

Abstract
N‐Glycans direct protein function, stability, folding and targeting, and influence immunogenicity. While most glycosidases that process N‐glycans cleave a single sugar residue at a time, enzymes from glycoside hydrolase family 99 are endo‐acting enzymes that cleave within complex N‐glycans. Eukaryotic Golgi endo‐1,2‐α‐mannosidase cleaves glucose‐substituted mannose within immature glucosylated high‐mannose N‐glycans in the secretory pathway. Certain bacteria within the human gut microbiota produce endo‐1,2‐α‐mannanase, which cleaves related structures within fungal mannan, as part of nutrient acquisition. An unconventional mechanism of catalysis was proposed for enzymes of this family, hinted at by crystal structures of imino/azasugars complexed within the active site. Based on this mechanism, we developed the synthesis of two glycosides bearing a spiro‐epoxide at C‐2 as electrophilic trap, to covalently bind a mechanistically important, conserved GH99 catalytic residue. The spiro‐epoxyglycosides are equipped with a fluorescent tag, and following incubation with recombinant enzyme, allow concentration, time and pH dependent visualization of the bound enzyme using gel electrophoresis.
Keywords: activity-based probes, endomannosidase, GH99, glycosidase, inhibitors
Introduction
N‐Linked glycans are complex oligosaccharides linked to asparagine (Asn) residues in eukaryotic proteins.1 They play important roles in protein function, stability, folding and targeting and are essential for a range of cellular functions.2 Erroneous N‐glycan composition is associated with various diseases including viral infections, Alzheimer's disease and metastatic cancer.3, 4, 5 Assembly of the N‐glycan commences in the endoplasmic reticulum (ER) where the 14‐mer polysaccharide Glc3Man9GlcNAc2‐diphosphodolichol is coupled to the Asn residue of the target protein by the enzyme oligosaccharyl transferase. The glycan undergoes stepwise “trimming” of the non‐reducing end glucoside residues by α‐glucosidase I and II, after which α‐mannosidase I truncates the resulting oligomannoside.6 The resulting Man5GlcNAc2 structure is redecorated to yield complex N‐glycans. Because α‐glucosidases I and II play important roles in the early stages of glycan maturation, these enzymes were investigated as therapeutic targets to control diseases involving incorrect N‐glycosylation.7, 8, 9, 10 However, inhibition of these enzymes did not block N‐glycosylation: mouse lymphoma cells inhibited with the α‐glucosidase inhibitor castanospermine as well as mutant cell lines lacking α‐glucosidase II retained up to 80 % of normal N‐glycan maturation.11, 12, 13 Spiro and co‐workers identified endo‐1,2‐α‐mannosidase,14, 15 (later classified as a member of glucoside hydrolase family 99 (GH99); see http://cazypedia.org),16 residing in the Golgi apparatus, which circumvents inhibition of ER α‐glucosidase I and II. The enzyme cleaves glucose‐substituted mannose from the A‐branch of ER escaped immature N‐glycoproteins bearing Glc1–3Man9GlcNAc2, releasing Glc1‐3Man. The resulting Man8GlcNAc2 glycoprotein subsequently re‐enters the normal processing route in the Golgi apparatus.
Bacterial GH99 orthologs including Bacteroides thetaiotaomicron (Bt) and Bacteroides xylanisolvens (Bx) enzymes possess endo‐1,2‐α‐mannosidase activity, but are more appropriately described as endo‐1,2‐α‐mannanases, as they act on yeast mannan17 and exhibit a tenfold preference for mannan‐based substrates versus the equivalent glucose‐substituted mannans.18 Several imino/azasugar inhibitors for GH99 endomannosidase have been developed, including α‐glucopyranosyl‐1,3‐isofagomine (GlcIFG, 1, Figure 1) and α‐mannopyranosyl‐1,3‐isofagomine (ManIFG, 2). Due to a preference for a mannopyranosyl residue in subsite −2 GH99 endo‐1,2‐α‐mannanases show a greater affinity for 2 than for 1.18 Recently, mannoeuromycin (ManNOE, 3), which features a 2‐hydroxyl allowing interaction with the proposed general base/acid residue, has been reported as the most potent endo‐1,2‐α‐mannanase inhibitor for bacterial GH99 enzymes with K D values in the low nanomolar range.19 Additionally, fluorescent20 and fluorogenic18, 21 substrates have been developed for monitoring endo‐1,2‐α‐mannosidase/mannanase activity.
