

Abstract
Dysfunction of degradation machineries causes cancers, including hepatocellular carcinoma (HCC). Overexpression of cyclin D1 in HCC has been reported. We previously reported that autophagy preferentially recruits and degrades the oncogenic microRNA (miR)‐224 to prevent HCC. Therefore, in the present study, we attempted to clarify whether cyclin D1 is another oncogenic factor selectively regulated by autophagy in HCC tumorigenesis. Initially, we found an inverse correlation between low autophagic activity and high cyclin D1 expression in tumors of 147 HCC patients and three murine models, and these results taken together revealed a correlation with poor overall survival of HCC patients, indicating the importance of these two events in HCC development. We found that increased autophagic activity leads to cyclin D1 ubiquitination and selective recruitment to the autophagosome (AP) mediated by a specific receptor, sequestosome 1 (SQSTM1), followed by fusion with lysosome and degradation. Autophagy‐selective degradation of ubiquitinated cyclin D1 through SQSTM1 was confirmed using cyclin D1/ubiquitin binding site (K33‐238R) and phosphorylation site (T286A) mutants, lentivirus‐mediated silencing autophagy‐related 5 (ATG5), autophagy‐related 7 (ATG7), and Sqstm1 knockout cells. Functional studies revealed that autophagy‐selective degradation of cyclin D1 plays suppressive roles in cell proliferation, colony, and liver tumor formation. Notably, an increase of autophagic activity by pharmacological inducers (amiodarone and rapamycin) significantly suppressed tumor growth in both the orthotopic liver tumor and subcutaneous tumor xenograft models. Our findings provide evidence of the underlying mechanism involved in the regulation of cyclin D1 by selective autophagy to prevent tumor formation. Conclusion: Taken together, our data demonstrate that autophagic degradation machinery and the cell‐cycle regulator, cyclin D1, are linked to HCC tumorigenesis. We believe these findings may be of value in the development of alternative therapeutics for HCC patients. (Hepatology 2018;68:141‐154).
Abbreviations
- AP
autophagosome
- ATG5
autophagy‐related 5
- ATG7
autophagy‐related 7
- BECN1
Beclin 1
- BrdU
bromodeoxyuridine
- CDK
cyclin‐dependent kinases
- CQ
chloroquine
- DDW
double‐distilled water
- GSK3β
glycogen synthase kinase 3β
- HBV
hepatitis B virus
- HBx
hepatitis B virus X protein
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- IB
immunoblotting
- IgG
immunoglobulin G
- IHC
immunohistochemistry
- IP
immunoprecipitation
- LC3
microtubule‐associated protein 1 light chain 3
- LR
liver regeneration
- MEF
mouse embryo fibroblast
- miR
microRNA
- N.T.
nontreated
- PNS
post‐nuclear supernatant
- SQSTM1/P62
sequestosome 1
- TEM
transmission electron microscopy
- WT
wild type
Hepatocellular carcinoma (HCC) is the third‐most common cause of cancer‐related death worldwide.1 In the impaired liver, aberrant cell‐cycle progression of liver cells leads to proliferation and hepatocarcinogenesis (HCG).2 Cyclin D1 is a regulatory subunit of cyclin‐dependent kinases (CDK) 4 and 6. It is synthesized at the G1 phase and then binds with CDK4/6 to regulate the G1/S‐phase transition. Cyclin D1 is degraded in the cytoplasm when the cell cycle enters the S phase. Cyclin D1 promotes liver cell growth, and overexpression of cyclin D1 initiates HCC development by promoting cell‐cycle progression.3, 4 These findings imply that dysregulated cyclin D1 participates in HCC tumorigenesis. It has been reported that cyclin D1 becomes overexpressed and induces HCC through gene amplification.5 However, whether any unidentified mechanism regulates cyclin D1 leading to an increased risk of HCC occurrence remains unclear.
Proteasomal and autophagic machinery are two major degradation systems of ubiquitinated proteins in the cell.6 Autophagy recruits as well as recycles unnecessary and dysfunctional cellular components, including proteins, pathogens, damaged organelles, and microRNA (miRs).7, 8 Autophagic machinery is classified into nonselective and selective autophagy. During selective autophagy, various receptor proteins participating in the processes have been identified (sequestosome 1 [SQSTM1]/P62, neighbor of BRCA1 gene 1 [NBR1], NDP52 [CALCOCO2; calcium‐binding and coiled‐coil domain 2], and optineurin).9 These receptors contain a conserved motif known as a microtubule‐associated protein 1 light chain (LC3) interacting region (LIR), which is responsible for binding with LC3 and transporting specific cargos into the double‐membrane autophagosome (AP). Most of the selective cargo proteins are ubiquitinated proteins. The SQSTM1 protein participates in the formation of cytoplasmic inclusion. Accumulation of SQSTM1 protein in the cell indicates a deficiency of autophagic activity and is related to Alzheimer's disease and chronic liver disorders.10 Accumulating evidence indicates that selective autophagy contributes to the aberrant activation of the signaling pathways and oncogenic factors related to the tumorigenesis of various cancers.11, 12
Transgenic mice with mosaic deletion of autophagy‐related 5 (Atg5) or liver‐specific deletion of autophagy‐related 7 (Atg7) gene only developed tumors in the liver.13 Similarly, in human HCC tumor tissues, decreased ATG5 and Beclin 1 (BECN1) gene expression together with SQSTM1 protein accumulation were found in comparison with adjacent nontumor tissues.7 We previously revealed that autophagy prevents liver tumor formation through selective recruitment and degradation of oncogenic miR‐224. In summary, these findings strongly suggest that an autophagic degradation system may act as a safeguard to maintain the homeostasis of diverse cellular factors in the cell, thereby preventing tumorigenesis, including HCC. To further extend our findings, we hypothesized that this autophagy degradation system may regulate multiple oncogenic factors related to HCC tumorigenesis. In this study, we found that autophagy selectively recruits and degrades another cell‐cycle regulator known as “cyclin D1,” and we further clarified the underlying mechanism in HCC tumorigenesis.
