

Abstract
Aim
The aim of this study was to develop an algorithm to prompt early clinical suspicion of mucopolysaccharidosis type I (MPS I).
Methods
An international working group was established in 2016 that comprised 11 experts in paediatrics, rare diseases and inherited metabolic diseases. They reviewed real‐world clinical cases, selected key signs or symptoms based on their prevalence and specificity and reached consensus about the algorithm. The algorithm was retrospectively tested.
Results
An algorithm was developed. In patients under two years of age, kyphosis or gibbus deformity were the key symptoms that raised clinical suspicion of MPS I and in those over two years they were kyphosis or gibbus deformity, or joint stiffness or contractures without inflammation. The algorithm was tested on 35 cases, comprising 16 Hurler, 10 Hurler–Scheie, and nine Scheie patients. Of these 35 cases, 32 (91%) – 16 Hurler, nine Hurler–Scheie and seven Scheie patients – would have been referred earlier if the algorithm had been used.
Conclusion
The expert panel developed and tested an algorithm that helps raise clinical suspicion of MPS I and would lead to a more prompt final diagnosis and allow earlier treatment.
Keywords: Algorithm, Diagnosis, Kyphosis, Mucopolysaccharidosis, Symptoms
Abbreviations
- ENT
Ear, nose and throat
- HSCT
Haematopoietic stem cell transplantation
- MPS I
Mucopolysaccharidosis type I
Key notes.
Our aim was to develop an algorithm to prompt early clinical suspicion of mucopolysaccharidosis type I (MPS I).
We convened a working party of 11 experts in paediatrics, rare diseases and inherited metabolic diseases who developed the algorithm, based on key signs and symptoms.
The algorithm was tested on 35 patients, and 91% would have been referred earlier if the algorithm had been used.
Introduction
Studies have shown that the diagnosis and treatment of mucopolysaccharidosis type I (MPS I) is substantially delayed and that the time between the onset of symptoms and diagnosis has not improved 1. This diagnostic delay is particularly prominent in patients with attenuated forms of MPS I, with an estimated gap between symptom onset and diagnosis ranging from two to nine years 1, 2, 3. Given the progressive and multi‐systemic nature of this inherited genetic disease, diagnosis should be established as early as possible in the disease course for patients to receive optimal benefit from treatment.
Although effective newborn screening techniques are now available to facilitate early diagnosis, newborn screening is not yet widely available and there are no biomarkers that can predict phenotypic severity in those newborn infants identified by screening 4, 5, 6. In some cases, the clinical phenotype could be predicted by the genotype, based on mutations of the human alpha‐L‐iduronidase gene.
Importance of early diagnosis
Depending on the healthcare system, the first physician to see a patient could be a general practitioner and/or a paediatrician, and greater awareness could lead to a prompt specialist referral 7. In general, multi‐systemic and progressive signs and symptoms should raise suspicion of a genetic disorder, including a lysosomal storage disorder such as MPS I 8. The benefits of early treatment with haematopoietic stem cell transplantation (HSCT) in patients with severe MPS I include substantial changes in the disease course along with improved cognition 9, 10, 11. Early treatment of patients with MPS I can also slow disease progression and improve patient outcomes, including quality of life 12. Sibling studies have suggested that early initiation of treatment with enzyme replacement therapy substantially modifies the disease course in patients with attenuated MPS I 13, 14, 15. Furthermore, enzyme replacement therapy is recommended once diagnosis is confirmed 16. Finally, early diagnosis allows for early genetic counselling and informed family planning.
Existing treatments
Haematopoietic stem cell transplantation before 2.5 years of age is the recommended treatment for patients diagnosed with the most severe form of MPS I, the Hurler phenotype 16. In these patients, allogeneic HSCT stabilises cognitive skills, normalises liver and spleen volume, improves survival 17, reduces ventricular hypertrophy and maintains cardiac function 18. Individual patients with an intermediate phenotype may also be considered for HSCT if the benefits outweigh the risks and an optimal donor is available 16.
Enzyme replacement therapy with recombinant human alpha‐L‐iduronidase, also known as laronidase, is the specific treatment for patients with the attenuated disease phenotype who do not undergo HSCT 16. This treatment is also recommended for all diagnosed MPS I patients who have not yet received HSCT or whose graft has failed 16. Evidence from the literature has suggested that early treatment initiation is associated with better outcomes. A retrospective analysis of nine sibling cases with attenuated MPS I suggested that initiation of laronidase prior to symptom onset slowed or prevented the development of severe clinical manifestations 13. In a large cohort of patients with attenuated MPS I, treatment with laronidase resulted in disease stabilisation in the majority of patients after a mean follow‐up of 6.1 years 19.
Clinical profile of MPS I
In patients with severe MPS I, symptom onset occurs in the first months of life and the disease progresses more rapidly than in the attenuated form. Common signs and symptoms include kyphosis or gibbus, developmental delay, hernias, coarse facial features, corneal clouding, cardiac valve abnormalities, hepatomegaly 1 and airway‐related symptoms 20.
In patients with attenuated MPS I, presenting symptoms that may seem innocuous often become important as the disease progresses and these should therefore prompt suspicion of MPS I. They include hernias, cardiac valve abnormalities, recurrent ear, nose and throat (ENT) infections, joint contractures or stiffness, coarse facial features, corneal clouding and carpal tunnel syndrome 3, 21. Other frequently occurring symptoms, including hepatomegaly, hearing loss and changes in hair morphology 22, 23, may be revealed by a meticulous medical history and complete physical examination of all the organ systems that are potentially affected by the disease.
Until newborn screening programmes and early phenotypic biomarkers of phenotypes are universally available, it is very important that physicians become more aware of the early clinical signs and symptoms of MPS I.
The aim of this study was to develop an algorithm to prompt early clinical suspicion of MPS I by bringing together an international group of experts in this field.
Methods
Consensus process and rationale
An international working group of 11 experts met in 2016 to reach a consensus on an algorithm to prompt early clinical suspicion of MPS I. The working group represented experts in paediatrics, rare diseases and inherited metabolic diseases from nine countries: Poland, Belgium, Italy, United Arab Emirates, France, Germany, Denmark, The Netherlands and Czech Republic.
The algorithm was drafted during two one‐day meetings. During the first meeting, the experts shared their experience based on real‐world clinical cases in an attempt to identify the signs and symptoms that contribute to a standard clinical profile, both the key early referral signs and symptoms and the less specific but common signs and symptoms. Based on this information, they drafted an algorithm to help physicians recognise MPS I and refer patients to metabolic disease specialists in a timely manner. The experts then collected data from the clinical records of their most recently diagnosed MPS I patients. These data were used during the second meeting to refine the draft algorithm. Another set of data, from the patient cohort used to draft the first version of the algorithm, was subsequently collected to investigate the age when specific signs and symptoms were first recognised by the relevant healthcare professional and noted in the patients’ medical records. This information was used to reconstruct the patients’ natural history, and then, the algorithm was finalised and the experts reached a consensus based on this information. Table 1 shows the key signs and symptoms that were selected on the basis of their prevalence and specificity, considering the natural history data analysed.
Table 1.