Figure 1.
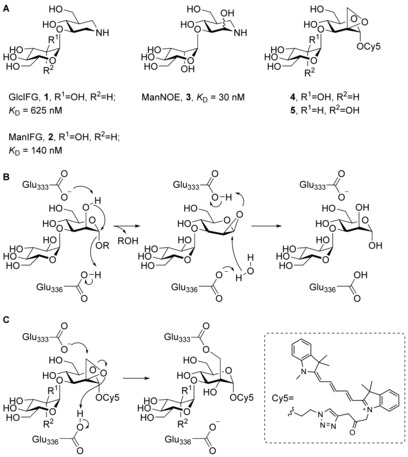
(A) Known GH99 endo‐1,2‐α‐mannosidase inhibitors (1–3) and fluorescent spiro‐epoxyglycosides 4 and 5 subject of this study. K D values are for B. thetaiotaomicron endo‐1,2‐α‐mannosidase (BtGH99). (B) The proposed catalytic mechanism for GH99 enzyme (amino acid numbering for B. xylanisolvens endo‐1,2‐α‐mannosidase (BxGH99)). (C) Anticipated covalent inhibition mechanism of GH99 enzymes.
Family GH99 endo‐1,2‐α‐mannosidases/mannanases cleave their substrate glycosides with retention of anomeric stereochemistry; however, instead of the classical Koshland double‐displacement mechanism for retaining enzymes,22 an unusual neighboring group participation hydrolytic mechanism was proposed in which a glutamate residue (Glu333 in BxGH99) acts as a general base assisting OH‐2 to displace the aglycon via a 1,2‐anhydro sugar that is subsequently hydrolyzed by water (Figure 1 B).23 In order to study enzyme function in biological settings, screen for inhibitors, as well as to further illuminate the catalytic reaction mechanism, the development of a mechanism‐based irreversible inhibitor would be of interest. Here, the synthesis is described of two putative covalent inhibitors 4 and 5, designed to, respectively inhibit eukaryotic GH99 endo‐1,2‐α‐mannosidases and bacterial endo‐1,2‐α‐mannanases and which vary in the nature of the pyranoside at the non‐reducing end (Figure 1 A, right). Both compounds contain a spiro‐epoxide at position C‐2 to serve as an electrophile to trap the general base residue. Inspection of the crystal structures of BxGH99 suggests that the general base will be situated close to the methylene group of the spiro‐epoxide, where it may open the ring via nucleophilic attack resulting in a covalent intermediate (Figure 1 C).23 The compounds are also equipped with a reporter tag, allowing active enzyme labeling by activity‐based protein profiling (ABPP)24 protocols, the efficiency of which is reported as well.
Results
Acceptor 7 was synthesized by 4,6‐silylidene protection of compound 6,25 followed by formation of the 2,3‐orthobenzoate and final treatment with acid (Scheme 1).26 Glucopyranoside donor 9 was synthesized from thiophenyl β‐glucopyranoside 8.27 While 4,6‐silylidene protection proceeded smoothly, elevated temperatures were required to install the TBS‐groups onto both secondary hydroxyl groups, presumably due to steric hindrance. Using a similar approach, thiophenyl α‐mannopyranoside 10 28 was converted to protected thiomannoside donor 11.
Scheme 1.