Materials and Methods
AP PURIFICATION
Hep 3B cells were treated with the inducer amiodarone for 24 hours followed by chloroquine (CQ) treatment for another 24 hours to block fusion of lysosome and AP. The extraction procedure was reported on elsewhere, and details are provided in the http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo section.7
CLINICAL SPECIMENS
HCC specimens were purchased from the Taiwan Liver Cancer Network and National Cheng Kung University Hospital (NCKUH; Tainan, Taiwan). Informed consent was obtained from all patients with approval by the Institutional Review Board (IRB), NCKUH, Tainan, Taiwan (IRB document number: A‐BR‐100‐156).
IMMUNOGOLD‐LABELED PROTEIN AND TRANSMISSION ELECTRON MICROSCOPY
The ultrathin sections of AP fraction on the nickel grids were treated with 10% H2O2 for 10 minutes. Grids were blocked for 30 minutes followed by anti‐cyclin D1, anti‐SQSTM1, or anti‐LC3 antibody treatment overnight. Grids were incubated with anti‐mouse immunoglobulin G (IgG) H&L (ab27242; Abcam, Cambridge, UK) or anti‐rabbit IgG H&L (ab105298; Abcam) secondary antibody for 1 hour. The sections on the grids were stained with saturated uranyl acetate and lead citrate and investigated under a transmission electron microscope (HITACHI‐7000).
Other details are available in the http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo section.
Results
HIGH CYCLIN D1 EXPRESSION WAS ASSOCIATED WITH LOW AUTOPHAGIC ACTIVITY IN HUMAN HCC TUMOR TISSUES AND CO‐OCCURRENCE OF THESE TWO EVENTS CORRELATED WITH POOR OVERALL SURVIVAL RATE OF HCC PATIENTS
To reveal the relationship between autophagic activity and cyclin D1 expression in HCC, 147 paired HCC specimens (tumor and nontumor parts) were analyzed (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). The representative HCC specimens from 1 patient showed that cyclin D1 expression and SQSTM1 accumulation were high, and BECN1 and ATG5 expression levels were low in the tumor tissue as compared to the adjacent nontumor tissue by immunohistochemistry (IHC) staining (Fig. 1A), and the quantification data showing statistical significance are depicted in Fig. 1B. Furthermore, high cyclin D1 expression and high SQSTM1 accumulation were detected in 11 of 20 HCC tumor tissue specimens analyzed by immunoblotting (IB; http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo, red numbers). SQSTM1 accumulation or low expression of BECN1 and ATG5 represent low autophagic activity. Further analysis revealed that cyclin D1 level was negatively correlated with BECN1 level and was positively correlated with SQSTM1 accumulation in these HCC specimens (Fig. 1C), indicating an inverse correlation between autophagy and cyclin D1. The multivariate analysis of the association of HCC clinicopathological parameters with cyclin D1, BECN1, and SQSTM1 levels revealed that tumor size positively correlated with the level of cyclin D1 (P = 0.032) or in combination with BECN1 (P = 0.041), SQSTM1 (P = 0.004) or BECN1, and SQSTM1 (P = 0.011; http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). We conducted Kaplan‐Meier survival analysis for cyclin D1 expression alone and by dividing the 147 HCC patients into a high‐risk group (high cyclin D1, low BECN1 expression, and high SQSTM1 accumulation in the tumor vs. the adjacent nontumor tissue) and a low‐risk group (the remaining HCC specimens), and our data showed that both the high cyclin D1 expression group (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo; P = 0.0317) and the high‐risk group (Fig. 1D; P = 0.0116) had worse overall survival rate among HCC patients. These results imply that low autophagic activity and high cyclin D1 expression were correlated in HCC tumorigenesis, and the co‐occurrence of these two events was associated with worse survival rate.
Figure 1.
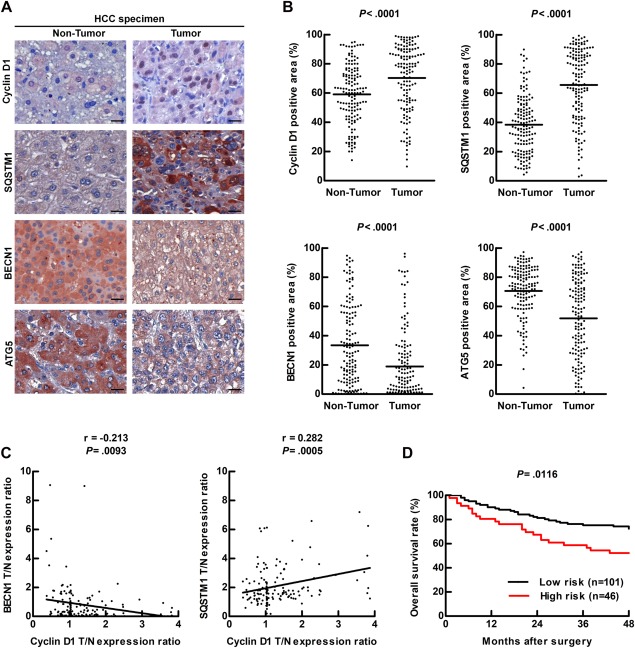
Low autophagic activity accompanied with high cyclin D1 expression was observed in HCC specimens. (A) Representative IHC images of HCC specimen sections show the expression levels of cyclin D1, SQSTM1, ATG5, and BECN1. Scale bar = 20 μm. (B) Quantification of the protein expression in 147 of HCC specimens was determined by defining ROI followed by HistoQuest analysis. A paired t test was used to compare two populations. (C) Correlation of cyclin D1 with SQSTM1 or BECN1 in 147 HCC specimens was analyzed, and linear regression coefficient and statistical significance are indicated. (D) Overall survival rate of HCC patients was determined by Kaplan‐Meier analysis and long‐rank test. High protein expression was defined by the expression in the tumor tissue versus the expression in the adjacent nontumor tissue > 1‐fold. The high‐risk group (n = 46) was defined as high‐cyclin D1, high‐SQSTM1, and low‐BECN1 expression. The remaining specimens were classified as the low‐risk group (n = 101). Abbreviations: ROI, region of interest; T/N, T: tumor tissue; N: adjacent nontumor tissue.