Key signs and symptoms prompting early clinical suspicion of mucopolysaccharidosis type I (MPS I) and timely referral of patients
| Presenting signs and symptoms | Expert comments and additional information from the literature |
|---|---|
| Kyphosis or gibbus deformity | Expert comments
|
| Joint stiffness or contractures |
Expert comments
Information from the literature |
| Recurrent or persistent hernia or hernia repair |
Expert comments
Information from the literature
|
| Recurrent ear, nose and throat (ENT) infections |
Expert comments
Information from the literature
|
| Coarse facial features or facial dysmorphism | Expert comments
|
| Developmental delay |
Expert comments
Information from the literature
|
| Hepatomegaly | Expert comments
|
| Cardiac valve disease |
Expert comments
Information from the literature |
| Growth velocity decline |
Expert comments
Information from the literature |
| Hearing loss not otherwise explained by ear infection or not directly related to ear infection | Expert comments
|
| Recurrent orthopaedic or ENT surgeries |
Expert comments
Information from the literature
|
| Corneal clouding |
Expert comments
Information from the literature
|
| Carpal tunnel syndrome |
Expert comments
Information from the literature
|
Results
Developing the algorithm
The proposed algorithm to prompt early clinical suspicion of MPS I and expedite referral to an appropriate specialist is shown in Figure 1. Highly specific signs and symptoms that prompt seeking medical advice were identified as the first clinical features for consideration. Clusters of less‐specific signs and symptoms were considered on the basis of their frequency, in combination with the most common specific signs and symptoms. Finally, age at presentation was included, as it could help to differentiate between the different forms of MPS I. Joint stiffness without inflammation and kyphosis or gibbus are highly specific presenting signs and symptoms that, on their own, are sufficient to raise suspicion of MPS I and prompt referral to a specialised centre for MPS testing (Fig. 2). A combination of two or more less‐specific signs and symptoms should also prompt referral to a specialised centre for further MPS testing. These include recurrent hernias, recurrent ENT infections, growth velocity decline, coarse facial features or facial dysmorphism, cardiac valve disease, developmental delay, recurrent orthopaedic or ENT surgery, hepatomegaly, carpal tunnel syndrome, corneal clouding and hearing loss that cannot be explained by an ear infection. The patient should be referred to the metabolic or genetic specialist without delay, even if just on the basis of a suspicion of MPS I.
Figure 1.
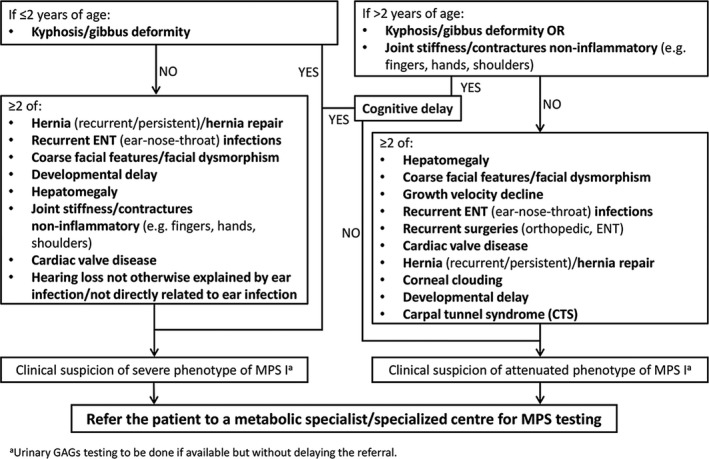
Algorithm to prompt early clinical suspicion of mucopolysaccharidosis type I (MPS I) and timely referral of patients.
Figure 2.
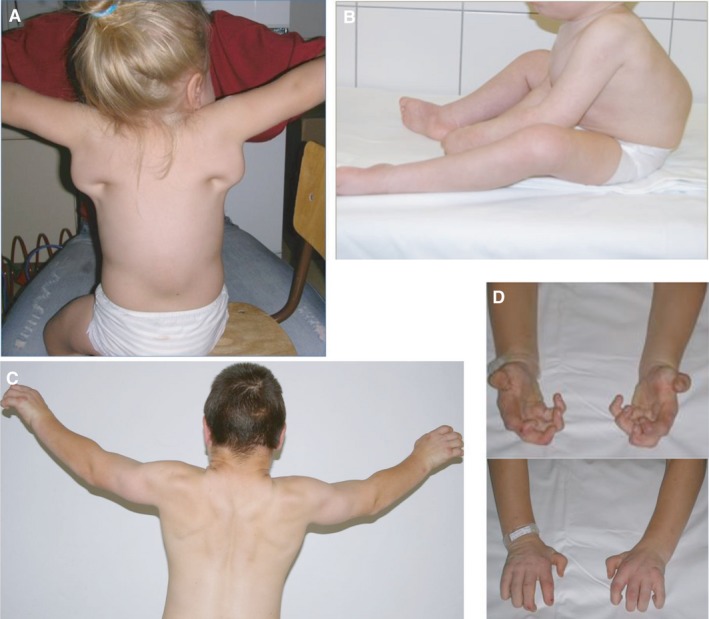
Characteristic signs and symptoms of mucopolysaccharidosis type I (MPS I) to prompt early suspicion of disease. (A) Joint stiffness or contractures in patient with Hurler syndrome. (B) Kyphosis or gibbus deformity in a seven‐month‐old patient with Hurler syndrome. (C) Joint stiffness or contractures in a patient with Scheie syndrome – shoulders. (D) Joint stiffness or contractures in a patient with Scheie syndrome – hands. Written informed consent was obtained from the patients, parents and carers for publication of these pictures.