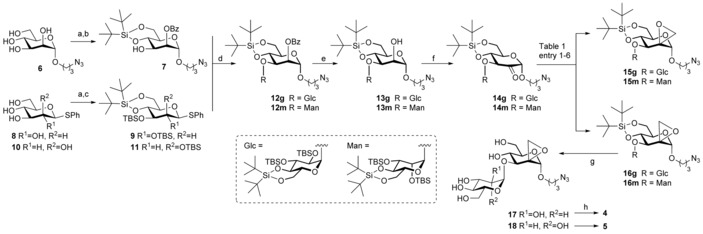
Synthesis of fluorescent spiro‐epoxyglycosides 4 and 5. Reagents and conditions: a) tBu2Si(OTf)2, 2,6‐lutidine, DMF, −50 °C; b) PhC(OMe)3, CSA, 2 h, then AcOH, H2O, 16 h, 57 % over 2 steps; c) TBSOTf, DMAP, pyridine, 60 °C, 16 h, yield 9: 85 % over 2 steps; yield 11: 81 % over 2 steps; d) donor 9 or 11, NIS, TMSOTf, DCM, 4 Å MS, −40 °C, 1 h, yield 12 g: 92 %; yield 12 m: 88 %; e) NaOMe, MeOH, DCM, yield 13 g: 95 %; yield 13 m: 86 %; f) DMP, DCM, yield 14 g: 98 %; yield 14 m: 96 %; g) TBAF, THF, 5 days, yield 17: 97 %; yield 18: 74 %; h) Cy5‐alkyne,47 CuSO4⋅5 H2O, sodium ascorbate, DMF, rt, 16 h, yield 4: 32 %; yield 5: 34 %.
Glycosylation of acceptor 7 by 9 or 11 was achieved in an N‐iodosuccinimide (NIS)/trimethylsilyl triflate (TMSOTf) mediated coupling at low temperature, affording 12 g or 12 m, respectively. Both glycosylations proceeded in excellent yield and stereoselectivity. Pedersen, Bols and co‐workers29 recently reported that silylidene‐protected mannosyl donors can be used for stereoselective β‐mannosylation. The contrasting selectivity obtained here is likely the result of the steric buttressing effect of the large silyl ether protecting groups at the C‐2‐ and C‐3‐hydroxyls of 7, consistent with the steric effects that large protecting groups and functionalities have in glycosylations of otherwise β‐selective benzylidene‐protected mannosyl donors.30 Thus, the β‐face of mannosyl donor 11 is shielded from attack by the incoming nucleophile. The glycosylation stereoselectivity of glucosyl donor 9 can be rationalized by its high reactivity. The “arming” silyl protecting groups allow this donor to readily form an oxocarbenium ion, which will likely take up a 4 H 3‐like conformation, which is preferentially attacked from the α‐face to provide the 1,2‐cis‐linked product.31, 32 Next, the benzoyl groups were deprotected under Zemplén conditions affording compounds 13 g and 13 m. The alcohols were then oxidized with Dess–Martin periodinane (DMP) to ketones 14 g and 14 m, which appeared to be in equilibrium with the corresponding hydrates.
Transformation of ketones 14 into their corresponding spiro‐epoxides was explored next (Table 1). Reaction of 14 g with diazomethane as methylenating agent33 resulted in the formation of the equatorial (15 g) and axial (16 g) methylenes in a 1:1 ratio and in good yields (entry 1). Their absolute configuration was determined by 1D‐NOE difference experiments (see Supplementary Information). Reaction of 14 m with diazomethane also resulted in a mixture of 15 m and 16 m, in a 3:1 ratio, in favor of the equatorial methylene group in almost quantitative yield (entry 2). We anticipated that a Corey–Chaykovsky epoxidation34 using stabilized dimethylsulfoxonium methylide would favor the formation of the equatorial methylenes 15 g and 15 m. Indeed, also in these cases both isomers were obtained, however the formation of axial methylenes was still favored in both cases (entries 3 and 4). Finally, using the more reactive dimethylsulfonium methylide, only the kinetically favored axial methylenes 16 g and 16 m were formed, albeit in moderate yields (entries 5 and 6). With spiro‐epoxides 16 g and 16 m in hand, global deprotection was accomplished by reaction with tetrabutylammonium fluoride (TBAF). Finally, a fluorescent Cy5 tag was installed at the azide handle using copper(I) catalyzed click chemistry, which after HPLC purification afforded spiro‐epoxyglycosides 4 and 5.
Table 1.