ACTIVATED AUTOPHAGY CORRELATED WITH DECREASED CYCLIN D1 EXPRESSION, CELL PROLIFERATION, AND CELL‐CYCLE ARREST AT THE G1 PHASE
Human hepatoma cell lines (Hep 3B and Huh 7) were treated with two autophagy inducers (rapamycin and amiodarone) for 24 hours. Both of the inducers increased LC3‐II expression and decreased cyclin D1 expression in a dose‐dependent manner (Fig. 2A). Decreased cyclin D1 expression was reversed when autophagic activity was suppressed by lentivirus‐mediated silencing of ATG5 or ATG7 gene expression in Hep 3B cells under autophagy induction (Fig. 2B). Similarly, cyclin D1 protein degradation was delayed in autophagy‐deficient mouse embryo fibroblast (MEF) MEF‐Atg5(–/–) cells compared to wild‐type (WT) MEF‐Atg5(+/+) cells (Fig. 2C). Surprisingly, we found that cyclin D1 mRNA levels were increased after autophagy induction, but there was no significant difference among these cells (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). These results suggest that autophagy‐induced suppression of cyclin D1 expression occurs at the posttranscriptional level. We also demonstrated that cell growth was suppressed (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo) and cell‐cycle arrest at the G1 phase (Fig. 2D) when autophagy was induced. To investigate cell‐cycle progression, we synchronized the cell cycle by starvation, followed by measurement of DNA synthesis (G0/G1 phase to S phase) using bromodeoxyuridine (BrdU) incorporation analysis. We demonstrated that the cell‐cycle progression decreased when autophagy was induced (Fig. 2E); however, this suppression was rescued by knockdown of either ATG5 or ATG7 genes in Hep 3B cells (Fig. 2E). Similarly, cell‐cycle progression was increased in MEF‐Atg5(–/–) cells compared to MEF‐Atg5(+/+) cells (Fig. 2F). In summary, our results imply that increased autophagic activity leads to cell‐cycle arrest, decreased cyclin D1 expression, and cell growth.
Figure 2.
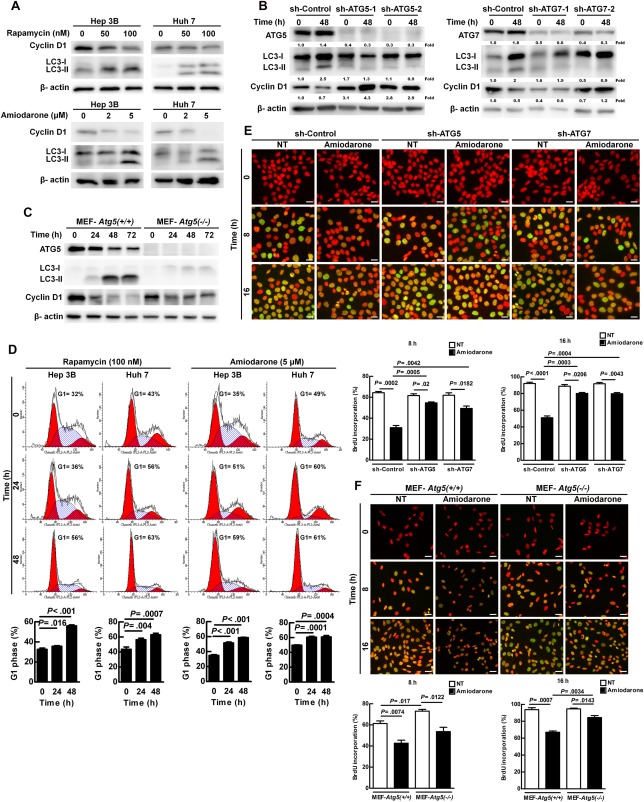
Increased autophagic activity suppressed cell growth, cell‐cycle progression, and cyclin D1 expression. (A) Hep 3B and Huh 7 cells were treated with various concentrations of rapamycin or amiodarone for 24 hours. Protein expression was examined by immunoblotting. (B) Hep 3B cells were infected with lentivirus harboring sh‐GFP (control), sh‐Atg5, or sh‐Atg7 sequence and then treated with amiodarone (5 μM) for 48 hours. The number under the individual band represents the fold changes of protein expression as compared to the control. (C) MEF‐Atg5(+/+) and MEF‐Atg5(–/–) cells were treated with amiodarone (15 μM) for various times. Protein expression was detected by immunoblotting. (D) Hep 3B and Huh 7 cells were treated with rapamycin (100 nM) or amiodarone (5 μM) for 0, 24, or 48 hours. Cell cycle was analyzed by flow cytometry analysis. (E,F) Hep 3B cells (harboring sh‐Control, sh‐Atg5, or sh‐Atg7), MEF‐Atg5(+/+), and MEF‐Atg5(–/–) cells were synchronized by serum‐free starvation for 24 hours, and then DNA synthesis was measured by BrdU incorporation after amiodarone treatment for 0, 8, and 16 hours. Scale bar = 20 μm. Quantification is shown in the diagram. Data represent means ± SEM (n = 5). Abbreviations: GFP, green fluorescent protein; sh, short hairpin.
We used amiodarone as the inducer in the experiments described below owing to its ability to induce autophagic activity, long‐lasting induction capability, and low toxicity in vivo.7, 14
ABUNDANT CYCLIN D1 WAS DETECTED IN THE AUTOPHAGOSOME WHEN LYSOSOME FUSION WAS BLOCKED BY CQ UNDER AUTOPHAGY INDUCTION CONDITIONS
We further revealed the colocalization of cyclin D1 with LC3 protein (AP marker) in Hep 3B cells after autophagy induction (Fig. 3A), suggesting that cyclin D1 is linked with AP. We then biochemically purified the AP from Hep 3B cells in the presence of CQ (a blocker of AP and lysosome fusion) to enrich the quantity of AP obtained. We observed large amounts of LC3‐II (AP marker) and cyclin D1 proteins in the purified AP fraction as compared to the postnuclear supernatant (PNS) fraction (Fig. 3B). We also observed abundant immunogold‐labeled cyclin D1 particles in the double‐membrane AP‐like vesicles under transmission electron microscopy (TEM; Fig. 3C). In summary, increased cyclin D1 existed in the double‐membrane AP‐like vesicles before fusing with the lysosome following autophagy induction.
