

Abstract
This work showcases the 1,3‐haloboration reaction of alkynes in which boron and chlorine add to propargyl systems in a proposed sequential oxazoliumborate formation with subsequent ring‐opening and chloride migration. In addition, the functionalization of these propargyl esters with dimethyl groups in the propargylic position leads to stark differences in reactivity whereby a formal 1,1‐carboboration prevails to give the 2,2‐dichloro‐3,4‐dihydrodioxaborinine products as an intramolecular chelate. Density functional theory calculations are used to rationalize the distinct carboboration and haloboration pathways. Significantly, this method represents a metal‐free route to highly functionalized compounds in a single step to give structurally complex products.
Keywords: alkynes, boranes, boron, haloboration, Lewis acid
The activation of carbon–carbon double and triple bonds by main group compounds has been a staple motif in the synthesis of a plethora of new element–carbon bonds such as C−C,1 C−H,2 C−N,3 C−B,4 and C−O5 bonds amongst many others.6 Seminal work by Wrackmeyer et al. showcased a powerful methodology using trivalent boranes in conjunction with “activated” alkynes, that is, M‐C≡C‐R, where M=Si, Ge, Sn, Pb inter alia.7 In these early cases, 1,1‐carboboration reactions were observed whereby a 1,2‐alkyl/aryl shift occurs between the distal and proximal carbons of the alkyne with the concomitant 1,2‐shift of the R group from boron to carbon (Scheme 1, top). Further to this, Erker has demonstrated extensive use of the carboboration mechanism to affect a number of complex rearrangement processes such as benzannulations8 and cyclizations,9 amongst others.10
Scheme 1.

Background and overview to this work.
More recent work in such elementoboration reactions are seen through the synthetically useful haloboration reaction whereby a halogen, predominantly chlorine, is installed typically through a 1,1‐ or 1,2‐haloboration to yield the corresponding halovinylboronic ester (Scheme 1, top). The formation of these species has been generated through the use of simple haloboranes such as BX3 (X=Cl, Br), or borocations developed by Ingleson et al. In the case of borocations, stereoselective control is observed to give predominantly the syn‐addition product.11 Interestingly, a similar study showcased a sequential alkyne addition to affect a formal 1,4‐haloboration whereby phenylacetylene undergoes a 1,2‐addition when exposed to [LutBCl2][AlCl4] which, upon addition of various 1‐trimethylsilylalkynes, undergoes a subsequent 1,2‐carboboration to yield the diene product.11d All such haloboration reactions are convenient synthetic protocols to append functional groups to olefins, specifically in the formation of tri‐ and tetra‐substituted alkenes through subsequent cross‐coupling reactions of the boronic ester.12 Another aspect of the reaction outlined within is the installation of a pendant alkyl chloride (Scheme 1, bottom), which has countless uses within organic chemistry from reactions with acetylides, alkoxides, and cyanates, as well as Grignard chemistry.
Although a significant amount of research has focused on the sterically encumbered, strong Lewis acid, B(C6F5)3, as well as others of a similar nature,13 other commercially available boranes have seemingly been absent from recent studies. Hence, this work aims to reinvigorate the use of such boranes in a range of synthetically imperative transformations.
Herein, we show how subtle adaptations to the alkyne starting material can dramatically alter the reactivity with the borane reagent to give the stereoselective trans‐product of a formal 1,3‐haloboration, or alternatively a complex 1,1‐carboboration mechanism to yield a stable dichlorodihydrodioxaborinine heterocycle all in very good to excellent conversions. Importantly, these reagents are then well positioned to undergo further functionalization such as cross‐couplings14 or allylations.15
Initial investigations of the commercially available PhBCl2 used the model substrate 1 a in a 1:1 stoichiometric ratio to yield a single product in near quantitative yields as observed using in situ multinuclear NMR spectroscopy. Detailed NMR spectroscopy (HSQC, HMBC) revealed the proposed structure of 3 a, which interestingly is the product of a formal 1,3‐haloboration reaction. These encouraging initial results then led us to expand the substrate scope to a series of phenyl substituted propargyl esters (Figure 1, Scheme 2). It was observed that in most cases the target haloboration product could be clearly identified with conversions greater than 95 % at ambient temperature with reaction times of 8 h (3 a), 18 h (3 b,c), and 48 h (3 d).
Figure 1.

Propargyl ester substrates used in this work.
Scheme 2.