If available, urinary glycosaminoglycan testing may be performed to facilitate the diagnosis of MPS. However, as false‐negative results may be obtained depending on the technique used, careful interpretation of the urinary glycosaminoglycan testing results is required to avoid delays in diagnosis.
When referring to this algorithm, several points need to be considered. First, as 6000–8000 different rare diseases have been identified, 75% of which manifest in childhood 30, it is not realistic to expect paediatricians and general practitioners to know about the specific signs or symptoms related to each disorder or to be able to use algorithms that may lead to an early diagnosis of every disorder. However, as MPS I is a treatable disease and timely initiation of therapy is paramount, early recognition and referral to the appropriate specialists should be prompted. The presented algorithm aims to increase the knowledge of paediatricians and general practitioners about the cluster of early signs and symptoms of MPS I, which could lead to timely collaboration with a metabolic disease specialist, a clinical geneticist or another specialist with expertise in the management of rare genetic disorders and thus achieve an optimal outcome for the patient. Second, the algorithm should be tailored according to the healthcare system in the individual countries, as different medical specialists may be using this algorithm. Lastly, if urinary glycosaminoglycan test results are negative, but the patient demonstrates other convincing symptoms of MPS, the healthcare physician should seek an appropriate referral for the patient.
Testing of the algorithm
Data from 35 MPS I cases – 16 Hurler, 10 Hurler–Scheie and nine Scheie patients – were used to test and finalise the proposed algorithm. For each patient, information on gender, age at diagnosis, phenotype and presenting signs and symptoms was captured along with the patient's age when a specific sign or symptom was first recognised by a healthcare professional and noted in the medical records. The specialty of the individual who recognised the sign or symptom was also noted.
Notably, if the proposed algorithm had been used for these patients by first‐line medical doctors such as paediatricians and general practitioners, it would have allowed an earlier diagnosis in 32/35 (91%) of cases: 16 Hurler, nine Hurler–Scheie and seven Scheie patients. Other types of healthcare professionals, such as orthopaedic specialists, ENT specialists and geneticists, would also have referred the patients if the algorithm had been implemented.
Discussion
Mucopolysaccharidosis type I is a multi‐organ, progressive disorder for which effective treatments are now available. Newborn screening for MPS I is starting to be included in newborn screening programmes, but until newborn screening is universally available, it is imperative that healthcare practitioners do not miss early opportunities for diagnosis. The benefits of timely and appropriate treatment are optimised with early diagnosis and include the prevention of irreversible organ damage, reduction of complications, possible improvement of quality of life, avoidance of unnecessary surgeries and treatments, mitigation of detrimental impacts on the patient and his or her family arising through uncertainty, the social aspects of an undiagnosed illness and family planning and counselling. These benefits highlight the need to evoke clinical suspicion of MPS I in patients who present with key clinical features of kyphosis or gibbus, or joint stiffness or contractures without inflammation and to better educate paediatricians, general practitioners and other specialists about the disease. In this regard, evaluating symptom and sign clusters rather than individual signs could assist in the early diagnosis of MPS I. Data from an observational study published in 2017 provided further support for using selected signs and symptoms to prompt suspicions of MPS, including MPS I, and the important role paediatricians play in facilitating an early diagnosis 31. Limitations of the algorithm included the limited number of patients in each subgroup and the retrospective nature of the testing, which was only performed on data from patients with confirmed MPS I.