Transformation of ketones 14 g and 14 m into their corresponding spiro‐epoxides.
| Entry | s.m. | Conditions | 15:16 | Yield [%][a] |
|---|---|---|---|---|
| 1 | 14 g | CH2N2, EtOH, 0 °C | 1:1 | 78 |
| 2 | 14 m | 3:1 | 97 | |
| 3 | 14 g | SOMe3I, nBuLi, THF, 60 °C | 1:5 | 83 |
| 4 | 14 m | 1:2 | 85 | |
| 5 | 14 g | SMe3I, NaH, DMSO, THF, −10 °C | 0:1 | 50 |
| 6 | 14 m | 0:1 | 53 |
[a] Combined yield after column chromatography. s.m.=starting material.
The ability of 4 and 5 to label recombinant Bt‐ and BxGH99 endo‐1,2‐α‐mannanase was evaluated (Figure 2 A). The compounds label both enzymes in a concentration‐dependent manner, at concentrations as low as 100 nm. Previous studies indicated a preference for a mannosyl residue at the −2 subsite of both enzymes.18 However, no difference in potency of labeling was observed. Studies on the effect of the pH dependence on labeling revealed that both spiro‐epoxyglycosides label the enzymes maximally at pH 6–8, corresponding to the pH optimum of GH99 enzymatic activity (Figure 2 B).23 Notably, more than one band is evident, suggesting enzyme degradation under reducing SDS‐PAGE conditions or alternatively that multiple labelling events may be occurring. Next, labeling of wild‐type (WT) BxGH99 was compared to analogous active‐site mutants (Figure 2 C). While WT enzyme is labeled by spiro‐epoxyglycosides 4 and 5 within 5 minutes, the general base mutant E333Q and the catalytic acid mutant E336Q were not labeled in the same time period with these compounds, suggesting that labeling is indeed activity‐based, and is consistent with reaction occurring in a mechanism‐based manner. However, incubation for longer times resulted in labeling of the mutant enzymes, indicating that either the spiro‐epoxide is susceptible to ring opening by the mutant catalytic residues, or that other residues may also be involved in covalent labelling. Denaturation of BtGH99 and BxGH99 completely abrogated labeling by spiro‐epoxyglycosides 4 and 5, indicating that labeling requires the natively folded enzyme (Figure 2 D).
Figure 2.
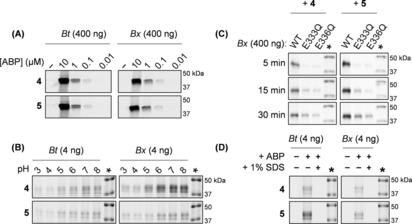
Fluorescent labeling of GH99 endomannanases. (A) Detection limit of Bt and Bx GH99 endomannanases (left and right, respectively), labeled with various concentrations of fluorescent spiro‐epoxyglycosides 4 or 5. (B) Effect of pH on labeling of Bt and Bx GH99 enzymes with 4 or 5. (C) Labeling of wild‐type and mutant BxGH99 with 4 or 5 (left or right, respectively) for 5, 15 or 30 minutes. (D) Effect of denaturation with 1 % (w/v) SDS and boiling on labeling of Bt and Bx GH99 enzymes (left and right, respectively) with 4 or 5. The marker is annotated with an asterisk (*).