Reaction between PhBCl2 and 1 to give 1,3‐haloboration products 3. Values are given as in situ NMR conversions. Solid‐state structure of compound 3 c, C: grey, H: white, N: blue, O: red, B: yellow‐green, Cl: green. Thermal ellipsoids shown at 50 % probability (inset).
Fortunately, the storage of a saturated CH2Cl2/hexane solution of 3 c at −40 °C produced a crop of crystals suitable for X‐ray diffraction. The structure was unambiguously determined to be indeed the product of a formal 1,3‐haloboration agreeing with spectroscopic analyses (Figure 2). Metrics of the solid‐state structure are as expected with the stereochemical conformation being determined as the trans product. Earlier work by Erker showcased the ability of vinylboranes to undergo photoinduced interconversion between the E/Z conformers upon exposure to UV light;16 thus, it was hoped similar reactivity could be observed here to yield the intramolecular chelate. However, no such species could be detected in the 11B NMR post‐irradiation, leaving the spectra identical to that of the non‐irradiated product 3.
Figure 2.

Stacked in situ spectra for the reaction between propargyl ester 1 a or 1 aD and PhBCl2 to yield a) 3 a (1H); b) 3 aD (1H); c) 3 a (2H); d) 3 aD (2H).
Conducting the reaction between PhBCl2 and 1 a in a variation of solvents ([D6]benzene, [D8]toluene, CH2Cl2, C6H5Cl) appeared to make no difference in reactivity with all showing almost quantitative conversion within 9 hours. Additionally, trialling other boron reagents such as BCl3 as well as the borocation [PhClB(2‐DMAP)][AlCl4]11a were unsuccessful with a mixture of products prevailing as observed in the resultant multinuclear NMR spectra.
To gain some insight into the proposed mechanism, isotopic labeling studies were performed. The terminal position of the alkyne was deuterated selectively using an amine appended resin (WA50) in accordance with the literature.17 Tracking the reaction progress using both 1H and 2H NMR spectroscopy whilst comparing the in situ data of the protic versus deuterated compounds shed light on the fate of the terminal alkynyl hydrogen atom and hence the reaction mechanism (see below, Scheme 4). A 1H resonance at δ=6.6 ppm is observed in 3 a for the proton on the carbon adjacent to boron, which is evidently absent in 3 aD (Figure 2). Additionally, following the 2H NMR spectra of the reactions using 1 a and 1 aD clearly shows the alkyne resonance at δ=2.5 ppm diminishing in intensity with the commensurate appearance of the previously identified new resonance at δ=6.6 ppm.
Scheme 4.

Proposed mechanism for the divergent elementoboration of 1 and 2 using PhBCl2.
Further derivatization of the starting materials to include methyl groups in the propargylic position was undertaken to yield compounds 2 a–2 c (Figure 1). Upon exposure of 2 to a stoichiometric amount of PhBCl2, new resonances in the 1H and 11B NMR spectra were noted after 8 h at 45 °C, which, interestingly, were not consistent with the 1,3‐haloboration products 3 from reagents 1. Indeed, a broad singlet resonance was observed in the 1H NMR spectrum at ca. δ=3.8 ppm alongside a sharp singlet resonance in the 11B NMR spectrum at ca. δ=8 ppm indicating the formation of a chelating dioxaborinine type structure as seen in Scheme 3. This was further expounded by means of the 13C NMR spectra with the presence of a new sp 3 carbon adjacent to boron presenting a resonance at ca. δ=40 ppm versus 120 ppm for the adjacent sp2 carbon in 3. Storing 4 a–c as a saturated CH2Cl2/hexane solution produced a number of colorless crystals suitable for X‐ray diffraction, which indeed determined the molecular structure to be the product of a formal 1,1‐carboboration reaction (Figure 3). Of particular note is the regioselectivity of this reaction with the product predominating as the selective transfer of the aryl group over the chloride fragment.18 This migration pattern could be confirmed once again through detailed 2D NMR spectroscopy to affirm the molecular connectivity revealed in the solid‐state structure (see the Supporting Information).
Scheme 3.

Reaction between PhBCl2 and 2 to give 1,1‐carboboration products 4. Yields are given as isolated yields.
Figure 3.

Solid‐state structure of compounds 4 a–c, C: grey, H: white, O: red, B: yellow‐green, Cl: green, F: pink. Thermal ellipsoids shown at 50 % probability.