Figure 3.
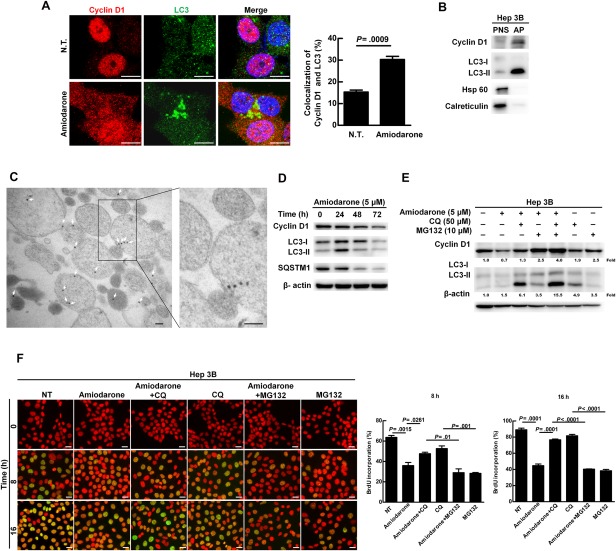
Increased autophagy caused cyclin D1 degradation and the existence of cyclin D1 in the AP. (A) Hep 3B cells were treated with or without amiodarone (5 μM) for 24 hours. The representative confocal images show cyclin D1 and LC3 protein. Scale bar = 10 μm. Quantification of colocalization of cyclin D1 and LC3 was conducted by randomly counting 30 cells. The data represent means ± SEM. (B) Hep 3B cells were treated with amiodarone (5 μM) for 24 hours followed by CQ (50 μM) treatment for another 24 hours. The proteins in the PNS and AP were analyzed by immunoblotting for the expression levels of cyclin D1, LC3, Hsp 60 (mitochondria marker), and calreticulin (endoplasmic reticulum marker). This experiment was repeated three times. (C) Immunogold‐labeled cyclin D1 (20‐nm gold bead) in the purified double‐membrane APs was detected under TEM. Scale bar = 100 nm. (D) Hep 3B cells were treated with amiodarone (5 μM) for various times. (E) Hep 3B cells were treated with amiodarone (5 μM) for 48 hours. CQ (50 μM) and MG132 (10 μM) were added at 24 hours after amiodarone treatment for another 24 hours. The number under the individual band represents the fold changes as compared to the control (set as 1). (F) Hep 3B cells were synchronized and DNA synthesis was measured under the same conditions as shown in Fig. 2E by amiodarone, CQ, and/or MG132 treatment for 0, 8, and 16 hours. Scale bar = 20 μm. Quantification is shown in the diagram. Data represent means ± SEM (n=5). Abbreviation: Hsp 60, heat shock protein 60.
AUTOPHAGIC DEGRADATION MACHINERY PARTIALLY PARTICIPATES IN DEGRADATION OF CYCLIN D1 PROTEIN AND REGULATES CELL DNA SYNTHESIS
Lipidated LC3‐II and SQSTM1 proteins associate with the outer and inner AP membrane, and LC3‐II protein increases at the early stage of autophagy induction, but starts to decrease after AQP fuses with lysosome.15 In our study, we showed that activation of autophagy triggered the progression of autophagic flux within 72 hours as shown by the expression levels of LC3‐II and SQSTM1 proteins reaching a peak at 24 hours then declining from 48 to 72 hours. Under the same conditions, cyclin D1 protein, but not mRNA, expression gradually decreased (Fig. 3D and http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). We using CQ to block degradation, and the decrease of cyclin D1 expression after autophagy induction for 48 hours was blocked in the presence of CQ (Fig. 3E). Similarly, cyclin D1 degradation was also blocked in the presence of MG132 (proteasomal inhibitor; Fig. 3E), and an additive effect was observed when the cells received combination treatment with CQ and MG132 (Fig. 3E). We further clarified the effect of CQ and MG132 on cell‐cycle progression by BrdU staining. Our results showed that amiodarone‐triggered suppression of cell‐cycle progression was abolished by CQ treatment (Fig. 3F), but not by MG132 treatment (Fig. 3F). Altogether, our data indicate that autophagic degradation machinery is partially involved in cyclin D1 protein degradation and regulation of cell‐cycle progression.
UBIQUITINATION OF CYCLIN D1 IS REQUIRED FOR AP RECRUITMENT
Autophagic degradation has been defined as selective and nonselective. The former recognizes ubiquitinated proteins through specific receptors, which assist the selected cargo to interact with LC3‐II protein on the AP membrane followed by lysosome fusion and degradation.9 We found that the interaction between LC3 and cyclin D1 increased when autophagy was induced for 24 hours (at the stage of AP formation; Fig. 4A, left panel). Concomitantly, the level of poly‐ubiquitinated cyclin D1 also increased (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). To clarify whether cyclin D1 ubiquitination is required for AP recruitment when autophagy was induced, we transfected WT (p‐cyclinD1‐HA‐WT) or ubiquitination site‐deficient cyclin D1 plasmid DNA (p‐cyclinD1‐HA‐K33‐238R) into Hep 3B cells (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). The cells harboring the mutant cyclin D1 K33‐238R gene showed abolished ubiquitination and degradation when autophagy was induced (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). The interaction between LC3 and cyclin D1 protein was abrogated in the K33‐238R mutant cyclin D1–transfected cells compared to the WT cyclin D1 cells (Fig. 4A, right panel), and in the purified AP fraction, only WT cyclin D1 was detected (Fig. 4B). Furthermore, it has been reported that glycogen synthase kinase 3β (GSK3β)‐mediated cyclin D1 phosphorylation at Thr‐286 induces translocation of cyclin D1 from the nucleus to cytoplasm and induces ubiquitination‐mediated proteolysis.16 We demonstrated that phosphorylation of GSK‐3β was activated under autophagy induction (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo), and Thr‐286‐phosphorylated cyclin D1 was detected in the purified AP (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). We also found that the cyclin D1 mutant (T286A) abolished ubiquitination of cyclin D1 and lost the capability to interact with LC3 when autophagy was induced (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). Our results imply that both the phosphorylation and ubiquitination sites of cyclin D1 are essential for its selective recruitment and binding with LC3 following induction of autophagy.