To further evaluate whether covalent inhibition of BtGH99 and BxGH99 is activity‐based, the processing of human α‐galactosidase A (GLA) by these enzymes was investigated (Figure 3 A). GLA contains three N‐glycosylation sites, of which two are decorated with oligo‐mannose structures, and one contains complex oligosaccharides low in mannose content.35, 36 We have previously demonstrated that fluorescent α‐galacto‐cyclophellitol aziridines such as TB340 covalently label GLA in activity‐based manner.37 Here, GLA was pre‐labeled with TB340 to enable fluorescent detection on gel. Without additives, GLA gives a distinct major band at ≈50 kDa (Figure 3 B, lane 1). Incubation of GLA with BtGH99 results in demannosylation of the two high‐mannose N‐glycans of GLA, resulting in a shift of the GLA band into lower bands at ≈42 kDa (lane 2). This shift in molecular weight is similar to the shift observed when GLA is incubated with Endo‐H (lane 4), which causes demannosylation of high‐mannose N‐glycans by cleaving within the chitobiosyl core leaving a residual GlcNAc on Asn. Treatment of GLA with PNGase‐F (lane 5), which cleaves most N‐glycans leaving Asn, results in a band of a lower molecular weight, most likely as a result of complete deglycosylation of all three N‐glycans. When BtGH99 was pre‐incubated with 4 or 5, demannosylation of TB340‐labeled GLA was mostly inhibited (lane 3), indicating that binding of 4 and 5 occurs in the BtGH99 active site. An identical experiment was conducted with BxGH99 wildtype and the E333Q and E336Q mutant enzymes (Figure 3 C). Similar to BtGH99, wildtype BxGH99 is able to process the N‐glycans decorating the surface of the enzyme, giving rise to a shift in molecular weight (lane 2), similar to processing by Endo‐H (lane 8). Pre‐incubation of WT BxGH99 by 4 or 5, prior to consecutive incubation with TB340‐labeled GLA resulted in an observed absence of glycan processing (lane 3), indicating that binding of 4 and 5 abrogates enzymatic activity. Interestingly, while mutants E333Q and E336Q are labeled by spiro‐epoxyglycosides 4 and 5 after prolonged reaction times, they are evidently unable to process TB340‐labeled GLA (lanes 4–7).
Figure 3.
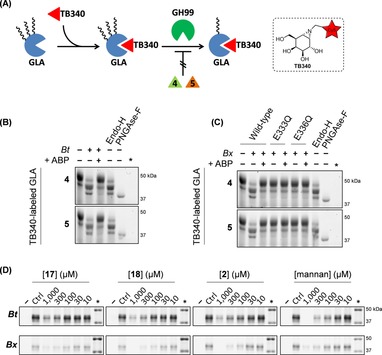
(A) Schematic representation of processing of human α‐galactosidase GLA by GH99 endomannosidase. GLA is pre‐labeled by fluorescent TB340, and contains high‐mannose N‐glycans which can be truncated by endomannosidase, resulting in a decrease in GLA molecular weight. Activity‐based labeling of endomannosidase by spiro‐epoxyglycosides 4 or 5 (prior to incubation with GLA) blocks its activity, and is therefore unable to process GLA. (B) BtGH99 wild‐type demannosylates GLA, causing a shift in molecular weight for the protein bands. Pre‐labeling BtGH99 wild‐type with 4 or 5 abrogates GLA demannosylation. Endo‐H cleaves high‐mannose structures, PNGase‐F cleaves full N‐linked glycan (leaving Asp‐GlcNAc). (C) BxGH99 wild‐type demannosylates GLA, while BxGH99 pre‐labeled with 4 or 5 is unable to do so. BxGH99 active‐site mutants E333Q and E3336Q are unable to process GLA. (D) Fluorescent labeling of BtGH99 (top) and BxGH99 (bottom) by 4 or 5 competed by different concentrations of 17, 18, ManIFG (2) and yeast mannan. The marker is annotated with an asterisk (*).
Finally, the inhibitory potencies of 2, 17, 18 and yeast mannan from S. cerevisiae (an α‐1,6‐linked mannose backbone branched with α‐1,2 and α‐1,3 mannoses)38 towards BtGH99 were investigated using spiro‐epoxyglycoside 5 as fluorescent read‐out (Figure 3 D). The enzyme was first pre‐incubated with the competitor for 30 min at 37 °C, followed by labeling with 1 μm 5. Compounds 17 and 18 both show a concentration‐dependent competition of fluorescent labeling in the range of 10‐1,000 μm, although full competition with labeling could not be achieved under these conditions. Similarly, the azasugar ManIFG (2) gave concentration‐dependent competition but again full competition was not achieved. However, pre‐incubation by yeast mannan achieved full competition with labeling, suggesting that processing of spiro‐epoxyglycoside 5 by BtGH99 endomannanase is specific and activity based. A similar competition experiment was performed for BxGH99, and it was shown that while pre‐incubation with 17 did not fully abrogate labeling, pre‐incubation with 1000 μm of 18 provided full competition, possibly hinting at a slight preference for a mannosyl residue in subsite −2. Additionally, yeast mannan showed concentration dependent (albeit incomplete) competition, and azasugar 2 fully competed with labeling at 1,000 μm, suggesting that processing of spiro‐epoxyglycoside 5 by BxGH99 is specific and activity‐based.
Discussion
Epoxide‐based probes have been investigated as mechanism‐based inhibitors of a range of glycosidases. Early work led to the development of epoxyalkyl glycosides,39 which were initially proposed as reagents that could specifically label the nucleophile of retaining glycosidases, however X‐ray crystallography later revealed labelling of both acid/base and nucleophile residues.40 In one classic study, conduritol C epoxide, which was originally believed to label the nucleophile of E. coli LacZ β‐galactosidase, was subsequently shown to covalently label the acid/base catalyst.41 Work from our laboratory has investigated related pyranose‐mimicking cyclophellitol epoxides and aziridines and shown that these typically exhibit excellent selectivity for labelling the nucleophile of assorted α‐ and β‐glycosidases.42 We have shown that introduction of a reporter tag (e.g. biotin or a fluorescent dye) onto these small molecule inhibitors affords chemical probes that enable quantification of activity,43, 44 and have distinct advantages over techniques such as transcription analysis and antibody‐based detection. We report here the first activity‐based probes for detection of GH99 enzymes, which were designed based on the proposed mechanism of this enzyme. In this proposed mechanism a 1,2‐anhydro‐epoxide intermediate is formed by general base assisted deprotonation of O2 by a carboxylate residue.23 Our design strategy includes a reactive C2 spiro‐epoxide that can potentially covalently label the general base (acting as a nucleophile), and includes a fluorescent label for visualization. Gel‐based analysis of labelled bacterial GH99 endo‐1,2‐α‐mannanases demonstrated concentration dependent labelling which occurs in a pH dependent manner consistent with the pH optimum of enzyme activity. Labelling could be competed by various substrates and inhibitors, providing evidence that it is active‐site directed. While mutation of the key general base and general acid residues inactivated the enzyme towards processing of natural substrate N‐glycans in GLA, the mutants could be labelled with the spiro‐epoxyglycosides, albeit with reduced potency. Collectively, our data suggests that these spiro‐epoxides do result in labelling at the active site, presumeably through the catalytic general base. However, the high reactivity of the primary epoxide means that labelling is most likely not exclusive at a single residue. While endo‐1,2‐α‐mannanase has a preference for mannosyl residues at the −2 binding subsite, there was minimal differences in the efficiency of labelling for spiro‐epoxides bearing either a mannosyl or glucosyl residue. We believe these compounds represent an important first step in devising probes that take advantage of the unique mechanism proposed for this family. Future studies will seek to better understand the mode of labelling by identifying the covalently labelled residue(s) by X‐ray crystallography or MS based techniques. By analogy to previously described irreversible cyclophellitol activity‐based probes,43 we propose these fluorescent spiro‐epoxyglycosides could ultimately lead to chemical tools for functional investigation of GH99 endo‐1,2‐α‐mannosidase/mannanases, both as isolated species and in tissue extracts.
Experimental Section
Chemicals were purchased from Acros, Sigma Aldrich, Biosolve, VWR, Fluka, Merck and Fisher Scientific and used as received unless stated otherwise. Tetrahydrofuran (THF), N,N‐dimethylformamide (DMF) and toluene were stored over molecular sieves before use. All reactions were performed under an argon atmosphere unless stated otherwise. TLC analysis was conducted using Merck aluminum sheets (Silica gel 60 F254) with detection by UV absorption (254 nm), by spraying with a solution of (NH4)6Mo7O24⋅4 H2O (25 g L−1) and (NH4)4Ce(SO4)4⋅2 H2O (10 g L−1) in 10 % sulfuric acid, followed by charring at ≈150 °C. Column chromatography was performed using Screening Device b.v. silica gel (particle size of 40–63 μm, pore diameter of 60 Å) with the indicated eluents. For reversed‐phase HPLC purifications an Agilent Technologies 1200 series instrument equipped with a semi‐preparative column (Gemini C18, 250×10 mm, 5 μm particle size, Phenomenex) was used. 1H NMR and 13C NMR spectra were recorded on a Brüker AV‐400 (400 and 101 MHz, respectively) or a Brüker DMX‐600 (600 and 151 MHz, respectively) spectrometer in the given solvent. Chemical shifts are given in ppm (δ) relative to the residual solvent peak or tetramethylsilane (0 ppm) as internal standard. High‐resolution mass spectrometry (HRMS) analysis was performed with a LTQ Orbitrap mass spectrometer (Thermo Finnigan), equipped with an electronspray ion source in positive mode (source voltage 3.5 kV, sheath gas flow 10 mL min−1, capillary temperature 250 °C) with resolution R=60 000 at m/z 400 (mass range m/z 150–2000) and dioctyl phthalate (m/z 391.28428) as lock mass. The high‐resolution mass spectrometer was calibrated prior to measurements with a calibration mixture (Thermo Finnigan). ManIFG was prepared as previously reported.18 Recombinant expression of B. thetaiotaomicron (Bt) and B. xylanisolvens (Bx) GH99 was achieved as previously described.23 Recombinant α‐galactosidase (GLA) was purchased from Genzyme (Cambridge, MA, USA). The α‐galactosidase ABP TB340 was synthesized as described earlier.37 Yeast mannan from S. cerevisiae was purchased from Sigma.
Synthesis and characterization
(4 aR ,6S,7S,8R,8 aS )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐hydroxyhexahydropyrano[3,2‐d][1,3,2]dioxasilin‐7‐yl benzoate (7): Compound 6 45 (1.00 g, 3.80 mmol) was co‐evaporated with dry toluene and dissolved in dry DMF (38 mL). The resulting solution was cooled to −50 °C and SitBu2(OTf)2 (1.11 mL, 3.42 mmol, 0.9 EQ) and 2,6‐lutidine (0.44 mL, 3.80 mmol) were added. The reaction was stirred at −50 °C for 30 minutes and subsequently quenched with brine (400 mL). The aqueous layer was extracted with Et2O (4×100 mL). The combined organic layers were washed with 1 m aqueous HCl (2×100 mL), H2O (100 mL), and brine and dried over Na2SO4. The solvents were removed under reduced pressure and the crude product was purified by gradient column chromatography (EtOAc/pentane, 1:4 to 1:2). The 4,6‐silydene product was obtained as white solid (970 mg, 70 %). 1H NMR (400 MHz, CDCl3): δ=4.81 (d, J=1.4 Hz, 1 H), 4.11 (dd, J=10.0, 5.0 Hz, 1 H), 4.07–4.00 (m, 2 H), 3.96 (t, J=10.2 Hz, 1 H), 3.86–3.76 (m, 2 H), 3.69 (td, J=10.0, 5.0 Hz, 1 H), 3.50 (ddd, J=10.0, 6.3, 5.2 Hz, 1 H), 3.40 (td, J=6.6, 2.0 Hz, 2 H), 1.94–1.82 (m, 2 H), 1.06 (s, 9 H), 1.00 ppm (s, 9 H). 