When comparing the divergent elementoboration observed here, it is proposed that the inclusion of non‐H groups in the propargylic position must play a critical role in which pathway is undertaken in this reaction. Mechanistically we propose that an initial 1,2‐trans‐oxyboration step occurs to yield the zwitterionic dioxolium borate.19 During the formation of this 5‐membered dioxolium intermediate (I, Scheme 4), when simple hydrogen atoms occupy the R2 position, the formation of 3 is slightly more favorable compared to when methyl groups are included in the R2 position. Conversely, if more bulky methyl groups are included, then the chloride migration pathway is less favored over 1,2‐aryl group migration resulting in the generation the intramolecular chelate 4. These experimental findings are supported through in silico studies (see below and the Supporting Information). Additionally, when using compound 5, which features a combination of H and Me in the propargyl position, a more complex transformation is observed when monitoring the reaction coordinate over time. Analyzing the in situ 1H and 11B NMR spectra suggests that, after initial combination of PhBCl2 with 5 in a 1:1.2 ratio, the haloboration product 6 prevails, as observed by the characteristic broad singlet at δ=6.35 ppm alongside the formation of a resonance at ca. δ=5.2 ppm for the proposed vinyl and methylene protons, respectively (see the Supporting Information). Over time, these resonances reduce in intensity giving way to a new broad singlet at δ=3.36 ppm, consistent with the generation of the proton in the adjacent to the borane in the chelating structure 7. In addition, new resonances appear for the newly formed vinyl proton quartet at δ=5.45 ppm, and the methyl doublet at δ=1.90 ppm. This assertion is bolstered when observing the in situ 11B NMR spectra over time whereby the expected singlet at about δ=55.1 ppm forms after 1 h at ambient temperature, which reduces in intensity over time yielding another singlet resonance at ca. δ=9.1 ppm, again indicating the reversible formation of 6 en route to 7 (Scheme 5).
Scheme 5.
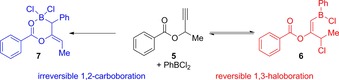
Conversion of 5 to 6 and 7 via a proposed reversible 1,3‐haloboration or 1,2‐carboboration mechanism.
To shed light on the divergent reactivity realized in this work, DFT calculations were performed. The reaction pathways for haloboration (upper) and carboboration (lower) are displayed in Figure 4. Pleasingly, the formation of products 3 and 4 proceeds in line with the proposed Scheme 4 via the key dioxolium intermediate I. Once the intermediate I is formed, chloride migration is the transition state for the haloboration reaction and phenyl migration is the rate‐determining transition state for the carboboration reaction. It is clear from Figure 4, upon comparison of the pathways, that carboboration (R2=Me) is strongly thermodynamically preferred over haloboration (R2=H). Intriguingly, however, we found that the hypothetical carboboration product (see the Supporting Information, Table S1) for R2=H is >30 kcal mol−1 more stable than the R2=H haloboration product obtained experimentally, raising the question: why does the carboboration reaction not occur for R2=H?
Figure 4.
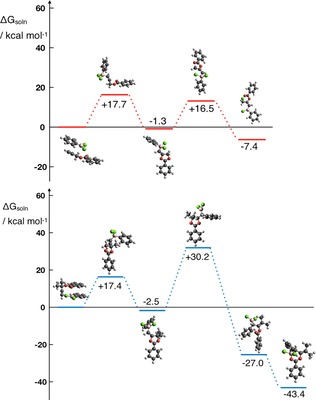
Free energy diagram comparing the mechanism of the haloboration (red) and carboboration (blue) reactions yielding products 3 and 4 respectively.
After 1,2‐trans‐oxyboration and formation of the intermediate dioxolium intermediate I, two pathways are available: 1) chloride migration that results in a metastable haloboration product or 2) phenyl migration that yields the thermodynamically favored carboboration product. The carboboration reaction of 1 a is disfavored for two reasons: first, following the kinetically favored pathway, chloride migration occurs yielding the haloboration product and reversion back to the dioxolium I is strongly hindered by a high reverse barrier (ΔG gas= +28.9 kcal mol−1) from the product. Second, in order to yield the carboboration product from I, a prohibitively large barrier for phenyl migration (ΔE gas=+37.20 kcal mol−1) must be overcome (see the Supporting Information, sections 3.2.3 and 3.2.4). For comparison, the phenyl migration barrier for compounds 2 a (R2=Me) is 9 kcal mol−1 lower than for 1 a (R2=H). Hence, for 1 a (R2=H), the kinetically favored haloboration product is formed. Conversely, the haloboration product of 2 a (R2=Me) has a comparatively low reverse barrier from the product (ΔG gas=+23.3 kcal mol−1) and also a low barrier for phenyl migration (ΔE gas=+28.2 kcal mol−1) in comparison to that for 1 a (R2=H). Hence, any haloboration product formed from starting materials 2 a may revert to I fleetingly and phenyl migration will occur. Once phenyl migration occurs, there is a strong free energy incentive to form the thermodynamically preferred chelate 4, which has a very prohibitive >+70 kcal mol−1 barrier (ΔG sol) to revert to I.