Figure 4.
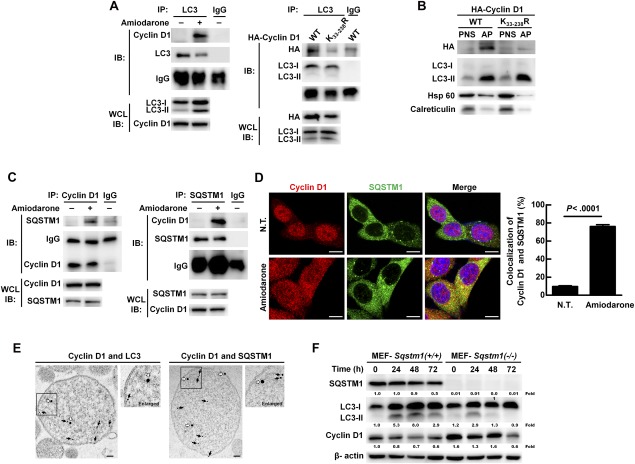
SQSTM1 is a receptor for selective recruitment and degradation of ubiquitinated cyclin D1 by the autophagic machinery in Hep 3B cells. (A) Hep 3B cells were treated with or without amiodarone (5 μM) for 24 hours (left panel). Hep 3B cells transfected under the same conditions as shown in http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo (right panel). Total protein extraction was immunoprecipitated by the anti‐LC3 antibody followed by IB. (B) Hep 3B cells were transfected under the same conditions as shown in http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo. These two cell lines were treated with amiodarone (5 μM) for 24 hours followed by CQ (50 μM) treatment for another 24 hours. Proteins in the PNS and AP of two cell lines were analyzed by IB. (C) Hep 3B cells were treated with or without amiodarone (5 μM) for 24 hours, and total protein extraction was immunoprecipitated by anti‐cyclin D1 or anti‐SQSTM1 antibody. (D) Hep 3B cells were treated with or without amiodarone (5 μM) for 24 hours. The representative confocal images show cyclin D1 and SQSTM1 protein. Scale bar = 10 μm. Quantification of cyclin D1‐SQSTM1 colocalization was performed by counting 30 cells, and data represent means ± SEM. (E) Coexistence of immunogold‐labeled cyclin D1 (20‐nm gold beads; white arrows) and immunogold‐labeled LC3 or SQSTM1 (12‐nm gold beads; black arrows) in the purified APs was examined under TEM. Scale bar = 100 nm. (F) MEF‐Sqstm1(+/+) and MEF‐Sqstm1(–/–) cells were treated with amiodarone (15 μM) for the times as indicated. Protein expression was measured by IB. Abbreviations: HA, hemagglutinin; Hsp 60, heat shock protein 60; WCL, whole cell lysate.
SQSTM1 RECEPTOR IS REQUIRED FOR CYCLIN D1 SELECTIVELY BINDING WITH LC3 ON THE AP AND REGULATION OF CELL‐CYCLE PROGRESSION
Diverse receptor proteins participate in selective autophagic degradation. Autophagy was induced in Hep 3B cells for 24 hours (the stage of AP formation) and the candidate receptor proteins, SQSTM1, NBR1, NDP52, and optineurin, were screened to detect binding with cyclin D1. Among the screened receptors, only SQSTM1 interacted with cyclin D1 by immunoprecipitation (IP) assay when autophagy was induced (Fig. 4C and http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). Concomitantly, we detected increased colocalization of SQSTM1 and cyclin D1 protein (Fig. 4D) and confirmed the coexistence of cyclin D1 with LC3 or SQSTM1 in the purified APs under TEM by immunogold labeling (Fig. 4E). We also found that the cyclin D1 protein degradation was prolonged in the MEF‐Sqstm1(–/–) cells compared to the MEF‐Sqstm1(+/+) cells (Fig. 4F), and the interaction between cyclin D1 and LC3 was only detected in MEF‐Sqstm1(+/+) after induction of autophagy (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). Furthermore, cell‐cycle progression increased in MEF‐Sqstm1(–/–) cells under autophagy induction for 8 and 16 hours (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). Altogether, our results imply that SQSTM1 is the receptor responsible for the interaction of cyclin D1 and LC3 in the AP and regulation of cell‐cycle progression.
ACTIVATED AUTOPHAGY LEADS TO DECREASED CELL PROLIFERATION, AS WELL AS REDUCED COLONY AND TUMOR FORMATION THROUGH DEGRADATION OF UBIQUITINATED CYCLIN D1
Initially, we demonstrated that activated autophagy had no effect on Hep 3B cell viability by flow cytometry analysis (Fig. 5A). Instead, it suppressed cell proliferation, and reduced colony and tumor formation (Fig. 5B‐D). This autophagy‐related suppression of tumorigenesis was counteracted by replenishment with the mutant, cyclin D1 (cyclin D1‐K33‐238R; Fig. 5B‐E), indicating that ubiquitinated cyclin D1 is required for autophagy‐mediated suppression of cell proliferation and tumorigenesis.
Figure 5.
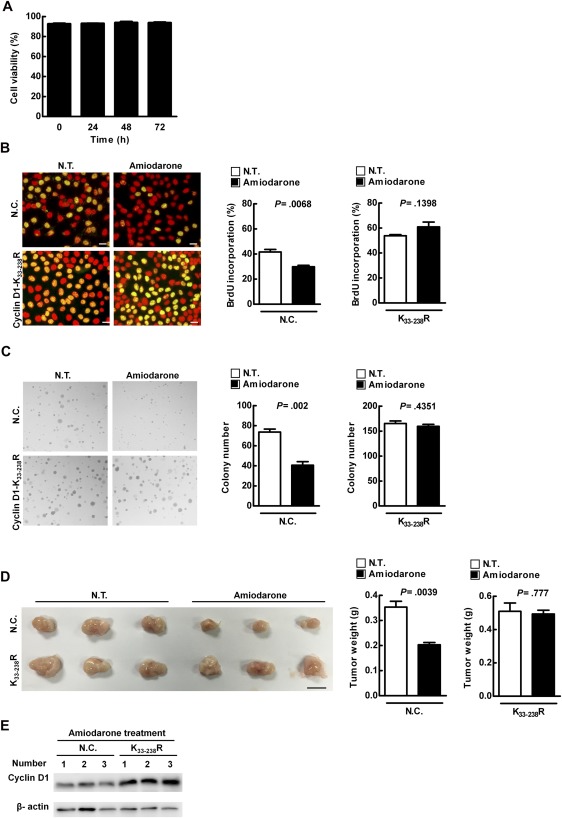
Activated autophagy leads to suppression of cell proliferation, as well as reduced colony and tumor formation, through regulation of cyclin D1 expression in Hep 3B cells. (A) Hep 3B cells were treated with amiodarone (5 μM) for various times, followed by propidium iodide (0.04 mg/mL) labeling. Cell viability was evaluated by flow cytometry analysis. (B‐E) Hep 3B cells were transfected with HA‐vector or mutant cyclin D1 plasmid (p‐cyclin D1‐HA‐K33‐238R). Cell proliferation was determined by BrdU incorporation assay in (B). Scale bar = 20 μm. Quantification is shown in the diagram. Data represent means ± SEM (n = 5). Soft agar assay was conducted and colony formation was measured at day 14 posttreatment in (C). This experiment was conducted in triplicate and repeated three times. Cells (1 × 107) were inoculated s.c. into NOD/SCID mice (n = 3). Mice were sacrificed and tumor weight was measured in (D). Scale bar = 1 cm. Protein expression level was evaluated by IB in (E). Abbreviations: HA, hemagglutinin; N.C., HA‐vector transfection; NOD/SCID, nonobese diabetic/severe combined immunodeficient; N.T., without amiodarone; s.c., subcutaneously.
HIGH CYCLIN D1 EXPRESSION ASSOCIATED WITH LOW AUTOPHAGIC ACTIVITY WAS DETECTED IN THE LIVER TUMORS OF TWO MURINE MODELS
To validate our findings in vivo, we checked the status of cyclin D1 expression as well as autophagic activity during liver tumor formation in two murine models. The first model is a viral‐related hepatitis B virus X protein (HBx) transgenic mice model (Fig. 6A, upper panel), in which a liver tumor is induced by HBx protein at 16 months. The second model is a nonviral rat orthotopic liver tumor model (Fig. 6A, lower panel), in which the inoculated rat hepatoma N1‐S1 cells induce liver tumor formation within 1 week.7 In these two models, low BECN1 expression accompanied with high SQSTM1 accumulation as well as high cyclin D1 expression were observed in liver tumor tissues compared to nontumor tissues (Fig. 6B,C), indicating that low autophagic activity, together with high cyclin D1 expression, is associated with liver tumor formation.
Figure 6.

High cyclin D1 expression accompanied with low autophagic activity was observed in murine models, and increase of autophagic activity suppressed liver tumor formation in an orthotopic rat model. (A) Representative images show liver without tumor from 6‐month‐old transgenic mice (6 M), liver with tumor from 16‐month‐old transgenic mice (16 M; upper panel), and normal liver and liver with tumor of 4‐week‐old rats from day 7 postinoculation (lower panel). Arrows point to the tumors. Scale bar = 1 cm. (B,C) Representative IHC images showed the protein expression levels in liver tissues. Scale bar=20 μm. The protein expression was quantified by ROI followed by HistoQuest analysis. (D) Rat hepatoma N1‐S1 cells were orthotopically inoculated into rat livers. The representative image shows the liver and the arrows point to the tumors. Scale bar = 1 cm. Tumor formation was quantified by measuring the tumor weight. (E) Representative confocal images show cyclin D1 and LC3 protein in tumor tissues. Scale bar = 10 μm. (F) Representative IHC images show the protein expression levels in N1‐S1 hepatoma cell–induced tumor tissues. Scale bar = 20 μm. Quantification of protein expression was determined by HistoQuest software analysis. (C‐F) Data represent means ± SEM (n = 5). Abbreviation: ROI, region of interest.
PHARMACOLOGICAL INDUCERS OF AUTOPHAGY EFFECTIVELY SUPPRESSED TUMOR FORMATION IN TWO MURINE MODELS
We further clarified whether increased autophagic activity could lead to regression of liver tumor in the rat orthotopic model. Our data demonstrate that the weight of the liver tumor decreased by around 60% in the amiodarone treatment group compared to that in the double‐distilled water (DDW) control group (Fig. 6D). LC3‐II expression and the number of double‐membrane APs was increased in tumor tissues of amiodarone‐treated rats compared to DDW‐treated rats (n = 5; http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). We also detected increased colocalization of cyclin D1 and LC3, together with decreased expression of cyclin D1 and SQSTM1, in tumors of the inducer‐treated rats compared to those of the DDW group (Fig. 6E,F). We further used the inducers, amiodarone and rapamycin, in the hepatoma Hep 3B xenograft tumor model. Similarly, we detected significant regression of tumors (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo) and increased colocalization of cyclin D1 and LC3 (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo), together with decreased expression of cyclin D1, SQSTM1, and Ki67 (cell proliferation marker), in tumors (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo) of inducer‐treated mice compared to those of the untreated group (nontreated; N.T.). Altogether, these data demonstrated that increased autophagic activity suppressed liver tumor formation through degradation of cyclin D1.
Discussion
In this study, high cyclin D1 expression accompanied with low autophagic activity was correlated with liver tumor formation and poor survival in HCC patients. Mechanistically, under autophagy induction conditions, cyclin D1 is ubiquitinated followed by proteasomal and autophagy degradation. However, the latter process is mediated by SQSTM1 leading to cyclin D1 degradation, cell‐cycle arrest, and low cell proliferation rate (Fig. 7A). We further suggest that in HCC patients, low autophagic activity accompanied with high cyclin D1 accumulation contributes to induction of cell‐cycle progression, cell proliferation, and liver tumor formation (Fig. 7B).
Figure 7.
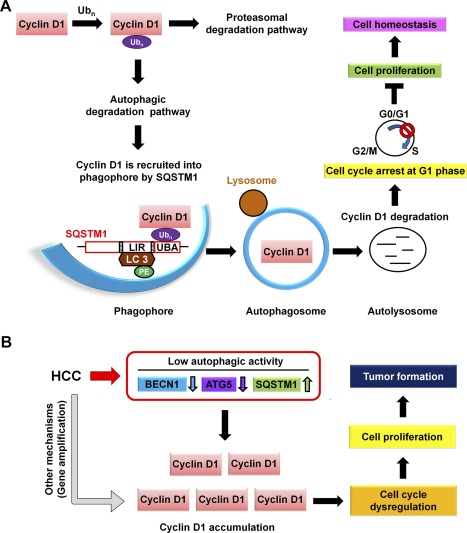
A schematic hypothetical model shows how autophagy selectively regulates cyclin D1 to affect cell proliferation and tumorigenesis in the normal cell and HBV‐ and HCV‐related HCC. (A) Under normal conditions, stress‐induced autophagy triggers posttranslational modification of cyclin D1 by ubiquitination. Ubiquitinated cyclin D1 is either degraded by proteasome or selectively recruited into the AP mediated by the receptor, SQSTM1, followed by fusion with lysosome for degradation during autophagy progression. Suppression of cell‐cycle progression results from cyclin D1 degradation by autophagy to maintain cell homeostasis. (B) Low autophagic activity was detected in HBV‐ and HCV‐related HCC patients. Low autophagic activity causes accumulation of cyclin D1 and promotes cell proliferation and tumor formation. Abbreviation: UBA, ubiquitin associated.
During hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, autophagy is induced to sustain the replication of the viruses.17 However, in the present study, we detected low autophagic activity in HBV‐ and HCV‐associated HCC tumor tissues. One possibility is that HBV HBx or HCV nonstructural 4B protein suppresses the late‐stage autophagic process by impairing fusion with lysosomes in virus‐associated HCC tumorigenesis.18, 19 Another possibility is that HBV genomic DNA may randomly integrate into the host chromosome and disrupt autophagy‐related genes during the latent stages of viral infection.20, 21 Nevertheless, the mechanism by which autophagy switches from high level (proviral replication) to low level (antiviral replication) to promote HCC tumorigenesis during liver virus infection warrants further investigation.
Cyclins and CDKs are ubiquitinated and degraded through the proteasomal pathway to maintain cell‐cycle progression.22 Loukil et al. reported that autophagy is a regulator of cyclin A2 degradation beyond the metaphase of the cell cycle in proliferating cells.23 We show that both of the autophagic and proteasomal degradation systems degrade cyclin D1, as demonstrated by MG132 and CQ treatment. However, the subsequent suppression of the cell‐cycle progression was only rescued by CQ treatment, but not by MG132 (Fig. 3F). This discrepancy indicates that the autophagic degradation pathway seems to be more specific for regulating cyclin D1–mediated cell‐cycle progression. It is also possible that there are undetermined factors controlling cell DNA synthesis, which are regulated by autophagic, but not proteasomal, degradation machinery. Altogether, autophagic degradation machinery and cell‐cycle regulator cyclin D1 are linked with the homeostasis of cell‐cycle progression in HCC.
Both GSK‐3β and dual‐specificity tyrosine phosphorylation regulated kinase 1B (DYRK1B) are involved in cyclin D1 phosphorylation and protein turnover.24 In this study, induction of autophagy increased GSK‐3β phosphorylation as well as detected phosphorylated cyclin D1 (Thr‐286) in the purified AP (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). We further abolished the Thr‐286 phosphorylation site of cyclin D1 (T286A) and confirmed that it is essential for cyclin D1 ubiquitination and binding with LC3 (http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). Our findings suggest that induction of autophagy leads to cyclin D1 turnover in a GSK‐3β–dependent manner. Under these conditions, the cell cycle was blocked at the G0/G1 phase accompanied with suppression of cell proliferation, but without evident cell death (Fig. 5). However, Yan et al. used low‐dose bafilomycin A1 to block autophagic flux, which also triggered cyclin D1 turnover, but in a DYRK1B‐dependent fashion, leading to the caspase‐independent death of HCC cells.25 Based on our findings and other reports, it appears that either induction or suppression of autophagy activation could trigger cyclin D1 turnover, but through different signaling pathways, which further determine the fate of the cells. Altogether, suppression of cell‐cycle progression through autophagy‐regulated cyclin D1 turnover may have potential as an effective therapy for HCC.
Autophagy deficiency was shown to result in multiple liver tumors in a mouse model.13 We found that in HCC patient tumors, autophagic activity was low, and an increase of autophagic activity effectively suppressed liver tumor formation in the animal models (Fig. 6D and http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo). In contrast, Toshima et al. reported that autophagy promotes cell proliferation and liver undergoes rapid protein turnover for remodeling after partial hepatectomy. They revealed that autophagy sustains epigenetic regulation to prevent senescence during liver regeneration (LR).26 In summary, these data indicate that autophagy plays different roles in LR and HCC tumorigenesis.
Amiodarone as an autophagy inducer could reduce liver injury and promote LR after partial hepatectomy, and amiodarone‐treated patients showed no evident toxicity.27, 28 Similarly, our results demonstrate that amiodarone is an antitumor agent against liver tumor formation. However, Kowalik et al. reported that amiodarone‐induced autophagy stimulated the proliferation of keratin 19 (a progenitor cell marker) preneoplastic lesions, leading to HCC progression at a very early stage in a rat model.29 Tian et al. reported that autophagy switches from protection to promotion of HCC tumorigenesis at the late stages by suppressing the expression of multiple tumor suppressors, including p53.30 To understand the possible anti‐HCC effect of amiodarone in the general population, we conducted a retrospective analysis of case‐control data comprising 32,625 patients sampled from a nation‐wide, population‐based claims database in Taiwan, and revealed a prophylactic effect after long‐term regular amiodarone usage. In contrast, short‐term exposure to amiodarone was associated with a higher risk of HCC incidence.31
Our result indicates that the protective effects of amiodarone may occur only after approximately 2 years of exposure (data not shown). A pharmaco‐epidemiological study using a larger sample size is needed to confirm this hypothesis. Altogether, autophagy plays different roles in LR and HCC tumorigenesis, amiodarone‐induced autophagy may affect HCG differently; therefore, we should be cautious about the timing of treatment.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo.
Supporting Information 1
Acknowledgments
We thank Dr. N. Mizushima (Tokyo Medical and Dental University, Japan) and Dr. T. Yoshimori (Osaka University, Japan) for providing MEF‐Atg5(+/+), MEF‐Atg5(–/–), MEF‐Sqstm1(+/+), and MEF‐Sqstm1(–/–) cell lines; Dr. E. Dmitrovsky (Dartmouth‐Hitchcock Medical Center, USA) for providing cyclin D1 plasmids; and Drs. Kuen‐Jer Tsai and Ya‐Chun Hsiao for conducting fluorescence‐activated cell sorting–like tissue cytometry, image acquisition, and data analysis.
Potential conflict of interest: Nothing to report.
Supported by a grant from the Ministry of Science and Technology (MOST‐104‐2320‐B‐006‐021‐MY3).
REFERENCES
- 1. Singh S, Singh PP, Roberts LR, Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2014;11:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bisteau X, Caldez MJ, Kaldis P. The complex relationship between liver cancer and the cell cycle: a story of multiple regulations. Cancers (Basel) 2014;6:79‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Che Y, Ye F, Xu R, Qing H, Wang X, Yin F, et al. Co‐expression of XIAP and cyclin D1 complex correlates with a poor prognosis in patients with hepatocellular carcinoma. Am J Pathol 2012;180:1798‐1807. [DOI] [PubMed] [Google Scholar]
- 4. Deane NG, Parker MA, Aramandla R, Diehl L, Lee WJ, Washington MK, et al. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res 2001;61:5389‐5395. [PubMed] [Google Scholar]
- 5. Woo HG, Park ES, Thorgeirsson SS, Kim YJ. Exploring genomic profiles of hepatocellular carcinoma. Mol Carcinog 2011;50:235‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McEwan DG, Dikic I. The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol 2011;21:195‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, et al. Autophagy suppresses tumorigenesis of hepatitis B virus‐associated hepatocellular carcinoma through degradation of microRNA‐224. Hepatology 2014;59:505‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 2015;16:461‐472. [DOI] [PubMed] [Google Scholar]
- 9. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011;7:279‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol 2010;32:431‐436. [DOI] [PubMed] [Google Scholar]
- 11. Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ, et al. Autophagy and SQSTM1 on the RHOA(d) again: emerging roles of autophagy in the degradation of signaling proteins. Autophagy 2014;10:201‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol 2010;12:781‐790. [DOI] [PubMed] [Google Scholar]
- 13. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev 2011;25:795‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009;4:e7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbash O, Diehl JA. SCF(Fbx4/alphaB‐crystallin) E3 ligase: when one is not enough. Cell Cycle 2008;7:2983‐2986. [DOI] [PubMed] [Google Scholar]
- 17. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV‐ and HCV‐associated hepatocellular carcinoma. Nat Rev Cancer 2013;13:123‐135. [DOI] [PubMed] [Google Scholar]
- 18. Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014;10:416‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang L, Tian Y, Ou JH. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog 2015;11:e1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fallot G, Neuveut C, Buendia MA. Diverse roles of hepatitis B virus in liver cancer. Curr Opin Virol 2012;2:467‐473. [DOI] [PubMed] [Google Scholar]
- 21. Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, et al. Viral‐human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014;25:335‐349. [DOI] [PubMed] [Google Scholar]
- 22. Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors 2006;24:13‐19. [DOI] [PubMed] [Google Scholar]
- 23. Loukil A, Zonca M, Rebouissou C, Baldin V, Coux O, Biard‐Piechaczyk M, et al. High resolution live cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci 2014. [DOI] [PubMed] [Google Scholar]
- 24. Zou Y, Ewton DZ, Deng X, Mercer SE, Friedman E. Mirk/dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J Biol Chem 2004;279:27790‐27798. [DOI] [PubMed] [Google Scholar]
- 25. Yan Y, Jiang K, Liu P, Zhang X, Dong X, Gao J, et al. Bafilomycin A1 induces caspase‐independent cell death in hepatocellular carcinoma cells via targeting of autophagy and MAPK pathways. Sci Rep 2016;6:37052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toshima T, Shirabe K, Fukuhara T, Ikegami T, Yoshizumi T, Soejima Y, et al. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology 2014;60:290‐300. [DOI] [PubMed] [Google Scholar]
- 27. Lin CW, Chen YS, Lin CC, Chen YJ, Lo GH, Lee PH, et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep 2015;5:15807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guiu B, Colin C, Cercueil JP, Loffroy R, Guiu S, Ferrant E, et al. Pilot study of transarterial chemoembolization with pirarubicin and amiodarone for unresectable hepatocellular carcinoma. Am J Clin Oncol 2009;32:238‐244. [DOI] [PubMed] [Google Scholar]
- 29. Kowalik MA, Perra A, Ledda‐Columbano GM, Ippolito G, Piacentini M, Columbano A, et al. Induction of autophagy promotes the growth of early preneoplastic rat liver nodules. Oncotarget 2016;7:5788‐5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, et al. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ 2014;22:1025‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Su VY, Hu YW, Chou KT, Ou SM, Lee YC, Lin EY, et al. Amiodarone and the risk of cancer: a nationwide population‐based study. Cancer 2013;119:1699‐1705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29781/suppinfo.
Supporting Information 1