

Abstract
Although mutational inactivation of E‐cadherin (CDH1) is the main driver of invasive lobular breast cancer (ILC), approximately 10–15% of all ILCs retain membrane‐localized E‐cadherin despite the presence of an apparent non‐cohesive and invasive lobular growth pattern. Given that ILC is dependent on constitutive actomyosin contraction for tumor development and progression, we used a combination of cell systems and in vivo experiments to investigate the consequences of α‐catenin (CTNNA1) loss in the regulation of anchorage independence of non‐invasive breast carcinoma. We found that inactivating somatic CTNNA1 mutations in human breast cancer correlated with lobular and mixed ducto‐lobular phenotypes. Further, inducible loss of α‐catenin in mouse and human E‐cadherin‐expressing breast cancer cells led to atypical localization of E‐cadherin, a rounded cell morphology, and anoikis resistance. Pharmacological inhibition experiments subsequently revealed that, similar to E‐cadherin‐mutant ILC, anoikis resistance induced by α‐catenin loss was dependent on Rho/Rock‐dependent actomyosin contractility. Finally, using a transplantation‐based conditional mouse model, we demonstrate that inducible inactivation of α‐catenin instigates acquisition of lobular features and invasive behavior. We therefore suggest that α‐catenin represents a bona fide tumor suppressor for the development of lobular‐type breast cancer and as such provides an alternative event to E‐cadherin inactivation, adherens junction (AJ) dysfunction, and subsequent constitutive actomyosin contraction. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: lobular breast cancer, α‐catenin (CTNNA1), E‐cadherin, mouse model, Rho/Rock
Introduction
Invasive lobular carcinoma (ILC) is a luminal ER‐positive breast cancer subtype that represents approximately 10–15% of all breast cancers. In contrast to the larger group of invasive ductal carcinoma, not otherwise specified (IDC‐NOS), loss of E‐cadherin (CDH1) in ILC has been identified as the key underlying cause of tumor development and progression 1, 2. ILC is characterized by non‐cohesive infiltrative growth patterns and diffuse dissemination to uncommon sites such as the gastrointestinal tract, ovary, and peritoneum (reviewed in 3). These pathological features are mostly due to inactivation of E‐cadherin, absence of functional classical cadherin redundancy, and a subsequent loss of epithelial cell–cell adhesion (reviewed in 4). In ILC, loss of cadherin‐based junctions is tolerated because of tumor suppressor inactivation or activation of oncogenes that render mammary tumor‐initiating cells resistant to anoikis 2, 5, 6. Acquisition of anoikis resistance is a critical feature of disseminating ILC cells and is instigated by p120‐catenin (p120)‐dependent autocrine activation of RhoA–Rock1, leading to constitutive actomyosin contraction in E‐cadherin‐mutant ILC 7, 8.
E‐cadherin controls epithelial homeostasis through formation of the adherens junction (AJ), which directly transduces cues to the actin cytoskeleton via αE‐catenin (from here on, α‐catenin) 9. While the exact mechanisms remain a matter of debate, it appears that α‐catenin connects the AJ with the actin cytoskeleton through binding of β‐catenin and actin filaments (reviewed in 10). Interestingly, a minority of ILC cases with a clear lobular phenotype have retained E‐cadherin expression without truncating or frame shift mutations or CDH1 promoter methylation, indicating that functional inactivation of the adherens junction must have occurred through means other than somatic loss or epigenetic silencing of E‐cadherin.
Proper functioning α‐catenin is essential for cell–cell adhesion through control of actin dynamics (reviewed in 11). Next to formin‐dependent radial actin filament formation 12, 13, 14, 15, α‐catenin also inhibits actin branching by competing with the Arp2/3 complex for actin binding 16. Moreover, α‐catenin can enhance p120‐catenin binding to E‐cadherin, thereby facilitating junctional stability 17. Studies in different organ systems have suggested that α‐catenin might function as a tumor suppressor. For instance, α‐catenin loss in the skin or cerebral cortex of mice caused epidermal and cerebral hyperproliferation 14, 18, 19. Second, loss of α‐catenin is a prognostic factor for poor survival of breast and other cancers (reviewed in 20). Finally, several studies have identified inactivating CTNNA1 mutations in breast cancer cell lines 21, 22 and a case of diffuse gastric cancer 23.
Here, we examined whether loss of α‐catenin in non‐invasive breast cancer cells expressing a functional AJ leads to the acquisition of lobular and pro‐metastatic features. We found that somatic inactivating CTNNA1 mutations are linked to ILC and observed that α‐catenin loss leads to E‐cadherin‐expressing invasive cancer cells that depend on constitutive actomyosin contraction for their anchorage independence. Finally, we present in vivo data suggesting that α‐catenin loss induces lobular features and invasive growth characteristics.
Materials and methods
Analysis of TCGA and METABRIC datasets
Allelic frequencies for all available somatic mutations in the individual samples (available for 18 out of 19 tumors) were collected from TCGA and METABRIC databases. Allelic values with a difference smaller than 0.01 were binned together for plotting histograms. Probability density functions were generated by kernel density estimates. All calculations and distributions were generated by R software version 3.3.3 and ggplot2 package version 2.2.1.
Cell lines and cell culture
Mouse cell lines Trp53Δ/Δ‐3 (KP6) and Trp53Δ/Δ‐7 (WP6) were derived from tumors that developed in either K14cre;Trp53 F/F or Wcre;Trp53 F/F female mice and cultured as described previously 2, 24. MCF7 was obtained from the American Type Culture Collection (Manassas, VA, USA), STR type verified by PCR, and cultured as described previously 25. Trp53Δ/Δ‐3 cells rendered knockout for E‐cadherin using CRISPR‐Cas9 have been described previously 26.
Constructs, viral production, and transduction
siRNA sequences targeting mouse (5'‐GTCACATGCTTCACTCAAA‐3') and human (5'‐GTCACTGTTCGTCACTCAA‐3') α‐catenin were cloned as shRNA in the lentiviral vector pFUTG as described previously 7. The shRNA against mouse α‐catenin was positioned in the 5'‐UTR. Lentiviral particles were produced in COS‐7 cells and used for transduction as described previously 7. To induce knockdown, cells were treated with 2 mg/ml doxycycline (1:5000, D9891; Sigma‐Aldrich, Zwijndrecht, The Netherlands) for at least 3 days.
Western blotting
Samples were lysed in sample buffer and proteins were separated and western blotted as described previously 27. The antibodies used were Gapdh (1: 2000; Millipore, Amsterdam, The Netherlands; Mab374), Akt1 (1:1000; Santa Cruz, Heidelberg, Germany; Sc‐1618), α‐catenin (1:2000; Sigma‐Aldrich; C2081), E‐cadherin (1:2000; BD Biosciences, Vianen, Utrecht, The Netherlands; 610182), p120‐catenin (1:2000; BD Biosciences; 610134), β‐catenin (1:2000; BD Biosciences; 610154), phospho‐cofilin (Ser3) (1:2000; Cell Signaling, Leiden, The Netherlands; 3311), and cofilin (1:500; Cell Signaling; 3312). All blots were incubated for 30 min with either rabbit anti‐goat‐PO (DAKO, Heverlee, Belgium; P160), goat anti‐rabbit‐PO (Bio‐Rad, Veenendaal, The Netherlands; 170‐6515) or goat anti‐mouse‐PO (Bio‐Rad; 170‐6516) secondary antibodies. Images were acquired on an Amersham Imager 600 (Amersham, ‘s‐Hertogenbosch, The Netherlands).
Immunofluorescence, confocal microscopy, and phase contrast microscopy
Cells were cultured on collagen I (BD Biosciences; 354236)‐coated coverslips, fixed with 100% methanol on ice, permeabilized using 0.3% Triton X‐100/PBS, and blocked with 4% BSA fraction V (GE‐Healthcare, Hoevelaken, Utrecht, The Netherlands; K45‐001) in PBS. Samples were incubated with antibodies against α‐catenin (1:1000; Sigma‐Aldrich; C2081), or clone 15D1 (1:100; Enzo Life Sciences, Farmingdale, NY, USA; Alx‐804‐101), anti‐E‐cadherin‐TRITC (1:300; BD Biosciences; 612130), anti‐p120‐catenin‐TRITC (1:300; BD Biosciences; 610137) or mouse anti‐β‐catenin (1:50; BD Biosciences; 610154) overnight at 4 °C. After washing, slides were incubated with secondary antibodies: goat anti‐mouse Alexa‐405 (1:600; Invitrogen, Landsmeer, The Netherlands; A31553) or antibodies goat anti‐rabbit Alexa‐405 (1:600; Invitrogen; A31556) for 1 h at room temperature. DNA was stained with ToPro‐3 (Invitrogen); coverslips were mounted using Vectashield (Vector Laboratories, Amsterdam, The Netherlands; X1215) and analyzed using a Zeiss LSM 700 confocal laser microscope using a 63× 1.4 NA objective (Zeiss, Breda, The Netherlands). Phase contrast pictures were produced using a Leica DMI 4000B microscope (20× objective) (Leica, Rijswijk, The Netherlands).
Anoikis resistance and pharmacological inhibition
Anoikis resistance was determined and defined as described previously 7. Survival of control cells was normalized to 1. For inhibition studies, cells were treated with: 0.02 μg/ml C3 transferase (Cytoskeleton Inc, Heerhugowaard, The Netherlands; CT04‐A), 10 μm Y27632 (Selleckchem, Huissen, The Netherlands; S1049), 1 μm GSK‐429286A (Selleckchem; S1474) or 3 μm blebbistatin (VWR, Radnor, PA, USA; 203390). Error bars depict the standard deviation. A two‐sided Student's t‐test was performed to calculate statistical significance; P values less than 0.05 were considered significant.
Isolation of primary mouse mammary epithelium cells
All animal experiments were performed according to institutional guidelines and national regulations under permit number AVD115002015263 and DEC2010.III.11.123. WAPcre;Trp53 F/F mice were generated and genotyped as described previously 24. Primary mouse mammary epithelium cells (MMECs) were obtained from gland numbers 3, 4, and 5 after removal of the intra‐mammary lymph nodes of 6‐ to 8‐week‐old female WAPcre;Trp53 F/F mice. Isolation of MMECS was based on described methods 28. Cell numbers were quantified and transferred into a 24‐well plate at 2 × 106 cells per well (Ultra‐Low Attachment Multiple Well Plate, CLS3471; Corning, Zwijndrecht, The Netherlands) for viral transduction.
Lentiviral transduction and transplantation of MMECs
Sequences targeting mouse α‐catenin (5′‐GTCACATGCTTCACTCAAA‐3') were cloned as shRNAs in the lentiviral vector pFUTG as previously described 7. Lentiviral particles were produced in COS‐7 cells and used for transduction as described in 7. MMECS were transduced with 1 × 107 TU of virus for 16 h in a final volume of 0.8 ml (MOI = 5) in primary growth medium (5 μg/ml insulin, 1 μg/ml hydrocortisone, 10 ng/ml mouse EGF, 10% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin G, 50 μg/ml gentamicin/G418 in DMEM/F12) containing 4 μg/ml polybrene (all Sigma). After 24 h, MMECs were washed five times in HBSS to remove live virus particles before transplantation.
2 MMECs (2 × 105) were transplanted using a cleared fat‐pad assay as previously described 29 in 3‐week‐old Hsd:Athymic Nude‐Foxn1 nu/nu (nude) mice. Recipient mice were anesthetized using isoflurane (IsoFlo; Le Vet Pharma, Oudewater, Utrecht, The Netherlands). Buprenorphine (100 μl; 0.03 mg/ml) was injected subcutaneously as analgesic treatment. After transplantation, the animals were either fed ad libitum with control chow or doxycycline‐containing chow (200 mg/kg; A153D00201; Sniff, Soest, Utrecht, The Netherlands).
Longitudinal tumor development
Onset of tumor growth was monitored longitudinally (n = 44 recipient mice) and mammary tumor volume was determined by digital pressure‐sensitive caliper measurements (Mitutoyo, Veenendaal, The Netherlands) and the following formula: 4/3 π × [(l + b)/4]3. Animals that developed tumors (n = 10) were euthanized if the mammary tumor reached a size of 1000 mm3 or in cases of severe discomfort otherwise.
Histological analysis
Formaldehyde‐fixed, paraffin‐embedded tissues were sectioned at 4 μm and stained with hematoxylin and eosin. For immunohistochemical staining, fixed sections were rehydrated and incubated with primary antibodies against α‐catenin (C2081; 1:1000; Sigma), E‐cadherin (612130; 1:150; BD Biosciences), and GFP (SC‐8334; 1:150; Santa Cruz). Endogenous peroxidases were blocked with 3% H2O2 and biotin‐conjugated secondary antibodies were used, followed by incubation with HRP‐conjugated streptavidin–biotin complex (DAKO). Substrate was developed with DAB (DAKO) and pictures were produced using a Nikon Eclipse E800 microscope with a Nikon DXM1200 digital camera (Nikon, Amsterdam, The Netherlands). Appropriate positive (expressing tissues) and negative controls (matched isotype and omitting primary antibody) were used throughout.
Results
Somatic CTNNA1 mutations in human breast cancer are associated with ILC and ducto‐lobular breast cancers
Approximately 10–15% of ILC cases retain E‐cadherin expression. Because inactivating mutations in α‐catenin were linked to cancer 21, 23, we performed a survey into the prevalence of CTNNA1 mutations in the publicly‐available online databases. Focusing on breast cancer, we found that a total of 19 unique somatic mutations were reported in the combined METABRIC and TCGA datasets (http://cancergenome.nih.gov) (Table 1 and supplementary material, Figure S1). Of these, five cases were diagnosed as ILC; two cases as mixed ductal and lobular (mixed IDL); one case was not classified; and the remaining 11 cases were registered as invasive ductal cancer, not otherwise specified (IDC‐NOS) (Table 1). Somatic inactivating CDH1 mutations were reported in five ILC cases (Table 1). We evaluated all 19 cases (blinded) to confirm the initial differential diagnosis and identified three additional cases that were initially diagnosed as IDC‐NOS but either showed a mixed ductal and lobular breast cancer phenotype or presented as ducto‐lobular tumors (Table 1 and Figure 1). Finally, we stratified the CTNNA1‐mutant cases based on mRNA expression. Intriguingly, we found a correlation between low CTNNA1 mRNA expression (< 40%), the presence of wild‐type CDH1 alleles, and a lobular, mixed, or ducto‐lobular breast cancer phenotype (Table 1). Cases that showed low CTNNA1 mRNA expression were accompanied by CTNNA1 splice site mutations (two cases), nonsense mutations (three cases), and a case with a somatic missense mutation in CTNNA1. One ILC case harbored an inactivating CTNNA1 mutation in the presence of a CDH1 mutation but was included due to borderline E‐cadherin mRNA expression (39%) and low α‐catenin expression (27%). We observed three cases from the METABRIC cohort that were diagnosed as mixed ductal and lobular, but with high CTNNA1 mRNA expression and unaffected CDH1 alleles. Based on the allelic frequencies, we generated plots to conduct an estimate on the clonal character of the analyzed tumors (supplementary material, Figure S2). The results in Figure 1, supplementary material, Figures S1 and S2, and Table 1 are in part based on data generated by the TCGA Research Network (http://cancergenome.nih.gov). In conclusion, these publicly available data suggested a correlation between α‐catenin inactivation and a lobular breast cancer phenotype.
Table 1.
Somatic functional inactivating CTNNA1 mutations and breast cancer phenotype
| Sample ID | CTNNA1 status | Diagnosis | CDH1 | |||
|---|---|---|---|---|---|---|
| AA change | Copy # | mRNA | Initial | Revised | ||
| TCGA‐EW‐A1OV‐01 | R129* | ShallowDel | 0% | IDC‐NOS | Mixed IDL | WT |
| MB‐3600 | X101_s | ShallowDel | 1% | IDC‐NOS | IDC‐NOS† | WT |
| TCGA‐AC‐A3TM‐01 | R54C | Diploid | 21% | ILC | Ducto‐lobular | MUT |
| MB‐4667 | Q887* | Diploid | 27% | ILC | PILC | MUT‡ |
| TCGA‐A2‐A1FX‐01 | X634_s | Gain | 37% | IDC‐NOS | Mixed IDL | WT |
| MB‐7157 | K733* | Diploid | 40% | IDC‐NOS | IDC‐NOS | WT |
| MB‐5431 | D904A | ShallowDel | 61% | IDC‐NOS | IDC‐NOS | WT |
| TCGA‐AN‐A046‐01 | R98* | Diploid | 64% | IDC‐NOS | IDC‐NOS | WT |
| MB‐6107 | R126Q | Diploid | 73% | ILC | ILC | MUT |
| MB‐5455 | R300C | Diploid | 78% | Mixed IDL | Mixed IDL | WT |
| MB‐5584 | R586C | Diploid | 78% | ND | ND | WT |
| TCGA‐BH‐A18U‐01 | S558 L | Gain | 81% | IDC‐NOS | IDC‐NOS | WT |
| MB‐6211 | A224T | Gain | 85% | IDC‐NOS | Ducto‐lobular | WT |
| TCGA‐A8‐A0A6‐01 | V688G | Diploid | 87% | ILC | ILC | MUT |
| MB‐0653 | D114H | Diploid | 91% | ILC (solid) | ILC (solid) | MUT |
| MB‐4855 | A885V | Gain | 92% | IDC‐NOS | IDC‐NOS | WT |
| MB‐5284 | P462L | Diploid | 92% | Mixed IDL | IDC‐NOS | WT |
| TCGA‐AN‐A0XW‐01 | E432Q | Diploid | 92% | IDC‐NOS | IDC‐NOS | WT |
| MTS‐T0697 | E406* | ShallowDel | ND | IDC‐NOS | IDC‐NOS | WT |
AA = amino acid; IDC‐NOS = invasive ductal carcinoma, not otherwise specified; ILC = invasive lobular carcinoma; PILC = pleomorphic ILC; IDL = invasive ductal and lobular; ND = no data; WT = wild type; MUT = mutant; _s = splice;
Mutations in bold are potentially pathogenic.
Contains signet ring cells;
40% mRNA expression.
Figure 1.
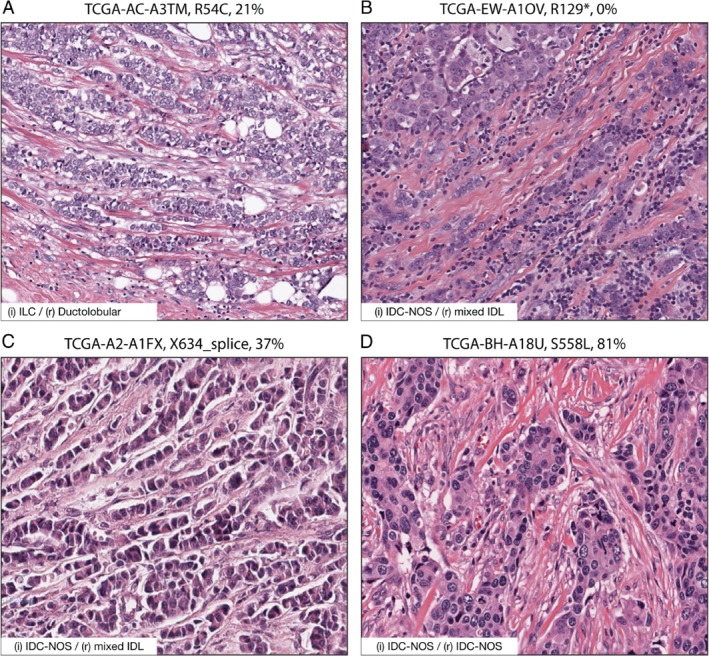
Somatic inactivating CTNNA1 mutations correlate with invasive lobular and ducto‐lobular mixed‐type breast cancer. (A–D) H&E processed samples harboring an inactivating CTNNA1 mutation and non‐mutated CDH1 alleles. Examples of a case diagnosed as ILC (A) and two cases that were initially diagnosed as IDC‐NOS, but that presented ducto‐lobular features (B, C) are shown. The case shown in D represents a typical IDC‐NOS and was diagnosed based on histological features and a positive E‐cadherin IHC analysis. Cancer study‐sample ID, CTNNA1 amino acid change (somatic mutation), and mRNA levels are indicated in the given order. The inset depicts initial (i) and revised (r) diagnosis. Samples can be cross‐referenced with Table 1; data were obtained from http://cancergenome.nih.gov. Scale bar = 100 μm.
Loss of α‐catenin leads to a dysfunctional epithelial adherens junction complex and a rounded cell morphology
To establish causality between loss of α‐catenin and AJ formation in non‐invasive breast cancer cell lines, we used mouse and human non‐invasive E‐cadherin‐expressing breast cancer cell lines (mouse Trp53Δ/Δ cells from mammary‐specific models 2, 24 and MCF7). These cell lines form mature epithelial AJs and show a classical cobblestone‐type morphology under 2D culture conditions 25. To enable inducible loss of α‐catenin, we used a doxycycline (Dox) inducible lentiviral knockdown (iKD) system and observed that addition of Dox induced a strong reduction in α‐catenin levels, while the expression of E‐cadherin, p120, and β‐catenin remained unaffected (Figure 2A and supplementary material, Figure S3A). Culturing iKD cells in the presence of Dox led to a rounded cell morphology, indicating a reduction of cell–cell and cell–matrix‐dependent adhesion (Figure 2B and supplementary material, Figure S3B). Loss of α‐catenin also induced a non‐cohesive growth pattern that prevented confluence and the formation of an epithelial sheet (Figure 2B and supplementary material, Figure S3B). Although we did not observe quantitative E‐cadherin expression protein differences using western blot, α‐catenin loss induced a marked change in E‐cadherin distribution and AJ formation in mouse and human breast cancer cells (Figure 2C and supplementary material, Figure S3C). At the sites of cell–cell adhesion, E‐cadherin, β‐catenin, and p120 still localized to the cell membrane, but maturation of the AJ appeared attenuated, which was characterized by aberrant localization of E‐cadherin and p120 in distinct puncta and clusters on the plasma membrane (Figure 2C, indicated by arrows). We furthermore observed an increase in the cytosolic localization of the core AJ members, suggesting active recycling of the AJ (Figure 2C, indicated by arrowheads). We next performed a reconstitution assay by introducing a non‐targetable and GFP‐tagged α‐catenin cDNA, which restored AJ formation and function, leading to the formation of a classical epithelial morphology in the presence of Dox (Figure 2).
Figure 2.
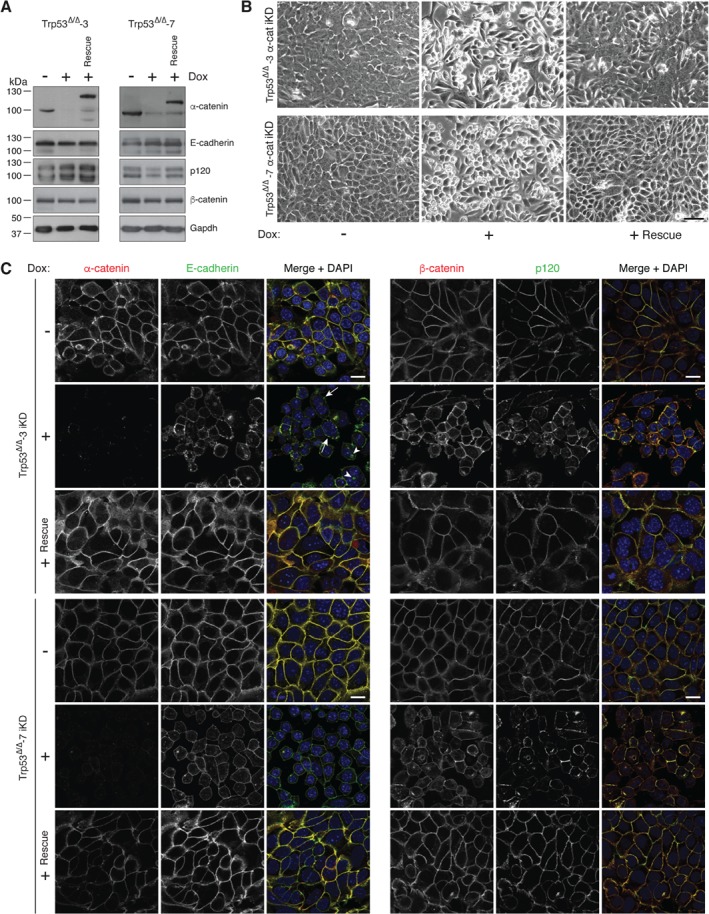
Loss of α‐catenin induces loss of epithelial cell morphology and leads to aberrant localization of AJ members. (A) Inducible knockdown of α‐catenin (α‐cat iKD) does not lead to inhibition of AJ complex member expression levels. Western blot showing the extent of α‐catenin iKD (+ Dox) on E‐cadherin, p120, and β‐catenin. Right lanes (+ Rescue) show the effects of an α‐catenin‐GFP cDNA reconstitution. Gapdh levels were used as loading control. (B) Loss of α‐catenin induces a rounded and non‐adherent cell morphology. Phase‐contrast images of α‐catenin iKD and rescue cell lines. Scale bar = 50 μm. (C) Dysfunctional formation of the AJ upon α‐catenin loss. Immunofluorescence images for the AJ complex members α‐catenin, E‐cadherin, p120, and β‐catenin in control (− Dox), α‐catenin iKD (+ Dox), and Rescue cells (+ Rescue) are shown. Note the distinct clustering of the AJ in membrane‐localized puncta upon α‐catenin loss (arrows) and the cytosolic localization of E‐cadherin (arrowheads). Scale bar = 10 μm.
Loss of E‐cadherin in Trp53Δ/Δ‐3 cells induced a comparable increase in anoikis resistance when compared with α‐catenin iKD, leading to approximately 70% viable cells in suspension after 4 days in culture (data not shown and 26). E‐cadherin knockout (KO) Trp53Δ/Δ‐3 cells grew as single cells in 2D (supplementary material, Figure S4A,B), probably due to the complete absence of cadherin‐dependent cell–cell adhesion (supplementary material, Figure S4C). Confirming previous studies, we observed that E‐cadherin knockout in mammary carcinoma cells leads to cytosolic and nuclear relocalization of p120‐catenin and a strong reduction in the expression of α‐catenin and β‐catenin (supplementary material, Figure S4C).
Loss of α‐catenin results in anoikis resistance of E‐cadherin‐expressing breast cancer cells
Because E‐cadherin loss underpins the acquisition of anoikis resistance and metastasis formation, we investigated the effect of α‐catenin iKD on anchorage independence and analyzed cell survival by FACS analysis in the presence of Dox for 4 days. While the majority of the mouse and human control iKD cell lines (no Dox) underwent anoikis, α‐catenin loss induced a significant 3‐ to 5‐fold increase in anoikis resistance (Figures 3A and 2B). Reconstitution with α‐catenin resulted in a full reversal of anoikis resistance to wild‐type levels (Figure 3A), which shows that α‐catenin loss is causal to the induction of anoikis resistance and indicates that α‐catenin exerts tumor suppressor activity in non‐invasive breast cancer cell lines.
Figure 3.
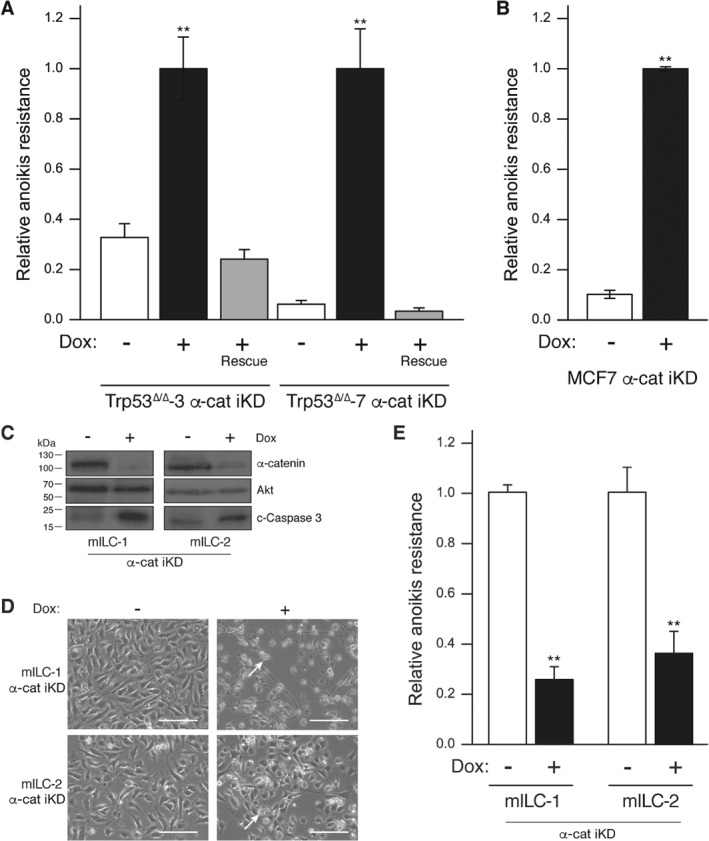
Loss of α‐catenin induces anoikis resistance in mouse and human E‐cadherin‐expressing cell lines and apoptosis in E‐cadherin‐mutant mouse ILC cells. Anoikis resistance was determined in mouse (A) and human (B) breast cancer cell lines upon α‐catenin iKD by FACS analysis using binding to annexin‐V and propidium iodide. Note the reversal to anoikis sensitivity upon reconstitution of α‐catenin in the mouse Trp53Δ/Δ cell lines (+ Dox Rescue; red bars). (C–E) Loss of α‐catenin in mouse ILC cells leads to apoptosis. Western blot (C) showing the extent of α‐catenin knockdown in two independent mouse ILC cells and the induction of cleaved caspase‐3 expression (c‐Caspase 3). Akt was used as loading control. Phenotypical consequences are shown in D. Note the apoptotic cells in the Dox‐treated cells (arrow). (E) Anoikis resistance was determined in the mILC cell lines upon α‐catenin iKD. Error bars represent the standard deviation of three independent experiments. **p = 0.001.
Interestingly, our data indicated that α‐catenin loss in an ILC cell type is not tolerated, because inducible Ctnna1 knockdown in two independent mouse ILC cell lines resulted in cell death through the induction of apoptosis (Figure 3C–E). These data suggest that α‐catenin exerts context‐dependent functions in cancer based on its localization and the presence of functional AJ.
Anoikis resistance upon loss of α‐catenin is dependent on an active Rho–Rock–actin pathway
Given that E‐cadherin‐mutant ILC depends on Rock activity for anchorage‐independent tumor growth and metastasis, we assayed the effect of α‐catenin loss on Rho–Rock pathway activity. Using serine phosphorylation of cofilin as a surrogate readout for the activity of Rho and Rock, we observed that α‐catenin loss resulted in an increase in phosphorylated cofilin in human and mouse breast cancer cells (Figure 4A), indicating activation through Rock1. Indeed, treatment with the pharmacological Rock inhibitor GSK‐429286A 30 decreased phosphorylation of cofilin in the presence of Dox and, surprisingly, reverted the rounded cell morphology when cells were cultured in 2D (Figure 4A, B). We observed identical results using the Rock inhibitor Y‐27632 (data not shown). Because α‐catenin null cells are unable to form mature AJs (Figure 1) and the α‐catenin‐depleted cells did not show restoration of epithelial junctions upon Rock inhibition (Figure 4B; right panels), our data suggested that detachment in 2D upon α‐catenin loss is due to inhibition of an integrin‐based cell–matrix adhesion, rather than a cadherin‐dependent homotypic interaction.
Figure 4.
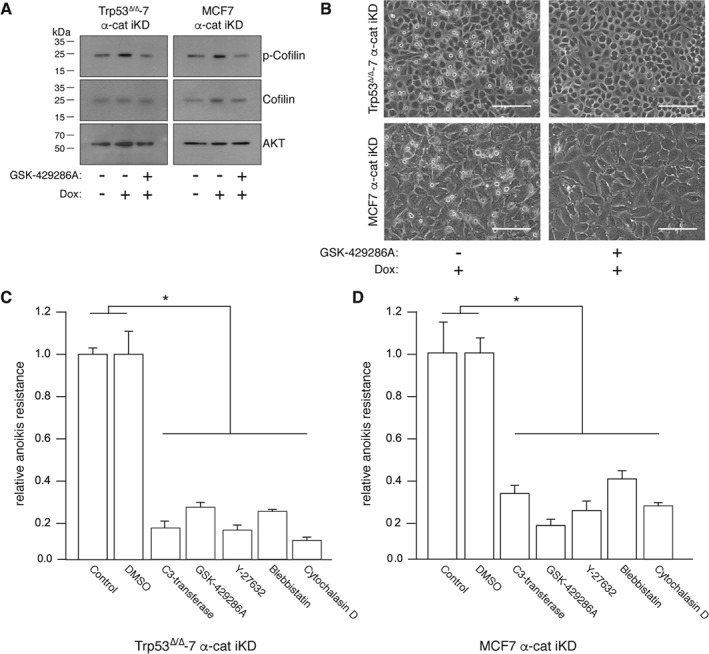
Rho–Rock actomyosin contractility controls anoikis resistance upon loss of α‐catenin. (A) Western blot showing cofilin and phosphorylated cofilin levels in α‐catenin iKD cell lines in the absence or presence of Dox and the Rock inhibitor GSK‐429286A. AKT levels were used as a loading control. (B) Inhibition of Rock in Dox‐treated α‐catenin iKD cells restores AJ‐independent cell spreading in 2D. Phase‐contrast pictures of α‐catenin iKD cells after inhibition of Rock using 1 μm GSK‐429286A. Scale bar = 30 μm. (C, D) Rho–Rock‐dependent actomyosin contraction controls anoikis resistance in the absence of α‐catenin. Anoikis resistance was determined by FACS analysis after culturing mouse (C) or human (D) breast cancer cells in the presence of Dox and the indicated inhibitors. Controls without inhibitors (control or DMSO) were set to 1. Error bars represent the standard deviation of three independent experiments. *p < 0.05.
To further investigate the dependency of the Rho–Rock pathway during anoikis resistance in α‐catenin iKD cells, we tested the effect of several inhibitors that act upstream or downstream of Rock. Pharmacological inhibition of Rho (C3 transferase), Rock (GSK‐429286A and Y‐27632), myosin II (blebbistatin), and F‐actin (cytochalasin D) all resulted in a robust reduction of anoikis resistance (Figure 4C, D), indicating that, similar to E‐cadherin‐mutant ILC, Rock‐dependent actomyosin contraction underpins anoikis resistance in the absence of α‐catenin.
Inducible α‐catenin loss leads to local tumor cell invasion and ducto‐lobular phenotypes in a transplantation‐based conditional mouse model of breast cancer progression
To address the functional consequences of α‐catenin loss in breast cancer, we developed a multi‐layered transplantation‐based mouse model. This model is based on orthotopic transplantation of mouse mammary epithelial stem cells (MMECs) from pre‐pubertal (unswitched) WAPCre;Trp53 F/F mice that are transduced with GFP‐tagged inducible shRNAs targeting Ctnna1. After transplantation, a normal mammary gland will develop and loss of Trp53 will occur under the influence of physiological prolactin‐dependent WAP promoter activation. During this period, mice are switched to a Dox‐containing diet 8 weeks post‐transplantation to trigger shRNA‐mediated α‐catenin loss (for an overview see Figure 5A). In this set‐up, we monitored longitudinal tumor growth and observed a significantly longer tumor‐free latency period in the Dox‐treated compared with the control animals (214 versus 181 days, respectively; p < 0.001). We next determined inclusion eligibility by performing immunohistochemistry (IHC) for GFP to exclude tumors that had not been transduced with the GFP‐tagged lentiviral construct. In total, we thus obtained five control and five Ctnna1‐iKD mammary carcinomas that were expressing GFP (out of the initial 44 transplanted animals).
Figure 5.
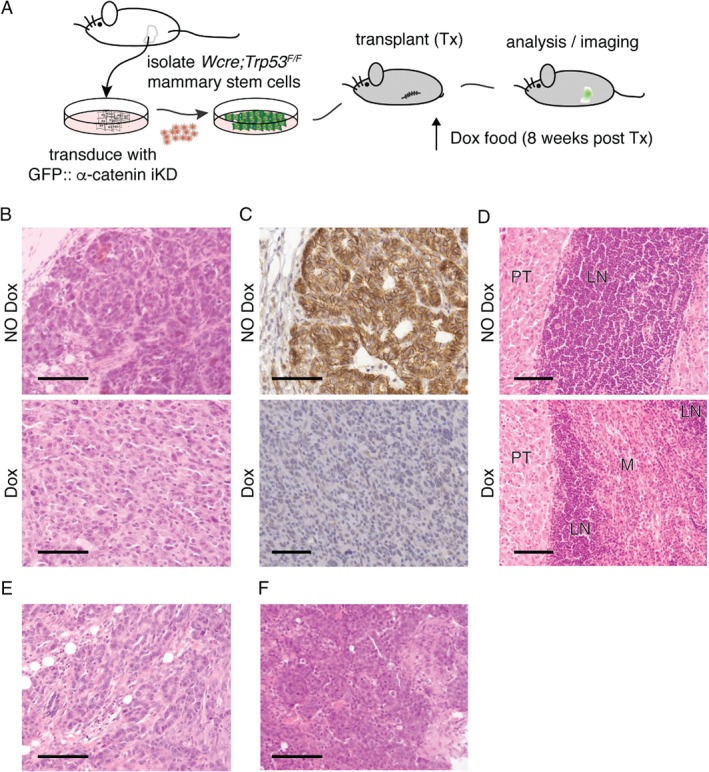
Inducible loss of α‐catenin leads to invasive mammary carcinoma in a conditional breast cancer progression mouse model. (A) Transplantation‐based models to study breast cancer progression. Schematic overview of the mammary stem cell (MSC) transplantation (Tx) system. Unswitched MSCs were harvested from conditional prepuberal female WAPcre;Trp53 F/F donor mice. After transduction with GFP‐tagged inducible knockdown (iKD) lentiviruses, WAPcre;Trp53 F/F::α‐catenin iKD MSCs were orthotopically transplanted into the cleared fat‐pad of recipient nude 8‐week‐old female mice. Induction of the second tumor suppressor ‘hit’ (loss of α‐catenin) was induced by feeding the mice doxycyclin (Dox)‐containing chow, and mice were monitored for tumor formation and progression. (B, C) Loss of α‐catenin leads to a switch from non‐invasive to invasive carcinoma. Experimental set‐up was as in A, and α‐catenin was inactivated in WAPcre;Trp53 F/F::α‐catenin iKD with doxycyclin 8 weeks post‐transplantation. Tumors were harvested, subtyped using hematoxylin/eosin (HE) (B), and stained for α‐catenin using immunohistochemistry (brown). Upper panels represent control mice (NO Dox); lower panels depict tumors from Dox‐fed mice (Dox). Note the marked acquisition of invasive growth (B and C top versus bottom panels) and the loss of α‐catenin in the Dox‐treated animals (C, bottom panel). (D) Loss of α‐catenin leads to invasion of blood vessels and regional lymph nodes. Tumors from a control (NO Dox, upper panel) and Dox‐fed mouse (Dox, bottom panel) that depict strong differences in local invasion of the inguinal lymph node (LN) are shown. PT = primary tumor; M = metastasis. (E, F) Loss of α‐catenin induces occasional lobular‐like phenotypical features. A representative example of trabecular ducto‐lobular growth patterns (E) and a lesion resembling solid‐type mouse ILC (F) are shown. Scale bars = 50 μm.
Carcinomas in the control cohort were either intermediate‐grade adenocarcinomas or high‐grade carcinosarcomas (Figure 5B) that were characterized by expansive growth patterns and large epithelial cells forming solid nests or irregular glands. However, while tumors that developed in the Dox‐treated mice were also predominantly carcinosarcomas or solid‐type carcinomas, inducible loss of α‐catenin resulted in a shift from non‐invasive to locally invasive tumors (Figure 5B). Compared with control mammary carcinomas, we observed that the invasive carcinomas of Dox‐treated mice showed loss of α‐catenin protein expression (Figure 5C). Also, in contrast to control mice that exclusively presented well‐circumscribed non‐invasive tumor margins, the Dox‐treated mice showed extensive local invasion including invasion of blood vessels and local lymph node involvement (Figure 5D). Although Ctnna1‐iKD mammary carcinomas showed extensive local invasion, we did not detect distant metastasis to lungs or lymph nodes. Interestingly, we observed focal lobular features in three Dox‐treated animals, i.e. relatively uniform tumor cells that were small in size growing in non‐cohesive clusters with sporadic ducto‐lobular trabecular growth patterns (Figure 5E), or features reminiscent of solid‐type mouse ILC (Figure 5F). In contrast to previous models in which E‐cadherin was concomitantly inactivated with p53 in cytokeratin 14 or WAP‐dependent Cre driver mice 2, 5, 24, we did not observe classical ILC invasive strands or ‘Indian file’‐type invasion patterns. Because we occasionally observed ducto‐lobular phenotypical features, our findings suggest that loss of α‐catenin represents an alternative route for the development of lobular‐type invasive breast cancer.
Discussion
Inactivation of the AJ as a whole is not the determining factor for the formation of lobular breast cancer. Whereas somatic ablation of E‐cadherin in the mammary gland results in metastatic mouse ILC 2, 5, 24, inactivation of the AJ through p120 loss induces the formation of invasive high‐grade and basal‐like IDC 25, 31. It appears that ILC may uniquely depend on Rho/Rock‐driven actin remodeling, given that p120 null mammary carcinoma cells and IDC‐derived human tumor cell lines (e.g. MDA‐MB‐231) do not rely on active Rock signals for their anchorage‐independent survival 7, 32. Second, although p120 regulates Rho signaling at the cell cortex, it is not directly physically connected to the actin cytoskeleton (for a review see 33), which may explain why loss of p120 in mice (although it leads to inactivation of the AJ and induces metastasis) does not result in a lobular tumor phenotype 25. Because p120 (CTNND1) is lost in approximately 30% of all IDC‐NOS breast cancers, but mutations and promotor methylation are virtually absent 34, we think that p120 is a tumor progression suppressor that is inactivated at the later stages of non‐lobular breast cancer.
In contrast, bi‐allelic inactivating mutations in CTNNA1 have been described in several breast cancer cell lines 21, 35. Because these cell lines are phenotypically luminal and express membrane‐bound wild‐type E‐cadherin, it strengthened our assumption that specific disruption of the link between E‐cadherin, α‐catenin, and actin may be sufficient to underpin lobular carcinoma etiology. Supporting this is a frame shift mutation in CTNNA1 that was described in a family with hereditary diffuse gastric cancer (HDGC) without detectable E‐cadherin defects 23. Similar to ILC, HDGC and plasmacytoid bladder cancer development is causally linked to mutational inactivation of E‐cadherin 36, 37, again implying that there may be a functional relationship between inactivation of α‐catenin, cell‐intrinsic actomyosin contraction, and the development and progression of ILC.
Despite the low overall prevalence of CTNNA1 mutations in breast cancer, we observed 19 somatic mutations in the publicly available TCGA database BioPortal, of which six cases showed potential inactivating CTNNA1 mutations in the presence of wild‐type CDH1. Given that α‐catenin is necessary for proper lateral E‐cadherin clustering and subsequent AJ maturation, and the fact that anoikis resistance could be fully reversed upon inhibition of the Rho/Rock pathway, it appears that inactivation of α‐catenin confers anchorage independence through constitutive activation of signals downstream of Rock. While we have not probed the underlying biochemistry, we hypothesize that the (bio)mechanical forces induced by activation of Rho/Rock‐dependent actomyosin contraction upon loss of α‐catenin can result in extracellular matrix (ECM)/integrin linkage detachment (comparable to the onset of mitosis), which may have substantial consequences for the migration and invasion of tumor cells. We therefore envisage that Rho activity is most likely caused by intracellular cytoskeletal tension upon cellular rounding due to the disruption of the E‐cadherin linkage to actin, which could potentially induce a positive feedback loop resulting in high Rho‐GTP, as has been shown previously by others 38, 39. Because of this, we think that it is likely that loss of α‐catenin in cancer may result in a double‐edged sword that drives tumor progression and development through constitutive activation of anchorage‐independent cell survival and cellular motility. As such, α‐catenin (like E‐cadherin) may represent a tumor suppressor that represents the same gene hypothesis, which states that specific mutations that provide a selective advantage during tumor initiation can also foster tumor progression 40.
Given the functional effect of α‐catenin on E‐cadherin form and function presented here, it could well be that the phenotypic output does not have to be clearly lobular per se. Our in vivo experiment yielded a non‐cohesive phenotype that lacked classical ILC phenotypical characteristics such as ‘Indian files’ and targetoid/periductal growth patterns.
First, loss of α‐catenin leads to erroneous localization and clustering of E‐cadherin, which is a possible cause of adhesive and propulsive differences of the tumor cells when compared with classical ILC. Second, p120‐catenin, which represents a key oncogene in classical ILC through its cytosolic translocation and indirect MRIP‐dependent activation of actomyosin contractions 7, is still largely retained at the plasma membrane, due to the expression of E‐cadherin. Given the cardinal functions of p120‐catenin in the control of RhoA 34, 41, this may also underlie the phenotypic differences in CTNNA1‐mutant versus affect classical ILC.
An alternative explanation for the differential phenotypes may be based on the fact that we have used mammary repopulating unit/progenitor cells for our in vivo experiments. Lentiviral integration of the Dox‐inducible Ctnna1 knockdown will probably overlap with the cancer‐initiating cell that undergoes WAPcre‐mediated Trp53 deletion, but it will likely also occur in p53 wild‐type cells in the mammary gland. If this is a myoepithelial/basal cell type that has a role in tissue homeostasis, α‐catenin loss may affect barrier function, with possible indirect effects on tumor outgrowth and phenotype. Finally, timing is different in the conditional E‐cadherin loss‐driven models and the current transplantation‐based α‐catenin knockdown set‐up. Whereas the E‐cadherin knockout models simultaneously ablate E‐cadherin and p53 either through cytokeratin 14 or WAPcre‐driven recombination, Ctnna1 inactivation in our transplantation setting might also occur shortly following p53 loss, with a potential impact on tumor morphology. We anticipate that, notwithstanding the fact that biochemical wiring of a CTNNA1‐mutant breast cancer is similar to classical ILC (i.e. functional AJ loss and constitutive actomyosin contraction), the above argument may in part also account for the ductal‐type appearance of these cancers. We think that this rationale will enable correct diagnosis, a conceptually essential prerequisite for future treatment of ILC 42 and other AJ‐related cancers such as HDGC and plasmacytoid bladder cancer.
In closing, we have shown that α‐catenin functions as a tumor progression suppressor in E‐cadherin‐expressing non‐metastatic tumor cells through loss of cell–cell and cell–matrix interactions and the acquisition of anchorage independence. Importantly, α‐catenin loss confers anoikis resistance through activation of Rho and Rock‐dependent actomyosin contraction, a hallmark of E‐cadherin‐mutant ILC. We thereby advocate loss of α‐catenin as a potential driving mechanism underpinning ILC tumor etiology. Because E‐cadherin is linked to the actin cytoskeleton through α‐catenin and ILC relies on disruption of the E‐cadherin–actin connection for sustained actomyosin contraction and tumor progression, we advocate that carcinomas driven by E‐cadherin loss should be considered ‘actin’ diseases. Our findings emphasize the potential clinical ramifications of Rock inhibition in the treatment of metastatic ILC and mixed‐type ducto‐lobular invasive breast cancer.
Author contributions statement
JSdG, MvA, MAKR, TE, EJV, and MTN performed the experiments. JSdG, JdR, and PWBD conceived the experiments. S‐FC and CC performed analyses on the METABRIC samples. PJvD diagnosed primary breast cancers. JJ aided in the execution of the mouse experiments. JSdG and PWBD wrote the manuscript. All authors approved the final version of the manuscript.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary figure legends
Figure S1. CTNNA1 genomic alterations in human cancer
Figure S2. Clonal characterizations of samples with CTNNA1 mutations
Figure S3. Loss of α‐catenin induces loss of epithelial cell morphology and leads to aberrant localization of AJ members in human breast cancer cells
Figure S4. Loss of E‐cadherin induces loss of epithelial cell morphology and dismantling of AJ members in mouse mammary carcinoma cells
Supporting information
Supplementary figure legends
Figure S1. CTNNA1 genomic alterations in human cancer. (A) The alteration frequency for mutations, amplifications, and deletions in CTNNA1 across multiple cancer subtypes. (B) Alteration frequency of CTNNA1 mutation in human breast cancer. Source for this figure: http://cancergenome.nih.gov/.
Figure S2. Clonal characterizations of samples with CTNNA1 mutations. The clonal characterizations of the samples with a CTNNA1 mutation as described in Table 1 are depicted. For 18 out of 19 samples, data for allele frequencies were available. The y‐axis represents mutation counts, the x‐axis allele frequencies, and the blue line the allele frequency of the CTNNA1 mutation. Above each plot, the sample ID is depicted with the total number of mutations in the sample. The sample IDs are color‐coded for the clonality result, with red depicting clonal, pink biclonal/uncertain, and grey non‐interpretable. Source for the TCGA data in this figure: http://cancergenome.nih.gov/.
Figure S3. Loss of α‐catenin induces loss of epithelial cell morphology and leads to aberrant localization of AJ members in human breast cancer cells. (A) Inducible knockdown of α‐catenin (iKD α‐cat) does not lead to inhibition of AJ complex member expression levels. Western blot showing the extent of α‐catenin iKD (+ Dox) on E‐cadherin, p120, and β‐catenin. AKT levels were used as loading control. (B) Loss of α‐catenin induces a rounded and non‐adherent cell morphology. Phase‐contrast images of control (− Dox) and α‐catenin knockdown cells (+ Dox). Scale bar indicates 50 μm. Immunofluorescence pictures of control (−) and α‐catenin knockdown (+) and rescue cell lines (+ Rescue). (C) Dysfunctional formation of the AJ upon α‐catenin loss. Immunofluorescence images for the AJ complex members α‐catenin, E‐cadherin, p120, and β‐catenin in control (− Dox) and α‐catenin iKD (+ Dox) are shown. Note the distinct clustering of E‐cadherin in membrane‐localized puncta (arrows) and the cytosolic localization upon α‐catenin loss (arrowheads).
Figure S4. Loss of E‐cadherin induces loss of epithelial cell morphology and dismantling of AJ members in mouse mammary carcinoma cells. (A) CRISPR‐Cas9 mediated E‐cadherin knockout in mouse Trp53Δ/Δ‐3 mammary carcinoma cells. Western blot showing the extent of E‐cadherin knockout (KO). AKT levels were used as loading control. (B) Loss of α‐catenin induces a non‐adherent cell morphology. Phase‐contrast images of control (scrambled guide RNA) and E‐cadherin knockdown cells. Size bar indicates 50 μm. (C) Dismantling of the AJ in E‐cadherin mutated cells. Immunofluorescence images for the AJ complex members E‐cadherin, α‐catenin, p120‐catenin, and β‐catenin in control and E‐cadherin knockout Trp53Δ/Δ‐3 cells are shown. Scale bar indicates 10 μm.
Acknowledgements
Special thanks go to the UMC Utrecht Cell Microscopy Center (CMC) for providing microscopy assistance. All members of the Derksen, Van Diest, Jonkers, and De Rooij laboratories are acknowledged for support and discussions. We are indebted to The Netherlands Cancer Institute mouse facility for support during the longitudinal tumor studies. This work was supported by a grant from The Netherlands Organization for Scientific Research (NWO‐VIDI 917.96.318), Foundation Vrienden UMC Utrecht (11.081), Dutch Cancer Society grants (KWF‐UU‐2011‐5230, KWF‐UU‐2014‐7201 and KWF‐2017‐10456), and the European Union's Horizon 2020 FET Proactive programme under the grant agreement No. 731957 (MECHANO‐CONTROL).
No conflicts of interest were declared.
References
- 1. Berx G, Cleton‐Jansen AM, Strumane K, et al E‐cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 1996; 13 : 1919–1925. [PubMed] [Google Scholar]
- 2. Derksen PWB, Liu X, Saridin F, et al Somatic inactivation of E‐cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006; 10 : 437–449. [DOI] [PubMed] [Google Scholar]
- 3. Vlug E, Ercan C, Wall E, et al Lobular breast cancer: pathology, biology, and options for clinical intervention. Arch Immunol Ther Exp 2013; 62 : 1–15. [DOI] [PubMed] [Google Scholar]
- 4. Bruner HC, Derksen PWB. Loss of E‐cadherin‐dependent cell–cell adhesion and the development and progression of cancer. Cold Spring Harb Perspect Biol 2018; 10 : a029330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boelens MC, Nethe M, Klarenbeek S, et al PTEN loss in E‐cadherin‐deficient mouse mammary epithelial cells rescues apoptosis and results in development of classical invasive lobular carcinoma. Cell Rep 2016; 16 : 2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Annunziato S, Kas SM, Nethe M, et al Modeling invasive lobular breast carcinoma by CRISPR/Cas9‐mediated somatic genome editing of the mammary gland. Genes Dev 2016; 30 : 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schackmann RCJ, van Amersfoort M, Haarhuis JHI, et al Cytosolic p120‐catenin regulates growth of metastatic lobular carcinoma through Rock1‐mediated anoikis resistance. J Clin Invest 2011; 121 : 3176–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van de Ven RAH, Tenhagen M, Meuleman W, et al Nuclear p120‐catenin contributes to anoikis resistance of lobular breast cancer through Kaiso‐dependent Wnt11 expression. Dis Model Mech 2015; 8 : 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol 2009; 1 : a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mège RM, Ishiyama N. Integration of cadherin adhesion and cytoskeleton at adherens junctions. Cold Spring Harb Perspect Biol 2017; 9 : a028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson WJ. Regulation of cell–cell adhesion by the cadherin–catenin complex. Biochem Soc Trans 2008; 36 : 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vasioukhin V, Bauer C, Yin M, et al Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell 2000; 100 : 209–219. [DOI] [PubMed] [Google Scholar]
- 13. Kobielak A, Pasolli HA, Fuchs E. Mammalian formin‐1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol 2004; 6 : 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silvis MR, Kreger BT, Lien W‐H, et al α‐Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 2011; 4 : ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bajpai S, Feng Y, Krishnamurthy R, et al Loss of alpha‐catenin decreases the strength of single E‐cadherin bonds between human cancer cells. J Biol Chem 2009; 284 : 18252–18259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drees F, Pokutta S, Yamada S, et al Alpha‐catenin is a molecular switch that binds E‐cadherin–beta‐catenin and regulates actin‐filament assembly. Cell 2005; 123 : 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Troyanovsky RB, Klingelhofer J, Troyanovsky SM. α‐Catenin contributes to the strength of E‐cadherin–p120 interactions. Mol Biol Cell 2011; 22 : 4247–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vasioukhin V, Bauer C, Degenstein L, et al Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha‐catenin in skin. Cell 2001; 104 : 605–617. [DOI] [PubMed] [Google Scholar]
- 19. Lien W‐H, Klezovitch O, Fernandez TE, et al αE‐Catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 2006; 311 : 1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benjamin JM, Nelson WJ. Bench to bedside and back again: molecular mechanisms of alpha‐catenin function and roles in tumorigenesis. Semin Cancer Biol 2008; 18 : 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hollestelle A, Elstrodt F, Timmermans M, et al Four human breast cancer cell lines with biallelic inactivating alpha‐catenin gene mutations. Breast Cancer Res Treat 2010; 122: 125–133. [DOI] [PubMed] [Google Scholar]
- 22. Bignell GR, Greenman CD, Davies H, et al Signatures of mutation and selection in the cancer genome. Nature 2010; 463 : 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Majewski IJ, Kluijt I, Cats A, et al An α‐E‐catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol 2013; 229 : 621–629. [DOI] [PubMed] [Google Scholar]
- 24. Derksen PWB, Braumuller TM, Van Der Burg E, et al Mammary‐specific inactivation of E‐cadherin and p53 impairs functional gland development and leads to pleomorphic invasive lobular carcinoma in mice. Dis Model Mech 2011; 4 : 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schackmann RCJ, Klarenbeek S, Vlug EJ, et al Loss of p120‐catenin induces metastatic progression of breast cancer by inducing anoikis resistance and augmenting growth factor receptor signaling. Cancer Res 2013; 73 : 4937–4949. [DOI] [PubMed] [Google Scholar]
- 26. Hornsveld M, Tenhagen M, van de Ven RA, et al Restraining FOXO3‐dependent transcriptional BMF activation underpins tumour growth and metastasis of E‐cadherin‐negative breast cancer. Cell Death Differ 2016; 23 : 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Derksen PWB, Keehnen R, Evers L, et al Cell surface proteoglycan syndecan‐1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood 2002; 99 : 1405–1410. [DOI] [PubMed] [Google Scholar]
- 28. Welm BE, Dijkgraaf GJP, Bledau AS, et al Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2008; 2 : 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeOme KB, Faulkin LJ Jr, Bern HA, et al Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland‐free mammary fat pads of female C3H mice. Cancer Res 1959; 19 : 515–520. [PubMed] [Google Scholar]
- 30. Goodman KB, Cui H, Dowdell SE, et al Development of dihydropyridone indazole amides as selective Rho‐kinase inhibitors. J Med Chem 2007; 50 : 6–9. [DOI] [PubMed] [Google Scholar]
- 31. Tenhagen M, Klarenbeek S, Braumuller TM, et al p120‐Catenin is critical for the development of invasive lobular carcinoma in mice. J Mammary Gland Biol Neoplasia 2016; 21 : 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soto E, Yanagisawa M, Marlow LA, et al p120 catenin induces opposing effects on tumor cell growth depending on E‐cadherin expression. J Cell Biol 2008; 183 : 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schackmann RCJ, Tenhagen M, van de Ven RAH, et al p120‐catenin in cancer – mechanisms, models and opportunities for intervention. J Cell Sci 2013; 126 : 3515–3525. [DOI] [PubMed] [Google Scholar]
- 34. van de Ven RAH, de Groot JS, Park D, et al p120‐catenin prevents multinucleation through control of MKLP1‐dependent RhoA activity during cytokinesis. Nat Commun 2016; 7 : 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hazan RB, Kang L, Roe S, et al Vinculin is associated with the E‐cadherin adhesion complex. J Biol Chem 1997; 272 : 32448–32453. [DOI] [PubMed] [Google Scholar]
- 36. Guilford P, Hopkins J, Harraway J, et al E‐cadherin germline mutations in familial gastric cancer. Nature 1998; 392 : 402–405. [DOI] [PubMed] [Google Scholar]
- 37. Al‐Ahmadie HA, Iyer G, Lee BH, et al Frequent somatic CDH1 loss‐of‐function mutations in plasmacytoid variant bladder cancer. Nat Genet 2016; 48 : 356–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pirone DM, Liu WF, Ruiz SA, et al An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA–ROCK signaling. J Cell Biol 2006; 174 : 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhadriraju K, Yang M, Alom Ruiz S, et al Activation of ROCK by RhoA is regulated by cell adhesion, shape, and cytoskeletal tension. Exp Cell Res 2007; 313 : 3616–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanahan D, Weinberg R. The hallmarks of cancer. Cell 2000; 100 : 57–70. [DOI] [PubMed] [Google Scholar]
- 41. Anastasiadis P, Moon S, Thoreson M, et al Inhibition of RhoA by p120 catenin. Nat Cell Biol 2000; 2 : 637–644. [DOI] [PubMed] [Google Scholar]
- 42. Ciriello G, Gatza ML, Beck AH, et al Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015; 163 : 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure legends
Figure S1. CTNNA1 genomic alterations in human cancer. (A) The alteration frequency for mutations, amplifications, and deletions in CTNNA1 across multiple cancer subtypes. (B) Alteration frequency of CTNNA1 mutation in human breast cancer. Source for this figure: http://cancergenome.nih.gov/.
Figure S2. Clonal characterizations of samples with CTNNA1 mutations. The clonal characterizations of the samples with a CTNNA1 mutation as described in Table 1 are depicted. For 18 out of 19 samples, data for allele frequencies were available. The y‐axis represents mutation counts, the x‐axis allele frequencies, and the blue line the allele frequency of the CTNNA1 mutation. Above each plot, the sample ID is depicted with the total number of mutations in the sample. The sample IDs are color‐coded for the clonality result, with red depicting clonal, pink biclonal/uncertain, and grey non‐interpretable. Source for the TCGA data in this figure: http://cancergenome.nih.gov/.
Figure S3. Loss of α‐catenin induces loss of epithelial cell morphology and leads to aberrant localization of AJ members in human breast cancer cells. (A) Inducible knockdown of α‐catenin (iKD α‐cat) does not lead to inhibition of AJ complex member expression levels. Western blot showing the extent of α‐catenin iKD (+ Dox) on E‐cadherin, p120, and β‐catenin. AKT levels were used as loading control. (B) Loss of α‐catenin induces a rounded and non‐adherent cell morphology. Phase‐contrast images of control (− Dox) and α‐catenin knockdown cells (+ Dox). Scale bar indicates 50 μm. Immunofluorescence pictures of control (−) and α‐catenin knockdown (+) and rescue cell lines (+ Rescue). (C) Dysfunctional formation of the AJ upon α‐catenin loss. Immunofluorescence images for the AJ complex members α‐catenin, E‐cadherin, p120, and β‐catenin in control (− Dox) and α‐catenin iKD (+ Dox) are shown. Note the distinct clustering of E‐cadherin in membrane‐localized puncta (arrows) and the cytosolic localization upon α‐catenin loss (arrowheads).
Figure S4. Loss of E‐cadherin induces loss of epithelial cell morphology and dismantling of AJ members in mouse mammary carcinoma cells. (A) CRISPR‐Cas9 mediated E‐cadherin knockout in mouse Trp53Δ/Δ‐3 mammary carcinoma cells. Western blot showing the extent of E‐cadherin knockout (KO). AKT levels were used as loading control. (B) Loss of α‐catenin induces a non‐adherent cell morphology. Phase‐contrast images of control (scrambled guide RNA) and E‐cadherin knockdown cells. Size bar indicates 50 μm. (C) Dismantling of the AJ in E‐cadherin mutated cells. Immunofluorescence images for the AJ complex members E‐cadherin, α‐catenin, p120‐catenin, and β‐catenin in control and E‐cadherin knockout Trp53Δ/Δ‐3 cells are shown. Scale bar indicates 10 μm.