Conclusion
A working group of international experts used data from real‐world clinical cases, together with data from published literature, to define the natural history of MPS I across the phenotypic spectrum. The proposed diagnostic algorithm that they have developed not only raises awareness of MPS I, but more importantly empowers individual physicians to identify potential cases and thus improve early diagnosis. Furthermore, the proposed algorithm is intended to shorten the interval between the first healthcare contact with the patient with symptoms and signs suggestive of MPS I and the final diagnosis with the overarching objective of improving outcomes for patients with this progressive, yet treatable disorder. In this regard, the role of the medical specialist, who is often a paediatrician or general practitioner, is of paramount importance for the timely recognition of suspected cases and collaboration with an appropriate specialist to ensure optimal outcomes. Continuous efforts are needed to engage the specialists involved in the primary care of these patients and to establish a referral network.
Funding
The MPS I European advisory boards were financially supported by Sanofi Genzyme. The company also supported the working group meetings and the writing and editing of this manuscript.
Conflicts of interest
The authors have various links with pharmaceutical companies, as listed in Appendix S1.
Supporting information
Appendix S1 Conflicts of interest.
Acknowledgements
We'd like to thank Alessia Piazza PhD, Excerpta Medica, for writing and editorial support and Meritxell Bernat Fuertes, Sanofi Genzyme for coordinating and supporting the experts during the project and for critically reviewing the manuscript.
References
- 1. Beck M, Arn P, Giugliani R, Muenzer J, Okuyama T, Taylor J, et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med 2014; 16: 759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Aco K, Underhill L, Rangachari L, Arn P, Cox GF, Giugliani R, et al. Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry. Eur J Pediatr 2012; 171: 911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vijay S, Wraith JE. Clinical presentation and follow‐up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr 2005; 94: 872–7. [DOI] [PubMed] [Google Scholar]
- 4. Clarke LA, Atherton AM, Burton BK, Day‐Salvatore DL, Kaplan P, Leslie ND, et al. Mucopolysaccharidosis type I newborn screening: best practices for diagnosis and management. J Pediatr 2017; 182: 363–70. [DOI] [PubMed] [Google Scholar]
- 5. de Ru MH, Bouwman MG, Wijburg FA, van Zwieten MC. Experiences of parents and patients with the timing of mucopolysaccharidosis type I (MPS I) diagnoses and its relevance to the ethical debate on newborn screening. Mol Genet Metab 2012; 107: 501–7. [DOI] [PubMed] [Google Scholar]
- 6. Peake RW, Marsden DL, Bodamer OA, Gelb MH, Millington DS, Wijburg F. Newborn screening for lysosomal storage disorders: quo vadis? Clin Chem 2016; 62: 1430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bruni S, Lavery C, Broomfield A. The diagnostic journey of patients with mucopolysaccharidosis I: a real‐world survey of patient and physician experiences. Mol Genet Metab Rep 2016; 8: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wraith JE. The mucopolysaccharidoses: a clinical review and guide to management. Arch Dis Child 1995; 72: 263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aldenhoven M, Wynn RF, Orchard PJ, O'Meara A, Veys P, Fischer A, et al. Long‐term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood 2015; 125: 2164–72. [DOI] [PubMed] [Google Scholar]
- 10. Poe MD, Chagnon SL, Escolar ML. Early treatment is associated with improved cognition in Hurler syndrome. Ann Neurol 2014; 76: 747–73. [DOI] [PubMed] [Google Scholar]
- 11. Yasuda E, Mackenzie W, Ruhnke K, Shimada T, Mason RW, Zustin J, et al. Long‐term follow‐up of post hematopoietic stem cell transplantation for Hurler syndrome: clinical, biochemical, and pathological improvements. Mol Genet Metab Rep 2015; 2: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muenzer J, Wraith JE, Clarke LA, the International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I . Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009; 123: 19–29. [DOI] [PubMed] [Google Scholar]
- 13. Al‐Sannaa NA, Bay L, Barbouth DS, Benhayoun Y, Goizet C, Guelbert N, et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet J Rare Dis 2015; 10: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gabrielli O, Clarke LA, Ficcadenti A, Santoro L, Zampini L, Volpi N, et al. 12 year follow up of enzyme‐replacement therapy in two siblings with attenuated mucopolysaccharidosis I: the important role of early treatment. BMC Med Genet 2016; 17: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laraway S, Breen C, Mercer J, Jones S, Wraith JE. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Mol Genet Metab 2013; 109: 315–6. [DOI] [PubMed] [Google Scholar]
- 16. de Ru MH, Boelens JJ, Das AM, Jones SA, van der Lee JH, Mahlaoui N, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis 2011; 6: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 2011; 50(Suppl 5): v49–59. [DOI] [PubMed] [Google Scholar]
- 18. Braunlin EA, Stauffer NR, Peters CH, Bass JL, Berry JM, Hopwood JJ, et al. Usefulness of bone marrow transplantation in the Hurler syndrome. Am J Cardiol 2003; 92: 882–6. [DOI] [PubMed] [Google Scholar]
- 19. Laraway S, Mercer J, Jameson E, Ashworth J, Hensman P, Jones SA. Outcomes of long‐term treatment with laronidase in patients with mucopolysaccharidosis Type I. J Pediatr 2016; 178: 219–26. [DOI] [PubMed] [Google Scholar]
- 20. Arn P, Bruce IA, Wraith JE, Travers H, Fallet S. Airway‐related symptoms and surgeries in patients with mucopolysaccharidosis I. Ann Otol Rhinol Laryngol 2015; 124: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas JA, Beck M, Clarke JT, Cox GF. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis 2010; 33: 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kloska A, Bohdanowicz J, Konopa G, Tylki‐Szymńska A, Jakóbkiewicz‐Banecka J, Czartoryska B, et al. Changes in hair morphology of mucopolysaccharidosis I patients treated with recombinant human alpha‐L‐iduronidase (laronidase, Aldurazyme). Am J Med Genet A 2005; 139: 199–203. [DOI] [PubMed] [Google Scholar]
- 23. Malinowska M, Jakóbkiewicz‐Banecka J, Kloska A, Tylki‐Szymańska A, Czartoryska B, Piotrowska E, et al. Abnormalities in the hair morphology of patients with some but not all types of mucopolysaccharidoses. Eur J Pediatr 2008; 167: 203–9. [DOI] [PubMed] [Google Scholar]
- 24. Arn P, Wraith JE, Underhill L. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr 2009; 154: 859–64. [DOI] [PubMed] [Google Scholar]
- 25. Arn P, Whitley C, Wraith JE, Webb HW, Underhill L, Rangachari L, et al. High rate of postoperative mortality in patients with mucopolysaccharidosis I: findings from the MPS I Registry. J Pediatr Surg 2012; 47: 477–84. [DOI] [PubMed] [Google Scholar]
- 26. Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis 2011; 34: 1183–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fesslová V, Corti P, Sersale G, Rovelli A, Russo P, Mannarino S, et al. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young 2009; 19: 170–8. [DOI] [PubMed] [Google Scholar]
- 28. Różdżyńska‐Świątkowska A, Jurecka A, Cieślik J, Tylki‐Szymańska A. Growth patterns in children with mucopolysaccharidosis I and II. World J Pediatr 2015; 11: 226–31. [DOI] [PubMed] [Google Scholar]
- 29. Cimaz R, Coppa GV, Koné‐Paut I, Link B, Pastores GM, Elorduy MR, et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatr Rheumatol Online J 2009; 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. EURORDIS . What is a rare disease? Available at: http://www.eurordis.org/sites/default/files/publications/Fact_Sheet_RD.pdf (accessed on June 1, 2018).
- 31. Colón C, Alvarez JV, Castaño C, Gutierrez‐Solana LG, Marquez AM, O'Callaghan M, et al. A selective screening program for the early detection of mucopolysaccharidosis: results of the FIND project ‐ a 2‐year follow‐up study. Medicine (Baltimore) 2017; 96: e6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Conflicts of interest.