Intriguingly, in line with the observed reversible behavior of the mono‐methylate 5 to form haloboration product 6 and then the carboboration product 7 (see Scheme 5), calculations show a reverse barrier from the product of ΔG gas=+25.8 kcal mol−1, which is intermediate between those found for R2=H and R2=Me and an intermediate barrier for phenyl migration of ΔE gas=+29.2 kcal mol−1. Hence, according to mechanistic and energetic considerations, the mono‐methylate should form a more kinetically stable haloboration product than bi‐methylate and is therefore more likely to be isolable under comparable reaction conditions, exactly in line with our findings. The phenyl migration barrier is lower for R2=Me, H than R2=H, so the carboboration product is more likely to be isolable, again in agreement with our observations in Scheme 5. These findings show that 3 and 6 are more kinetically stable than the haloborated product of 2 a (R2=Me), explaining why both 3 and 6 are observed. The relatively low barriers for phenyl migration to form 4 and 7 with the strong thermodynamic driving force explain why the carboboration products occur for R2=Me and R2=Me, H. Overall, these calculations reveal a remarkably subtle interplay of kinetic and thermodynamic factors that are acutely sensitive to the R2 groups and which cause profoundly different reaction products.
In summary, this work has shown both the formal 1,1‐carboboration as well as formal 1,3‐haloboration of alkynes can occur through simple tuning of the alkyne starting material being used. All of these multi‐step reactions proceed cleanly with high conversions and yields being noted in a one‐pot, atom efficient manner, garnering synthetically useful and functionally diverse compounds for further reactivity. In depth computational studies have helped elucidate the proposed mechanism that differentiates this divergent elementoboration.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.L.M. would like to acknowledge the EPSRC (EP/N02320X/1) for funding. B.S. wishes to acknowledge ARCHER UK National Supercomputing Service for support of this work and access through EPSRC grant (EP/L000202).
L. C. Wilkins, Y. Soltani, J. R. Lawson, B. Slater, R. L. Melen, Chem. Eur. J. 2018, 24, 7364.
References
- 1.
- 1a. Reddy J. S., Xu B.-H., Mahdi T., Fröhlich R., Kehr G., Stephan D. W., Erker G., Organometallics 2012, 31, 5638–5649; [Google Scholar]
- 1b. Hansmann M. M., Melen R. L., Rudolph M., Rominger F., Wadepohl H., Stephan D. W., Hashmi A. S. K., J. Am. Chem. Soc. 2015, 137, 15469–15477; [DOI] [PubMed] [Google Scholar]
- 1c. Soltani Y., Wilkins L. C., Melen R. L., Angew. Chem. Int. Ed. 2017, 56, 11995–11999; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12157–12161. [Google Scholar]
- 2.
- 2a. Chernichenko K., Madarász Á., Pápai I., Nieger M., Leskelä M., Repo T., Nat. Chem. 2013, 5, 718–723; [DOI] [PubMed] [Google Scholar]
- 2b. Hounjet L. J., Stephan D. W., Org. Process Res. Dev. 2014, 18, 385–391; [Google Scholar]
- 2c. Lawson J. R., Wilkins L. C., Melen R. L., Chem. Eur. J. 2017, 23, 10997–11000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Yin Q., Soltani Y., Melen R. L., Oestreich M., Organometallics 2017, 36, 2381–2384. [Google Scholar]
- 3. Wixey J. S., Ward B. D., Chem. Commun. 2011, 47, 5449–5451. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Piers W. E., in Advances in Organometallic Chemistry, Vol. 52, Academic Press, Cambridge, 2004, pp. 1–76; [Google Scholar]
- 4b. Melen R. L., Hansmann M. M., Lough A. J., Hashmi A. S. K., Stephan D. W., Chem. Eur. J. 2013, 19, 11928–11938; [DOI] [PubMed] [Google Scholar]
- 4c. Feldmann A., Kehr G., Daniliuc C. G., Muck-Lichtenfeld C., Erker G., Chem. Eur. J. 2015, 21, 12456–12464. [DOI] [PubMed] [Google Scholar]
- 5. Faizi D. J., Issaian A., Davis A. J., Blum S. A., J. Am. Chem. Soc. 2016, 138, 2126–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Bayne J. M., Stephan D. W., Chem. Soc. Rev. 2016, 45, 765–774; [DOI] [PubMed] [Google Scholar]
- 6b. Issaian A., Tu K. N., Blum S. A., Acc. Chem. Res. 2017, 50, 2598–2609. [DOI] [PubMed] [Google Scholar]
- 7. Wrackmeyer B., Coord. Chem. Rev. 1995, 145, 125–156. [Google Scholar]
- 8. Liedtke R., Tenberge F., Daniliuc C. G., Kehr G., Erker G., J. Org. Chem. 2015, 80, 2240–2248. [DOI] [PubMed] [Google Scholar]
- 9. Chen C., Harhausen M., Liedtke R., Bussmann K., Fukazawa A., Yamaguchi S., Petersen J. L., Daniliuc C. G., Fröhlich R., Kehr G., Erker G., Angew. Chem. Int. Ed. 2013, 52, 5992–5996; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6108–6112. [Google Scholar]
- 10. Kehr G., Erker G., Chem. Sci. 2016, 7, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Yao M.-L., Reddy M. S., Zeng W., Hall K., Walfish I., Kabalka G. W., J. Org. Chem. 2009, 74, 1385–1387; [DOI] [PubMed] [Google Scholar]
- 11b. Lawson J. R., Clark E. R., Cade I. A., Solomon S. A., Ingleson M. J., Angew. Chem. Int. Ed. 2013, 52, 7518–7522; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7666–7670; [Google Scholar]
- 11c. Ingleson M. J., in Fundamental and Applied Properties of Borocations, Synthesis and Application of Organoboron Compounds, Springer, Cham, 2015, 39–71; [Google Scholar]
- 11d. Warner A. J., Lawson J. R., Fasano V., Ingleson M. J., Angew. Chem. Int. Ed. 2015, 54, 11245–11249; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11397–11401; [Google Scholar]
- 11e. Lawson J. R., Fasano V., Cid J., Vitorica-Yrezabal I., Ingleson M. J., Dalton Trans. 2016, 45, 6060–6070. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Hara S., Dojo H., Takinami S., Suzuki A., Tetrahedron Lett. 1983, 24, 731–734; [Google Scholar]
- 12b. Satoh Y., Tayano T., Koshino H., Hara S., Suzuki A., Synthesis 1985, 1985, 406–408; [Google Scholar]
- 12c. Satoh Y., Serizawa H., Hara S., Suzuki A., J. Am. Chem. Soc. 1985, 107, 5225–5228; [Google Scholar]
- 12d. Sato M., Yamamoto Y., Hara S., Suzuki A., Tetrahedron Lett. 1993, 34, 7071–7074. [Google Scholar]
- 13. Lawson J. R., Melen R. L., Inorg. Chem. 2017, 56, 8627–8643. [DOI] [PubMed] [Google Scholar]
- 14. Wang G.-Z., Jiang J., Bu X.-S., Dai J.-J., Xu J., Fu Y., Xu H.-J., Org. Lett. 2015, 17, 3682–3685. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Brown H. C., Jadhav P. K., J. Am. Chem. Soc. 1983, 105, 2092–2093; [Google Scholar]
- 15b. Roush W. R., Hoong L. K., Palmer M. A. J., Straub J. A., Palkowitz A. D., J. Org. Chem. 1990, 55, 4117–4126. [Google Scholar]
- 16. Chen C., Voss T., Fröhlich R., Kehr G., Erker G., Org. Lett. 2011, 13, 62–65. [DOI] [PubMed] [Google Scholar]
- 17. Yamada T., Park K., Monguchi Y., Sawama Y., Sajiki H., RSC Adv. 2015, 5, 92954–92957. [Google Scholar]
- 18. Wilkins L. C., Lawson J. R., Wieneke P., Rominger F., Hashmi A. S. K., Hansmann M. M., Melen R. L., Chem. Eur. J. 2016, 22, 14618–14624. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Hansmann M. M., Melen R. L., Rominger F., Hashmi A. S. K., Stephan D. W., J. Am. Chem. Soc. 2014, 136, 777–782; [DOI] [PubMed] [Google Scholar]
- 19b. Hansmann M. M., Melen R. L., Rominger F., Hashmi A. S. K., Stephan D. W., Chem. Commun. 2014, 50, 7243–7245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary