

Abstract
Nephropathic cystinosis is characterized by abnormal intralysosomal accumulation of cystine throughout the body, causing irreversible damage to various organs, particularly the kidneys. Cysteamine, the currently available treatment, can reduce lysosomal cystine and postpone disease progression. However, cysteamine poses serious side effects and does not address all of the symptoms of cystinosis. To screen for new treatment options, a rapid and reliable high‐performance liquid chromatography–tandem mass spectrometry (HPLC‐MS/MS) method was developed to quantify cystine in conditionally immortalized human proximal tubular epithelial cells (ciPTEC). The ciPTEC were treated with N‐ethylmaleimide, lysed and deproteinized with 15% (w/v) sulfosalicylic acid. Subsequently, cystine was measured using deuterium‐labeled cystine‐D4, as the internal standard. The assay developed demonstrated linearity to at least 20 μmol/L with a good precision. Accuracies were between 97.3 and 102.9% for both cell extracts and whole cell samples. Cystine was sufficiently stable under all relevant analytical conditions. The assay was successfully applied to determine cystine levels in both healthy and cystinotic ciPTEC. Control cells showed clearly distinguishable cystine levels compared with cystinotic cells treated with or without cysteamine. The method developed provides a fast and reliable quantification of cystine, and is applicable to screen for potential drugs that could reverse cystinotic symptoms in human kidney cells.
Keywords: ciPTEC, cystine, cystinosis, HPLC‐MS/MS
Abbreviations
- ciPTEC
conditionally immortalized human proximal tubular epithelial cells
- NEM
N‐ethylmaleimide.
1. INTRODUCTION
Nephropathic cystinosis (MIM219800) is a rare, but severe genetic disorder characterized by intralysosomal accumulation of cystine in different cell types. It is caused by pathogenic mutations in CTNS, a gene that encodes for the lysosomal cystine/proton symporter cystinosin. Mutations in CTNS lead to the lysosomal accumulation of cystine throughout the body and cause irreversible damage to various organs, particularly the kidneys (Town et al., 1998). Cysteamine, the only treatment available to date, can reduce lysosomal accumulation of cystine and postpone the disease progression (Thoene, Oshima, Crawhall, Olson, & Schneider, 1976). In lysosomes, cysteamine acts in a disulfide exchange reaction with cystine, leading to the formation of cysteine and cysteine‐cysteamine molecules which can be transported out of lysosomes by the cysteine transporter and the PQLC2 transporter, respectively (Besouw, Masereeuw, van den Heuvel, & Levtchenko, 2013; Gahl, Thoene, & Schneider, 2002). However, cysteamine poses serious side effects and does not correct all symptoms associated with cystinosis.
To screen for new drugs to treat nephropathic cystinosis, a quantitative bioanalytical assay for cystine is a pre‐requisite. Measurement of cystine concentrations in leukocytes of patients is a clinical routine for diagnosis of cystinosis and monitoring cysteamine treatment (de Graaf‐Hess, Trijbels, & Blom, 1999; Garcia‐Villoria, Hernandez‐Perez, Arias, & Ribes, 2013). For drug screening and development, however, measuring the cystine content in cultured renal proximal tubule cells can also be of high value. In nephropathic cystinosis, kidneys are initially affected with generalized proximal tubular dysfunction, so called renal Fanconi syndrome (Elmonem et al., 2017). In addition, it has been reported that cystinosin expression is predominantly high in renal proximal tubules when compared with other segments of the nephron, signifying that cystinosin is crucial for proximal tubular cell function (Kalatzis, Nevo, Cherqui, Gasnier, & Antignac, 2004). Hence, measurement of cystine in renal proximal tubule cells can bring a new versatile tool to screen for potential drugs to reverse the cystinotic symptoms. Here, we used conditionally immortalized proximal tubular epithelial cells (ciPTEC) derived from urine samples of both healthy controls and cystinotic patients. Cystinotic ciPTEC are a well‐characterized human renal model of cystinosis, and have been demonstrated to have increased intracellular cystine levels when compared with healthy ciPTEC (Wilmer et al., 2011).
Critical steps must be considered in order to have reliable intracellular cystine measurements. Cystine is a biologically active aminothiol formed from the oxidation of two cysteine molecules via a disulfide bond formation. Since cysteine content of the cytosol greatly exceeds the cystine concentration in lysosomes (de Graaf‐Hess et al., 1999), oxidation of cysteine to cystine would lead to an undesirable increase in cystine concentrations. Moreover, disulfide exchange reactions of cystine with other sulfhydryl groups can lead to the loss of cystine in the cells. To avoid these artifactual oxidation–reduction reactions and in order to have an adequate measurement of cystine in the cells, derivatization with N‐ethylmaleimide (NEM) is required immediately during sample preparation (de Graaf‐Hess et al., 1999; Escobar et al., 2016). NEM, an alkylating agent, forms stable bonds with sulfhydryl containing molecules, enabling them to be permanently blocked and prevent disulfide bond formation.
Several methods have been developed to detect the levels of aminothiols in different biological samples (Bayram, Rimbach, Frank, & Esatbeyoglu, 2014; Donnelly & Pronovost, 2000; Du, Cao, & Fung, 2014; Hodakova, Preisler, Foret, & Kuban, 2015). Among these, high‐performance liquid chromatography (HPLC) with fluorescence detection is the most widely used (Steele, Ooi, & Munch, 2012). Graaf‐Hess et al. (1999) have applied such method for measuring cystine in leukocytes and fibroblasts of cystinotic patients. On the other hand, Escobar et al. (2016) have recently reported a rapid and reliable bioanalytical assay for detection of cystine and other related thiols in whole blood using ultra‐performance liquid chromatography coupled to tandem mass spectrometry (UPLC‐MS/MS). Unfortunately, these methods are hampered in use because of their high limit of detection and poor cystine recovery.
Here, we describe a fast and reliable method to measure cystine in both healthy and cystinotic ciPTEC using HPLC‐MS/MS (Figure 1). Furthermore, the method is applicable in screening potential drugs that could reverse cystinotic symptoms in vitro using human kidney cells.
Figure 1.
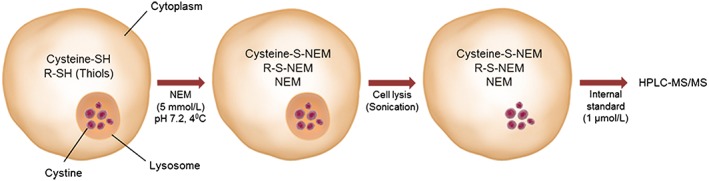
Schematic presentation of the method. N‐ethylmaleimide (NEM), an alkylating agent, forms stable bonds with cysteine and other thiols in cytosol, enabling them to be permanently blocked and prevent disulfide bond formation, while leaving cystine in lysosomes intact. This process is then followed by cell lysis and acid precipitation of proteins. This causes cystine to be present in the acid‐soluble fraction, which can be quantified by HPLC‐MS/MS
2. MATERIALS AND METHODS
2.1. Reagents and materials
Cystine, NEM and sulfosalicylic acid were purchased from Sigma‐Aldrich (Zwijndrecht, The Netherlands). Deuterated internal standard cystine‐D4 was purchased from Cambridge Isotope Laboratories (Tewksbury, https://en.wikipedia.org/wiki/Massachusetts, USA). Acetonitrile (HPLC‐S grade) was obtained from Biosolve (Valkenswaard, The Netherlands). Formic acid was purchased from Merck (Darmstadt, Germany). Ultrapure water was home‐purified on a Milli‐Q® Advantage A10 Water Purification Systems (Merck, The Netherlands).
2.2. Cell sample preparation
The ciPTEC were cultured and matured as described previously by Wilmer et al. (2010) in ciPTEC complete medium (DMEM/F12; Gibco/Invitrogen, Breda, The Netherlands). In short, cells were seeded at a density of 55,000 cells/cm2 in tissue culture flasks (25 cm2) and grown for 24 h at 33°C, 5% (v/v) CO2, for proliferation followed by 7 days of maturation at 37°C. Next, the cells were harvested using accutase and centrifuged at 250 g for 5 min at 4°C. The cell pellets were then quickly frozen in liquid nitrogen and stored at −80°C until quantification. Cell extracts were prepared by suspending the frozen pellets of ciPTEC in 100 μL of NEM solution (5 mmol/L NEM in 0.1 mol/L sodium phosphate buffer, pH 7.2) on ice. The cells were sonicated (Hielscher, UP50H; 1 cycle, 80% amplitude) on ice three times for 10 s with 20 s cooling intervals. Subsequently, 50 μL of 15% (w/v) sulfosalicylic acid was added to precipitate the proteins and the suspension was centrifuged at 20,000 g for 10 min at 4°C. Protein concentration was determined by the method of the Pierce™ BCA protein assay kit according to the manufacturer's protocol (ThermoFischer, The Netherlands), and the cystine concentration was measured using HPLC‐MS/MS.
2.3. Analytical sample pre‐treatment
Sample supernatants were thawed on ice and diluted (1:10) in 0.1% (v/v) formic acid in ultrapure water. Subsequently, 5 μL of internal standard solution cystine‐D4 (final concentration 1 μmol/L) was added to 95 μL of previously diluted supernatant. Finally, 100 μL of the clear supernatant was transferred into a glass injection vial and injected in the chromatographic system.
2.4. HPLC–MS/MS chromatographic system
The chromatographic system used consisted of a DUG14A degasser, two LC10‐ADvp‐μ pumps, a SIL‐HTC autosampler and a CTO‐10Avp oven (Shimadzu, Kyoto, Japan), coupled to a TSQ Quantum Discovery Max triple‐quadrupole mass spectrometer with electrospray ionization (ThermoFischer Scientific, San Jose, CA, USA). The mass spectrometer was operating with positive ionization in the selected reaction monitoring mode. Electrospray settings of the assay were a 3000 V spray voltage, a 323°C capillary temperature and the skimmer voltage was set at −5 V. The selected reaction monitoring mode with 0.15 s dwell times was used with argon as the collision gas at 1.4 mTorr. The tube lens offset was 102 V for both cystine and the internal standard cystine‐D4. Cystine was monitored at m/z 241.0 → 152.0 and 74.0 at −12 and − 28 V collision energies, and the internal standard cystine‐D4 at m/z 245.0 → 154.0 at −12 V collision energy. The mass resolutions were set at 0.7 for both separating quadrupoles. Separation conditions were selected to achieve an appropriate chromatographic retention by injecting 1.0 μL on an Atlantis dC18 column (100 × 2.1 mm, d.p. = 3.0 μm, Waters, Milford, USA) with a ChromSep Guard Polaris 3 C18A pre‐column (10 × 2.0 mm, d.p. = 3.0 μm, Agilent, Santa Clara, USA). The column temperature was maintained at 40°C and the autosampler at 4°C. Isocratic elution was performed at the rate of 0.5 mL/min with mobile phase A [0.1% (v/v) formic acid in ultrapure water], and B (acetonitrile). After injection, the percentage of solvent B was held at 0% (v/v) for 0.50 min, and was increased linearly to 100% (v/v) until 0.99 min. At 1.0 min the percentage of solvent B was decreased back to 0% (v/v) and finally the column was equilibrated for 3.0 min until starting the next injection. The whole eluate was transferred to the electrospray probe at 0.3–1.1 min using the MS diverter valve. Thermo Fisher Xcalibur software (version 2.0.7 SP1) was employed to acquire chromatography–mass spectrometric data and these data were further processed using Microsoft Excel (Office 2016, version 15.11.2).
2.5. Method validation
The analytical parameters assayed during the validation procedure were linearity, lower limit of quantification (LLOQ), limit of detection (LOD), precision and accuracy as well as matrix effect and stability of cell samples and standards.
2.5.1. Calibration
Individual stock solutions of cystine and the internal standard cystine‐D4 were prepared at a concentration of 20 μmol/L in 0.1% (v/v) formic acid in ultrapure water. The calibration samples were prepared daily in duplicate by serial dilution in 0.1% (v/v) formic acid in ultrapure water at concentrations ranging from 0.1 to 20 μmol/L of cystine. Least‐squares weighted linear regression was employed to acquire the calibration curve, using the ratio of the peak areas of both cystine and the internal standard cystine‐D4. The weighting factor was defined as the reversed square of the concentration (1/x 2). The LOD and LLOQ were defined as the lowest concentrations required to generate a signal‐to‐noise ratio of 3 and 10, respectively.
2.5.2. Accuracy and precision
The accuracy of the method was evaluated by means of the recovery test. To do this, cell extracts were spiked with three different concentration levels of cystine (i.e. low, 0.25; mid, 8.0; and high, 16.0 μmol/L), and were analyzed in three analytical runs on three separate days in triplicate. Nonspiked cell samples were also analyzed in each validation day in triplicate. In addition, to determine that the sample preparation (described above) does not result in cystine loss, recovery in whole cell samples was studied as well. For this purpose, cells were collected and spiked with the determined concentration of cystine (0.25, 8.0 and 16.0 μmol/L) before protein precipitation and the recovery was determined. The percentage recovery was calculated by subtracting the recovery value of the spiked cell samples from the nonspiked samples. To determine the precision of the method, spiked cell samples (the same samples used for accuracy determination) were analyzed within one validation batch (intra‐day) and among validation batches (inter‐day). The intra‐ and inter‐day run precisions were subsequently calculated as the percentage coefficient of variations (CV).
2.5.3. Stability
Stability of cystine was investigated in spiked cell samples (cell extracts and whole cell samples) under both short‐term (24 h at 4°C), and long‐term (three freeze–thaw cycles) storage conditions. For the long‐term stability test, spiked cell samples were frozen and thawed for three cycles (thawing at 20°C and freezing again at −80°C) within a period of minimum 1 month.
2.5.4. Matrix effect
The matrix effect was assessed at three different concentration levels (i.e. low, mid and high) by processing each sample and correcting the relative peak area of the spiked cell samples to that of the nonspiked samples. Thereafter, the areas were compared with the relative peak areas of cystine standard solutions.
2.6. Statistical analysis
Statistical differences between groups were assessed using One‐way Analysis of Variance (ANOVA) followed by Tukey's post‐hoc multiple comparisons tests using GraphPad Prism software 6.01. A p‐value <0.05 was considered to be statistically significant. Data is expressed as means ± standard deviation (SD) of three independent experiments performed in triplicate.
3. RESULTS
3.1. HPLC–MS/MS method
To obtain maximal sensitivity, the electrospray ionization–MS/MS settings were optimized for cystine and the internal standard cystine‐D4. The chromatographic method was adapted from Escobar et al. (2016) and optimized empirically based on MS response, peak shape and retention time. Acetonitrile and 0.1% (v/v) formic acid in ultrapure water resulted in a good MS response with a narrow cystine peak. With a total run time of 3.0 min, both cystine and the internal standard cystine‐D4 eluted at 0.63 min (± 0.003) without any peak interference in the 0.3–1.1 min time window. Representative chromatograms of extracted cells, with and without internal standard, are shown in Figure 2.
Figure 2.
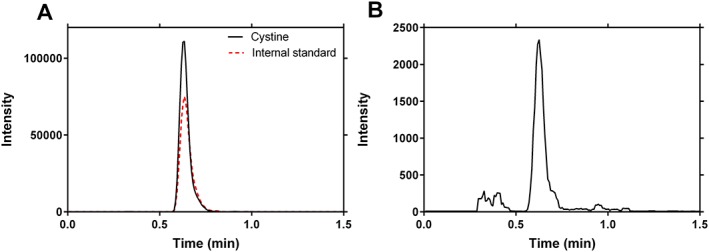
Selected reaction monitoring chromatograms of cystine. (a) Representative chromatogram obtained from cystinotic conditionally immortalized human proximal tubular epithelial cells (ciPTEC; 3.90 μmol/L) with internal standard cystine‐D4 (1 μmol/L), showing the retention time of cystine at 0.63 min (± 0.003). (b) Representative chromatogram obtained from healthy control ciPTEC (0.21 μmol/L)
3.2. Analytical method validation
3.2.1. Calibration
The relative response of cystine showed a good linearity within the concentrations tested, ranging from 0.1 to 20 μmol/L. For six independent calibrations, the concentrations were back‐calculated from the ratio of the peak areas of both cystine and the internal standard cystine‐D4. Deviations from the average of each level were ≤ 2.3% (data not shown), allowing linear regression analysis. The equation of the calibration curve revealed is y = 0.0034 (± 0.0008) + 0.0015 (± 0.00012)x with a coefficient of determination (R 2) value of 0.996 (± 0.002). Here, x is the cystine concentration (μmol/L) and y is the response of drug relative to the internal standard. The LOD and LLOQ of the assay were 0.05 and 0.1 μmol/L, respectively.
3.2.2. Accuracy and precision
The accuracy and precision of the method at three different concentrations are reported in Table 1. The method resulted in low intra‐ and inter‐day run variations (<15%) with accuracies ranging from 97.3 to 102.9% for both cell extracts and whole cell samples.
Table 1.
Cystine recovery (accuracy), precision and matrix effect measurements obtained after analysis of the spiked cell samples at three different concentrations
| Cystine concentration (μmol/L) | Recovery | Intra‐day run precision | Inter‐day run precision | Matrix effect | |
|---|---|---|---|---|---|
| Cell extracts ± SD (%) | Whole cells ± SD (%) | CV (%) | CV (%) | ± SD (%) | |
| 0.25 | 99.9 ± 11.2 | 101.7 ± 15.4 | 13.16 | 13.24 | 100.8 ± 10.5 |
| 8.0 | 101.4 ± 13.0 | 102.9 ± 8.3 | 7.30 | 7.42 | 101.7 ± 4.7 |
| 16.0 | 97.3 ± 7.9 | 98.7 ± 13.6 | 4.71 | 4.69 | 99.4 ± 2.8 |
Recovery and matrix effect in ± SD and precision in CV of three independent experiments performed in triplicate.
3.2.3. Stability and matrix effect
As shown in Table 2, cystine was sufficiently stable in spiked cell extracts and whole cell samples under both short‐term (24 h at 4°C) and long‐term (after one, two and three freeze–thaw cycles) storage conditions. The stability values ranged between 90.3 and 105.0%. No matrix effects could be observed at three different concentration levels (Table 1). The matrix factor ranged between 99.4 to 101.7%. Overall, the absence of matrix effect and high recovery and stability of the analyte contributed to a successful validation of the assay.
Table 2.
Cystine stability obtained from the analysis of spiked cell extracts and whole cell samples under short‐term (24 h at 4°C) and long‐term (one to three freeze–thaw cycles – thawing at 20°C and freezing again at −80°C for a period of minimum 1 month) storage conditions
| Cystine concentration (μmol/L) | Short‐term stability ± SD (%) 24 h at 4 °C | Long‐term stability ± SD (%) | ||
|---|---|---|---|---|
| One cycle | Two cycles | Three cycles | ||
| Cell extracts | ||||
| 0.25 | 110.6 ± 14.8 | 99.2 ± 17.7 | 99.2 ± 24.7 | 93.1 ± 22.7 |
| 8.0 | 107.8 ± 9.5 | 103.3 ± 6 | 103.2 ± 10.8 | 99.0 ± 11.2 |
| 16.0 | 103.8 ± 5 | 102.2 ± 5 | 101.0 ± 11.2 | 97.4 ± 16.2 |
| Whole cells | ||||
| 0.25 | 108 ± 25.9 | 90.3 ± 22.6 | 105.0 ± 22.9 | 97.2 ± 12.4 |
| 8.0 | 105.3 ± 10.6 | 97.4 ± 27.3 | 93.6 ± 21.1 | 97.6 ± 14.5 |
| 16.0 | 110.0 ± 14.5 | 96.2 ± 23.1 | 103.0 ± 13.9 | 96.4 ± 5.7 |
Data is expressed as mean ± SD of three independent experiments performed in triplicate.
3.3. Cystine levels in healthy and cystinotic cells
After a successful validation procedure, the new assay was used to detect cystine levels in both healthy and cystinotic ciPTEC (Figure 3). The results obtained by HPLC‐MS/MS showed a significantly higher level of cystine in cystinotic ciPTEC (3.90 μmol/L; 2.22 ± 0.43 nmol/mg protein) when compared with that of healthy control cells (0.21 μmol/L; 0.057 ± 0.022 nmol/mg protein). This is in accordance with the pathophysiology of the disease, the hallmark being abnormal intracellular accumulation of cystine. Upon treatment with a known cystine‐depleting agent, cysteamine (100 μmol/L), for 24 h, cystinotic cells showed a significant reduction in cystine levels (0.74 ± 0.051 nmol/mg protein). Such a concentration of cystine, however, was still significantly higher (13‐fold) than that found in healthy ciPTEC.
Figure 3.
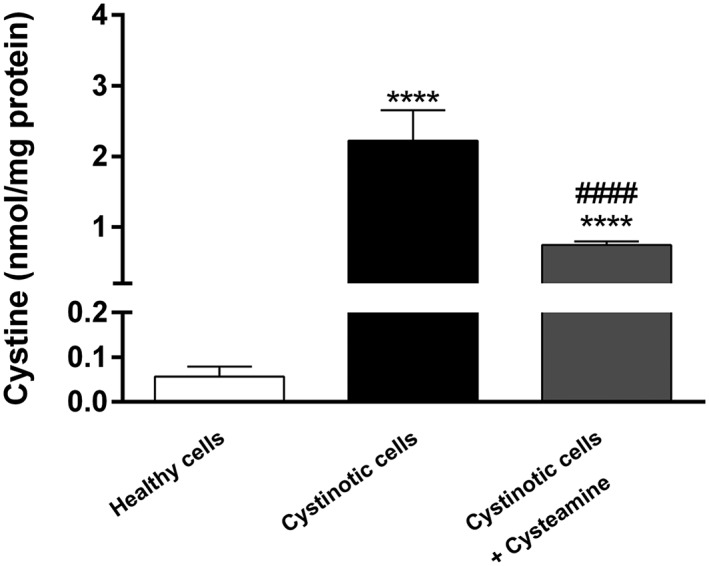
Cystinotic ciPTEC have increased intracellular cystine levels. Cystine levels (nmol/mg protein) measured in healthy cells, cystinotic cells (untreated) and cystinotic cells treated with 100 μmol/L cysteamine. HPLC‐MS/MS analysis showed significantly elevated levels of cystine in cystinotic cells when compared with those of healthy cells (**** p < 0.0001), while cysteamine was able to significantly reduce the levels of cystine in cystinotic ciPTEC (#### p < 0.0001 compared with cystinotic cells). Data is expressed as means ± SD of three independent experiments performed in triplicate
3.4. The effect of N‐ethylmaleimide
To demonstrate the added value of NEM, we measured the levels of cystine in cell samples treated either with or without NEM. The lack of NEM caused a significant increase in the amount of cystine in cystinotic ciPTEC (Figure 4). This shows that, in the absence of NEM, cysteine is readily oxidized into cystine, leading to a cumulative amount of cystine and a false‐positive quantification of its intracellular levels. In contrast, the absence of NEM had no effect on the levels of cystine in healthy ciPTEC. This is probably due to the fact that healthy ciPTEC contain very low levels of cystine and cysteine.
Figure 4.
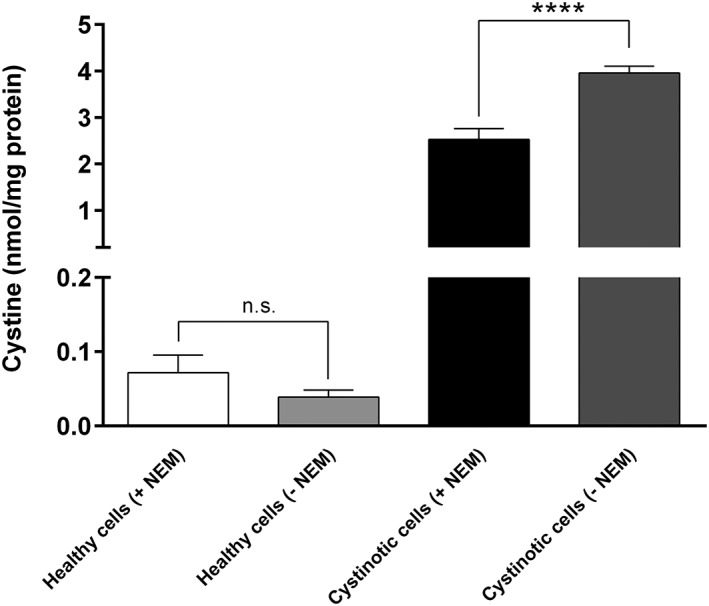
NEM is required to have reliable intracellular cystine measurements. Cystine levels (nmol/mg protein) measured in healthy and cystinotic ciPTEC in both the presence and absence of NEM (5 mmol/L). The absence of NEM did not appear to have any significant effect on the measurement of cystine in healthy cells. In cystinotic cells, however, the absence of NEM caused a significant increase in the concentration of cystine (**** p < 0.0001). Data is expressed as means ± SD of three independent experiments performed in triplicate
4. DISCUSSION
In recent years, various methods have been developed to quantify cystine levels in different biological samples. The HPLC with fluorescence detection assay described by Graaf‐Hess et al. is currently used to quantify cystine levels in cystinotic fibroblasts, leukocytes, zebrafish and ciPTEC (Wilmer et al., 2007, 2011; Elmonem et al., 2017). Nevertheless, the assay is hampered by its high limit of detection (0.3 μmol/L), low cystine recovery and tedious sample preparation procedure (de Graaf‐Hess et al., 1999).
The method we describe in this study allows for fast sample processing with excellent analytical performance. The assay comprises two simple steps of sample preparation: (a) treatment of cells with NEM followed by (b) acid precipitation and finally injection of the supernatant into the chromatographic system. In addition, the LOD (0.05 μmol/L) and LLOQ (0.1 μmol/L) of the method are low enough to allow an accurate cystine measurement both in healthy (0.21 μmol/L) and cystinotic (3.90 μmol/L) cells.
The present chromatographic method was adapted from the UPLC‐MS/MS assay described by Escobar et al. (2016). By comparing our HPLC‐MS/MS method with the UPLC‐MS/MS method, we have further optimized the method in terms of sensitivity (the volume of sample injected is, 1.0 vs 5.0 μL), and limit of detection for cystine (0.1 vs 0.34 μmol/L). Another important difference is that our method provided a better cystine recovery rate with low standard variations (97.3–102.9 vs 75–134%), contributing to a successful validation of the method.
One of the hallmarks of cystinosis is that patients accumulate lysosomal cystine owing to a defective cystinosin transporter. Cysteamine, a cystine depleting agent, was able to significantly reduce cystine accumulation in cystinotic ciPTEC but not to the level of control cells. This is in agreement with clinical observations where cysteamine treatment does not offer a cure but only postpones disease progression. Hence, there is a clear need to screen for new treatment options for cystinosis and therefore our method could be used as a screening tool. Moreover, the assay can also be applied in clinical studies as a diagnostic and biochemical follow‐up tool for cystinosis. Measurement of cystine concentrations in leukocytes of patients is a clinical routine for diagnosis of cystinosis and monitoring cysteamine treatment. The low detection limit of the method would enable us to accurately measure cystine concentrations in the leukocytes of healthy controls (0.04–0.13 nmol cystine/mg protein) and cystinotic patient (2.43 nmol cystine/mg protein; de Graaf‐Hess et al., 1999). Of note, control and cystinotic ciPTEC had an accumulation of cystine similar to that of the leukocytes (Figure 3), indicating ciPTEC as a good representative of cystinosis model.
5. CONCLUSION
A fast and reliable method to measure cystine in both healthy and cystinotic ciPTEC was developed. The method was fully validated and can be applied to screen for potential drugs to reverse cystinotic symptoms in vitro in human kidney cells.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
This work was financially supported by a grant from the Dutch Kidney Foundation (grant nr.150KG19) and the E‐Rare Joint Transnational Call for Proposals 2014, Novel Therapies for Cystinosis.
Jamalpoor A, Sparidans RW, Pou Casellas C, et al. Quantification of cystine in human renal proximal tubule cells using liquid chromatography–tandem mass spectrometry. Biomedical Chromatography. 2018;32:e4238 10.1002/bmc.4238
REFERENCES
- Bayram, B. , Rimbach, G. , Frank, J. , & Esatbeyoglu, T. (2014). Rapid method for glutathione quantitation using high‐performance liquid chromatography with coulometric electrochemical detection. Journal of Agriculture and Food Chemistry, 62(2), 402–408. [DOI] [PubMed] [Google Scholar]
- Besouw, M. , Masereeuw, R. , van den Heuvel, L. , & Levtchenko, E. (2013). Cysteamine: an old drug with new potential. Drug Discovery Today, 18(15–16), 785–792. [DOI] [PubMed] [Google Scholar]
- Donnelly, J. G. , & Pronovost, C. (2000). Evaluation of the Abbott IMx fluorescence polarization immunoassay and the bio‐rad enzyme immunoassay for homocysteine: comparison with high‐performance liquid chromatography. Annals of Clinical Biochemistry, 37(Pt 2), 194–198. [DOI] [PubMed] [Google Scholar]
- Du, F. , Cao, S. , & Fung, Y. S. (2014). A serial dual‐electrode detector based on electrogenerated bromine for capillary electrophoresis. Electrophoresis, 35(24), 3556–3563. [DOI] [PubMed] [Google Scholar]
- Elmonem, M. A. , Khalil, R. , Khodaparast, L. , Khodaparast, L. , Arcolino, F. O. , Morgan, J. , … Levtchenko, E. (2017). Cystinosis (ctns) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Science Reports, 7, 42583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar, J. , Sanchez‐Illana, A. , Kuligowski, J. , Torres‐Cuevas, I. , Solberg, R. , Garberg, H. T. , … Chafer‐Pericas, C. (2016). Development of a reliable method based on ultra‐performance liquid chromatography coupled to tandem mass spectrometry to measure thiol‐associated oxidative stress in whole blood samples. Journal of Pharmaceutical and Biomedical Analysis, 123, 104–112. [DOI] [PubMed] [Google Scholar]
- Gahl, W. A. , Thoene, J. G. , & Schneider, J. A. (2002). Cystinosis. New England Journal of Medicine, 347(2), 111–121. [DOI] [PubMed] [Google Scholar]
- Garcia‐Villoria, J. , Hernandez‐Perez, J. M. , Arias, A. , & Ribes, A. (2013). Improvement of the cystine measurement in granulocytes by liquid chromatograhy–tandem mass spectrometry. Clinical Biochemistry, 46(3), 271–274. [DOI] [PubMed] [Google Scholar]
- de Graaf‐Hess, A. , Trijbels, F. , & Blom, H. (1999). New method for determining cystine in leukocytes and fibroblasts. Clinical Chemistry, 45(12), 2224–2228. [PubMed] [Google Scholar]
- Hodakova, J. , Preisler, J. , Foret, F. , & Kuban, P. (2015). Sensitive determination of glutathione in biological samples by capillary electrophoresis with green (515 nm) laser‐induced fluorescence detection. Journal of Chromatography A, 1391, 102–108. [DOI] [PubMed] [Google Scholar]
- Kalatzis, V. , Nevo, N. , Cherqui, S. , Gasnier, B. , & Antignac, C. (2004). Molecular pathogenesis of cystinosis: Effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Human Molecular Genetics, 13(13), 1361–1371. [DOI] [PubMed] [Google Scholar]
- Steele, M. L. , Ooi, L. , & Munch, G. (2012). Development of a high‐performance liquid chromatography method for the simultaneous quantitation of glutathione and related thiols. Analytical Biochemistry, 429(1), 45–52. [DOI] [PubMed] [Google Scholar]
- Thoene, J. G. , Oshima, R. G. , Crawhall, J. C. , Olson, D. L. , & Schneider, J. A. (1976). Cystinosis. Intracellular cystine depletion by aminothiols in vitro and in vivo. Journal of Clinical Investment, 58(1), 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town, M. , Jean, G. , Cherqui, S. , Attard, M. , Forestier, L. , Whitmore, S. A. , … Antignac, C. (1998). A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nature Genetics, 18(4), 319–324. [DOI] [PubMed] [Google Scholar]
- Wilmer, M. J. , Kluijtmans, L. A. , van der Velden, T. J. , Willems, P. H. , Scheffer, P. G. , Masereeuw, R. , … Levtchenko, E. N. (2011). Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochimica et Biophysica Acta, 1812(6), 643–651. [DOI] [PubMed] [Google Scholar]
- Wilmer, M. J. , Saleem, M. A. , Masereeuw, R. , Ni, L. , van der Velden, T. J. , Russel, F. G. , … Levtchenko, E. N. (2010). Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Research, 339(2), 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmer, M. J. , Willems, P. H. , Verkaart, S. , Visch, H. J. , de Graaf‐Hess, A. , Blom, H. J. , … Levtchenko, E. N. (2007). Cystine dimethylester model of cystinosis: still reliable? Pediatric Research, 62(2), 151–155. [DOI] [PubMed] [Google Scholar]