identifies GATA1 mutations resulting in Diamond‐Blackfan anemia. Journal of Clinical Investigation, 122, 2439–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smetanina, N. S. , Mersiyanova I, V. , Kurnikova, M. A. , Ovsyannikova, G. S. , Hachatryan, L. A. , Bobrynina, V. O. , … Maschan, A. A. (2015). Clinical and genomic heterogeneity of Diamond Blackfan anemia in the Russian Federation. Pediatric Blood & Cancer, 62, 1597–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon, J. , Kamalammal, R. , Menezes, G. A. , Sait, M. Y. , Lohith, H. , & Ramalingam, R. (2014). A case of Diamond Blackfan Anemia (DBA) with mutation in ribosomal protein S19. Journal of Clinical and Diagnostic Research, 8, 179–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsangaris, E. , Klaassen, R. , Fernandez C, V. , Yanofsky, R. , Shereck, E. , Champagne, J. , … Dror, Y. (2011). Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population‐based cohort and identification of novel mutations. Journal of Medical Genetics, 48, 618–628. [DOI] [PubMed] [Google Scholar]
- Vlachos, A. , Ball, S. , Dahl, N. , Alter, B. P. , Sheth, S. , Ramenghi, U. , & Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference . (2008). Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. British Journal of Haematology, 142, 859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos, A. , Osorio, D. S. , Atsidaftos, E. , Kang, J. , Lababidi, M. L. , Seiden, H. S. , … Lipton, J. M. (2018). Increased prevalence of congenital heart disease in children with Diamond Blackfan anemia suggests unrecognized Diamond Blackfan anemia as a cause of congenital heart disease in the general population: A report of the Diamond Blackfan anemia registry. Circulation: Genomic and Precision Medicine, 11, e002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Yoshida, K. , Toki, T. , Sawada, T. , Uechi, T. , Okuno, Y. , … Ito, E. (2015). Loss of function mutations in RPL27 and RPS27 identified by whole‐exome sequencing in Diamond‐Blackfan anaemia. British Journal of Haematology, 168, 854–864. [DOI] [PubMed] [Google Scholar]
- Willig, T. N. , Draptchinskaia, N. , Dianzani, I. , Ball, S. , Niemeyer, C. , Ramenghi, U. , … Tchernia, G. (1999). Mutations in ribosomal protein S19 gene and diamond blackfan anemia: Wide variations in phenotypic expression. Blood, 94, 4294–4306. [PubMed] [Google Scholar]
- Zhang, J‐Y. , Jia, M. , Zhao, H‐Z. , Luo, Z‐B. , Xu, W‐Q. , Shen, H‐P. , & Tang, Y‐M. (2016). A new in‐frame deletion in ribosomal protein S19 in a Chinese infant with Diamond‐Blackfan anemia. Blood Cells, Molecules & Diseases, 62, 1–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL