

Abstract
Aims
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by ventricular arrhythmias and sudden death. Currently 60% of patients meeting Task Force Criteria (TFC) have an identifiable mutation in one of the desmosomal genes. As much overlap is described between other cardiomyopathies and ARVC, we examined the prevalence of rare, possibly pathogenic sarcomere variants in the ARVC population.
Methods
One hundred and thirty‐seven (137) individuals meeting 2010 TFC for a diagnosis of ARVC, negative for pathogenic desmosomal variants, TMEM43, SCN5A, and PLN were screened for variants in the sarcomere genes (ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNC1, TNNI3, TNNT2, and TPM1) through either clinical or research genetic testing.
Results
Six probands (6/137, 4%) were found to carry rare variants in the sarcomere genes. These variants have low prevalence in controls, are predicted damaging by Polyphen‐2, and some of the variants are known pathogenic hypertrophic cardiomyopathy mutations. Sarcomere variant carriers had a phenotype that did not differ significantly from desmosomal mutation carriers. As most of these probands were the only affected individuals in their families, however, segregation data are noninformative.
Conclusion
These data show variants in the sarcomere can be identified in individuals with an ARVC phenotype. Although rare and predicted damaging, proven functional and segregational evidence that these variants can cause ARVC is lacking. Therefore, caution is warranted in interpreting these variants when identified on large next‐generation sequencing panels for cardiomyopathies.
Keywords: ARVC, cardiomyopathy, genetics, sarcomere, whole‐exome sequencing
1. INTRODUCTION
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial disease characterized by fibrofatty replacement of the myocardium. This results in a predisposition to often life‐threatening ventricular arrhythmias and functional alterations to the right and left ventricles that can lead to the development of heart failure. A familial pattern of disease can be recognized in up to 50% of cases, predominantly inherited in an autosomal dominant manner.1 Reduced penetrance, and variable expressivity of presentation have complicated the identification of a genetic cause in some cases.
ARVC is primarily thought of as a disease of the desmosome, or the cell–cell junctions at the intercalated disk. Pathogenic variants in five genes (DSP, PKP2, DSG2, DSC2, and JUP) are classically thought to play a large role in ARVC pathogenesis.2, 3 In routine clinical practice, however, even in a well phenotyped population, up to 40% of ARVC cases still elude identification of a genetic root cause.2, 3 Because of the importance of identifying those at risk for sudden death, gene finding efforts have continued.
Increasingly, nondesmosomal genes have been implicated in ARVC pathogenesis, such as CDH2 and CTNNA3.4, 5 PLN has been identified as a causative factor in a significant portion of individuals with ARVC patients.3, 6 Additionally, recent reports have identified SCN5A mutations in a small percentage of ARVC patients.7 Overlap between ARVC and dilated cardiomyopathy has been well described, and pathogenic variants in sarcomere genes have been associated with DCM.8 Little data exists, however, on the prevalence of other cardiomyopathy‐associated genes in the ARVC population.9 In this study, we sequenced an expanded panel of cardiac genes among individuals with a clinical diagnosis of ARVC and without mutations in the ARVC‐associated genes PKP2, DSG2, DSP, DSC2, and JUP, or nondesmosomal TMEM43, SCN5A, and PLN. We aimed to (1) assess the prevalence and pathogenicity of sarcomere gene mutations in individuals with ARVC without an identified desmosomal pathogenic variant and (2) compare clinical characteristics and course between the two groups.
2. METHODS
2.1. Study population
Patients were eligible for inclusion if they were (1) diagnosed with ARVC based on the 2010 diagnostic Task Force Criteria (TFC) for ARVC,10 (2) were the family proband as defined as the first person in the family to come to clinical attention and gain a clinical diagnosis of ARVC, (3) lacked a pathogenic/likely pathogenic variant in any of the 5 desmosomal genes noted above, or TMEM43, PLN, or SCN5A, (4) underwent broad cardiomyopathy gene screening as described below, and (5) enrolled in the ARVC registries from the Johns Hopkins University and the Netherlands Heart Institute, a cardiovascular research institute with collaborative participation of all eight Dutch University Medical Centers. This study was approved by the JHSOM Institutional Review Board.
2.2. Molecular genetic screening
Sequencing and deletion/duplication analysis of the nine most common sarcomere genes (ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNC1, TNNI3, TNNT2, and TPM1, hereafter referred to as sarcomere genes) were completed through a variety of methods: clinical genetic testing, and research‐lab based panel and exome sequencing. Within the JHU registry, 92 patients underwent clinical cardiomyopathy panel sequencing and deletion/duplication analysis, and 18 patients consented to whole‐exome sequencing using the Ilumina HiSeq2000 platform. For exome sequencing, the human assembly GRCh37/hg19 was used as reference genome. In the Dutch registry, 27 patients were sequenced using a next‐generation sequencing (NGS) panel consisting of 209 genes (candidate genes and genes known to be involved in cardiomyopathy). Therefore, in total 137 probands underwent sequencing and deletion/duplication analysis of the sarcomere genes. Nucleic acid deviations were compared with the reference sequence for presence of variants in the sarcomere genes. All mutations were confirmed by Sanger sequencing after polymerase chain reaction amplification.
Potentially causal variants were identified using standard filtering criteria as follows. Variants were excluded if they had a minor allele frequency (MAF) >0.1% in the Exome Aggregation Consortium Browser (ExAC)11, 12 and/or if they were present in dbSNP 126, 129, and 131. Variants also were included if predicted damaging by Polyphen‐2 and below the MAF above.13 Variants were assessed and classified according to the 2015 American College of Medical Genetics classification criteria, as reported in Table 1.14 All patients and their families gave informed consent for genomic DNA sample collection, storage, and sequencing.
Table 1.
Variants in the sarcomere genes identified upon sequencing of 137 probands with ARVC
| Family | Gene | Exon | Amino acid change | Nucleotide change | % ExAC | Polyphen (2) | ACMG pathogenicity classification |
|---|---|---|---|---|---|---|---|
| 1 | MYH7 | 22 | p.847_847del | c.2541_2549del | 0.0000% | n/a | VUS |
| 2 | 21 | p.Arg1500Gln | c.4499G > A | 0.0025% | 0.999 | VUS | |
| 3 | 37 | p.Arg1846Cys | c.5536C > T | 0.0017% | 1.000 | VUS | |
| 4‡ | 20 | p.Arg723Cys | c.2167C > T | 0.0025% | 0.995 | P | |
| 5* | MYBPC3 | 4 | p.Gly148Arg | c.442G > A | 0.0042% | 0.070 | P |
| 6† | MYL3 | 4 | p.Arg154His | c.461G > A | 0.0025% | 0.861 | LP |
Note: Table 2 lists variants in sarcomere genes identified in the ARVC probands. ExAC frequencies as of 4/2017 are listed.11 Assessment of pathogenicity according to criteria put forth by the American College of Medical Genetics is listed (LP = likely pathogenic; P = pathogenic; VUS = variant of uncertain significance).14
‡Segregated with disease in relatives from multiple families. (ClinVar: http://www.ncbi.nlm.nih.gov/clinvar/variation/14095/).
*This variant is a low penetrance variant and frequently observed in the Dutch HCM population. It is a founder mutation and creates a cryptic splice acceptor site (P. Van Tintelen, personal communication). Prediction programs (Polyphen‐2) are not valid because of supposed pathogenic splice site effect. Aberrant splicing is demonstrated in two university hospitals. This individual also carried a variant in LMNA c.1003C > T; p.Arg335Trp that has been described as likely pathogenic.19
†This mutation has been demonstrated to cause a disturbed binding to myosin in vitro.16
2.3. Phenotype evaluation
All individuals were phenotyped via medical records for diagnostic criteria according to the 2010 TFC.10 A definite ARVC diagnosis was characterized by the presence of ≥2 major criteria, 1 major and 2 minor criteria, or 4 minor criteria. As indicated in Supplementary Table S1, none of the individuals met diagnostic criteria for hypertrophic cardiomyopathy of having a septal thickness of ≥1.5 cm.15
2.4. Statistical analysis
Statistical analyses were performed using SPSS (version 22.0). All continuous data were calculated as mean and categorical variables as numbers (percentages). Variables were compared using the Fisher's exact test for proportions, and chi‐square test. A P‐value of <0.05 was considered statistically significant.
3. RESULTS
3.1. Genetic screening
In total, 137 individuals meeting diagnostic criteria for ARVC underwent sequencing of the sarcomere genes. Sarcomere variants identified are described in Table 1. These variants were rare in ExAC (<0.1% as described above). A total of six variants were identified in 6/137 (4.3%) separate probands. Variants were identified in MYH7, MYBPC3, and MYL3. Variants in MYH7 included three missense mutations in MYH7 and one in‐frame deletion. There was one missense variant identified in MYBPC3 and one variant in MYL3. For most families, the proband was the only reported affected in the family. In one family (Individual #2 as described in Table 2), the variant was identified in her, and also in her mother who had met diagnostic criteria for ARVC with T wave inversions through V3 on ECG and over 500 PVCs on Holter monitoring. The variant identified in MYH7 was previously published in the literature segregating with HCM in multiple families.16 For the other four families, family screening was not completed or cardiac screening, including ECG, Holter monitoring, and either echocardiogram and/or cardiac MRI in first degree relatives was within normal limits. As noted in Table 1, many of these variants may not meet pathogenicity criteria for a pathogenic call when classified according to the ACMG criteria; however, would still be reported. Even those that may be classified as pathogenic for a diagnosis of HCM may not be reported as pathogenic for a diagnosis of ARVC given the lack of evidence for disease association.
Table 2.
Clinical characteristics of the 6 patients identified to have pathogenic sarcomere variants
| Clinical presentation | ECG or SAECG | Structure | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Family # | Sex | Age at presentation (years) | AA medication | Type of presentation | Cardiac Syncope | Repolarization abnormality | Depolarization abnormality | RV structural abnormality | LV dysfunction |
| 1 | M | 50 | + | Sympt | + | M | n/a | M | + |
| 2 | F | 14 | + | Sympt | + | M | – | M | + |
| 3 | M | 27 | + | Sympt | + | M | – | M | – |
| 4 | M | 26 | + | Sympt | + | m | – | M | + |
| 5 | M | 28 | + | Sympt | – | M | m | M | + |
| 6 | M | 36 | + | Sympt | – | m | – | M | – |
| Arrhythmia | Outcome | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Holter ectopy | EPS inducibility | Sustained VA | VT storm | ICD implant | ICD therapy | VT ablation | Heart failure | Death/cardiac transplant | Follow‐up (years) | TFC |
| n/a | + | + | + | + | + | + | + | + | 23 | 6(D) |
| 470 | + | + (At presentation) | – | + | + | + | + | – | 20 | 4(D) |
| n/a | + | + (At presentation) | + | + | + | + | – | – | 27 | 6(D) |
| 472 | + | + | – | + | + | + | – | – | 14 | 5(D) |
| n/a | + | + (At presentation) | – | + | + | – | – | – | 6 | 9(D) |
| 1181 | n/a | + | – | – | – | – | – | – | 13 | 4(D) |
AA = antiarrhythmic; B = borderline; D = definite; EP = electrophysiology; F = female; ICD = implantable cardiovertor defibrillator; LV dysfunction = LV EF ≤50%; M = male; m = minor abnormality as per the 2010 ARVC revised Task Force Criteria; M = major abnormality as per the 2010 ARVC Revised Task Force Criteria; Sympt = symptomatic with chest pain, dyspnea, palpitations, presyncope, or syncope; TFC = 2010 revised Task Force Criteria for the diagnosis of ARVC; VA = ventricular arrhythmias including VT and VF; VT /VF = ventricular tachycardia/ventricular fibrillation.
3.2. Clinical evaluation
All individuals met diagnostic criteria as described in Table 2.10 Compared to previously published ARVC patients carrying desmosomal mutations, individuals with sarcomere variants tended to be slightly older (36.8 years of age vs. 33.2 years of age), though not significant.2 Clinical characteristics of the study population were compared to previously published values2 in desmosomal mutations (definitions in Supplementary Table S2): gender, type of presentation, syncope, inducibility at electrophysiology study, premature ventricular contraction (PVC) count on 24‐hour ambulatory monitoring (Holter), ICD placement, appropriate ICD therapy, ventricular tachycardia (VT) storm, VT ablation, and left ventricular dysfunction (left ventricular ejection fraction below 50%), heart failure, and transplant. Individuals with ARVC carrying sarcomere variants were more likely to have undergone a VT ablation (P = 0.009), but otherwise had a similar disease presentation and course to desmosomal mutation carriers. Figure 1 shows the ECG and cardiac MRI short axis image of individual 3, showing characteristic T wave inversions across the precordium and dyskinetic basal right ventricular (RV) free wall with enhancement, suggestive of a diagnosis of ARVC. Supplementary Table S1 describes septal thickness; all values were way below the threshold for HCM.
Figure 1.
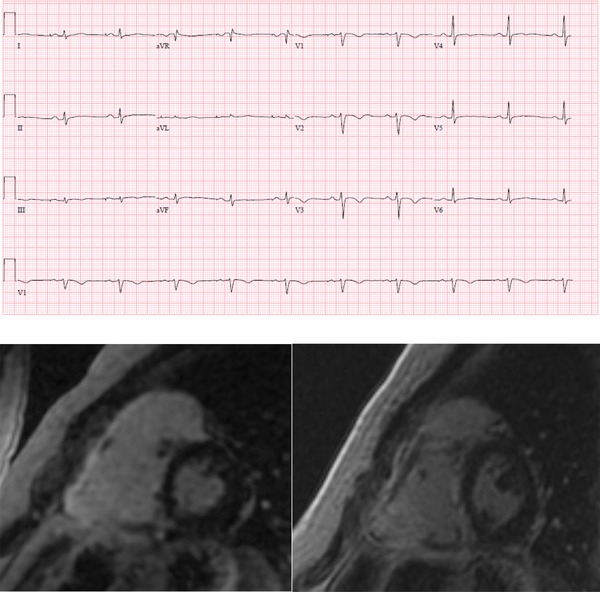
Electrocardiogram and cardiac MRI of proband #3 showing major criterion of T wave inversions across the precordium and dyskinetic base and enhancement of the RV wall [Color figure can be viewed at http://wileyonlinelibrary.com]
4. DISCUSSION
It has been well documented that approximately 40–60% of individuals meeting diagnostic criteria for ARVC have a mutation mainly in genes encoding the cardiac desmosome.2, 3 The availability of tools such as whole‐exome sequencing and expansion of number of genes included on clinical cardiomyopathy panels has opened the doors for further gene discovery, but has also introduced significant challenges in counseling patients for genetic testing, and in interpretation of variants for the clinician. Understanding the pathogenicity of a variant is critical in the identification of those at risk for sudden death and implementing risk‐stratifying cascade screening.
In this cohort, we describe that a small but significant percentage (4.3%) of individuals with ARVC may have putative likely pathogenic/pathogenic variants reported in the sarcomere genes. A similar yield has recently also been reported by others.9 Without systematic functional studies or extensive segregation in affected individuals on each of these rare novel variants, however, it is difficult to determine the role these variants play in the ARVC phenotype of individuals. Integration of segregation analysis in families with ARVC is also challenging given the well described reduced penetrance.3 In addition, mounting exome population data over the years have led to systematic reclassification of previously described pathogenic variants in the cardiomyopathies as uncertain, or even benign.17 Therefore, we as well as other centers are hesitant to immediately label these variants as “pathogenic” given the implications for cascade screening in these families. Given this, without further evidence, sarcomere variants should not be used to guide presymptomatic screening in families with ARVC.
Continued genetic overlap between the cardiomyopathies has been described, and it follows that structural heart disease with arrhythmias that primarily affects the RV may produce a phenotype mimicking ARVC.4, 10 Indeed, these individuals with sarcomere variants meet TFC for ARVC, and do not have significant differences in structural disease than desmosomal variant carriers. The cohort reported here is less likely to have a reported family history of disease than previous reported prevalences in desmosomal mutation carriers.2 They have no significant differences in phenotype by TFC, do not meet HCM criteria, but also, importantly, they do not have any significant differences in clinical course (other than being more likely to undergo catheter ablation, which is by physician judgment) than previously described individuals with ARVC with desmosomal mutations.2 These are important findings as it indicates that these individuals are not misdiagnosed, in fact, they have similar phenotype and clinical course, confirming that these individuals do indeed have ARVC.
This study is limited in that most individuals are the only one affected in their family, so familial segregation is not informative. Unfortunately, due to family choice, prospective information on family screening could not be obtained. Negative family history is not unusual in ARVC, as reduced penetrance is well described.3 Additionally, as with most cardiomyopathy variants, functional data are lacking for the majority of the variants identified here. These sarcomere variants may have a pathogenic role, since there is some (in silico) functional evidence, cosegregation with HCM in prior publications, and altered splicing for one of these variants.14, 18 At this time, however, as conclusive data of a role in ARVC are lacking, this analysis provides important information to clinicians who may order large‐scale sequencing panels that caution is advised when sarcomere variants are resulted in a patient with an ARVC phenotype. Even if a variant is reported as pathogenic/likely pathogenic due to limited functional data, these data suggest that sarcomere variants should not be interpreted or used clinically as pathogenic in ARVC families.
5. CONCLUSION
Despite these limitations, the results of our study highlight that a small proportion of patients exhibiting an ARVC phenotype may have variants identified in the sarcomere genes. This provides important additional evidence for clinical practice to recommend caution in interpretation of comprehensive cardiomyopathy gene testing results in genetic screening of ARVC patients. At this point, without further functional studies or strong segregation with disease in multiple families, these variants should not be used for clinical care. In addition, the identification of these variants, yet absence of evidence of a causative role, highlights the importance of cardiac genetics expertise in the interpretation of these large cardiomyopathy panels in families with inherited heart disease.
Further work would be of great interest to investigate the functional role of these variants in the function of the sarcomere and the desmosome, and in the pathogenesis of ARVC. Until then, these variants should not be utilized in clinical decision making, or family screening.
Supporting information
Supplementary Table S2. Definitions of clinical variables employed in this study
Supplementary Table S1 HCM echocardiogram measurements in the 6 probands
ACKNOWLEDGMENT
We are grateful to the ARVC patients and families who have made this work possible.
Murray B, Hoorntje ET, te Riele ASJM, et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC). J Cardiovasc Electrophysiol. 2018;29:1004–1009. 10.1111/jce.13621
J. Peter van Tintelen and Dennis Dooijes contributed equally to this study.
Whole‐exome sequencing was performed at the Baylor‐Hopkins Center for Mendelian Genomics, funded by the National Human Genome Research Institute (U54HG006542). The Johns Hopkins ARVD/C Program is supported by the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, Dr. Francis P. Chiaramonte Private Foundation, the Peter French Memorial Foundation, the Wilmerding Endowments, the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, and the St. Jude Medical Foundation. The work was financially supported by the Netherlands Cardiovascular Research Initiative, an initiative supported by the Dutch Heart Foundation (CVON2012‐10 PREDICT, CVON2014‐40 DOSIS). Dr. te Riele is supported by the Netherlands Heart Foundation (grant 2015T058); UMC Utrecht fellowship clinical research talent; and CVON‐PREDICT Young Talent Program.
Disclosures: None.
REFERENCES
- 1. Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation. 2005;112:3823–3832. [DOI] [PubMed] [Google Scholar]
- 2. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated mutation carriers. Eur Heart J. 2015;36:847–855. [DOI] [PubMed] [Google Scholar]
- 3. Groeneweg JA, Bhonsale A, James CA, et al. Clinical presentation, long‐term follow‐up, presentation, long‐term follow‐up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8:437–446. [DOI] [PubMed] [Google Scholar]
- 4. Mayosi BM, Fish M, Shaboodien G, et al. Identification of Cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10. [DOI] [PubMed] [Google Scholar]
- 5. van Hengel J CaloreM, Bauce B, et al. Mutations in the area composita protein αT‐catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–210. [DOI] [PubMed] [Google Scholar]
- 6. van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Te Riele AS, Agullo‐Pascual E, James CA, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non‐canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McNally EM, Mestroni L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ Res. 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Medeiros‐Domingo A, Saguner A, Magyar I, et al. Arrhythmogenic right ventricular cardiomyopathy: Implications of next‐generation sequencing in appropriate diagnosis. Europace. 2016;9:1063–1069. [DOI] [PubMed] [Google Scholar]
- 10. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Eur Heart J. 2010;31:806–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lek M, Karczewski KJ, Minikel EV, et al. Exome Aggregation Consortium. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: An empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Method. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elliott PM, Anastasakis A, Borger MA, et al. ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 16. Tesson F, Richard P, Charron P, et al. Genotype‐phenotype analysis in four families with mutations in beta‐myosin heavy chain gene responsible for familial hypertrophic cardiomyopathy. Hum Mutat. 1998;12:385–392. [DOI] [PubMed] [Google Scholar]
- 17. Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–170. [DOI] [PubMed] [Google Scholar]
- 18. Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG‐AMP variant‐interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016;98:1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stallmeyer B, Koopmann M. Schulze‐Bahr. Identification of novel mutations in LMNA associated with familial forms of dilated cardiomyopathy. Genet Test Mol Biomarkers. 2012;16:543–549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S2. Definitions of clinical variables employed in this study
Supplementary Table S1 HCM echocardiogram measurements in the 6 probands