13C NMR (101 MHz, CDCl3): δ=166.0, 133.4, 129.9, 129.7, 128.5, 98.1, 75.2, 72.0, 70.2, 67.4, 66.6, 64.6, 48.2, 28.8, 27.4, 27.0, 22.8, 20.0 ppm. IR (neat): =3524, 2934, 2886, 2097, 1732, 1717, 1558, 1472, 1267, 1095, 1072, 1026, 885, 826, 710, 654 cm−1. [α] (c 0.1, DCM): −16. HRMS (ESI) m/z: [M+Na]+ calcd for C24H37N3O7SiNa 530.22930, found 530.22907. The 4,‐6‐silydene compound (889 mg, 2.20 mmol) was dissolved in trimethyl orthobenzoate (5.7 mL) and CSA (102 mg, 0.44 mmol) was added. The reaction was stirred for 2 hours at room temperature and cooled to 0 °C. Aqueous AcOH (50 %, 20 mL) was added and the mixture was stirred overnight while the cooling bath was allowed to reach room temperature. The solution was poured into saturated aqueous NaHCO3 (50 mL) and the water layer was extracted with CH2Cl2 (3×50 mL). The combined organic layers were washed with NaHCO3 (50 mL) and dried over MgSO4. The solvents were removed under reduced pressure and the crude product was purified by gradient column chromatography (EtOAc/pentane, 1:99 to 1:10). The title product was obtained as colorless oil (922 mg, 82 %). 1H NMR (400 MHz, CDCl3): δ=8.15–7.98 (m, 2 H), 7.63–7.54 (m, 1 H), 7.51–7.41 (m, 2 H), 5.42 (dd, J=3.4, 1.6 Hz, 1 H), 4.88 (d, J=1.4 Hz, 1 H), 4.23–4.06 (m, 3 H), 3.99 (t, J=10.2 Hz, 1 H), 3.86–3.76 (m, 2 H), 3.59–3.49 (m, 1 H), 3.43 (t, J=6.6 Hz, 2 H), 1.99–1.81 (m, 2 H), 1.09 (s, 9 H), 1.02 ppm (s, 9 H). 13C NMR (101 MHz, CDCl3): δ=166.0, 133.4, 129.9, 129.7, 128.5, 98.1, 75.2, 72.0, 70.2, 67.4, 66.6, 64.6, 48.2, 28.8, 27.4, 27.0, 22.8, 20.0 ppm. IR (neat): =3524, 2934, 2886, 2097, 1732, 1717, 1558, 1472, 1267, 1095, 1072, 1026, 885, 826, 710, 654 cm−1. [α] (c 0.1, DCM): −16. HRMS (ESI) m/z: [M+Na]+ calcd for C24H37N3O7SiNa 530.22930, found 530.22907.
(4 aR ,6S,7R,8S,8 aR )‐2,2‐Di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)‐6‐(phenylthio)hexahydropyrano[3,2‐d][1,3,2]dioxasiline (9): Compound 8 27 (2.6 g, 9.5 mmol) was dissolved in dry DMF (100 mL) under Ar‐atmosphere. The mixture was cooled to −50 °C and 2,6‐lutidine (3.3 mL, 28.5 mmol) and SitBu2(OTf)2 (3.4 mL, 10.5 mmol) was added. The reaction was stirred for 2 hours at −50 °C and subsequently quenched with H2O (100 mL). The water layer was extracted with EtOAc (3×100 mL). The organic layers were combined and washed with H2O (2×200 mL) and brine (200 mL) and dried over MgSO4. The solvents were removed under reduced pressure and the crude product was purified by gradient column chromatography (EtOAc/pentane, 1:4 to 1:2). The 4,6‐silydene product was obtained as a white solid (3.58 g, 91 %). 1H NMR (400 MHz, CDCl3): δ=7.57–7.46 (m, 2 H), 7.38–7.28 (m, 3 H), 4.60 (d, J=9.7 Hz, 1 H), 4.21 (dd, J=10.2, 5.1 Hz, 1 H), 3.90 (t, J=10.2 Hz, 1 H), 3.68 (t, J=9.0 Hz, 1 H), 3.60 (t, J=8.7 Hz, 1 H), 3.51–3.37 (m, 2 H), 2.92 (s, 1 H), 2.77 (s, 1 H), 1.04 (s, 9 H), 0.98 ppm (s, 9 H). 13C NMR (101 MHz, CDCl3): δ=132.9, 131.7, 129.1, 128.3, 88.6, 77.8, 76.4, 74.5, 71.8, 66.1, 27.4, 27.0, 22.7, 19.9. IR (neat): =3241, 2932, 2858, 1695, 1471, 1058 cm−1. [α] (c 0.06, DCM): −57.0. HRMS (ESI) m/z: [M+Na]+ calcd for C20H32O5SSiNa 435.16319, found 435.16315. The 4,6‐silydene product (1.0 g, 2.42 mmol) was co‐evaporated with toluene (3×), dissolved in dry pyridine (5 mL) and cooled to 0 °C. DMAP (30 mg, 0.24 mmol) and TBSOTf (3.33 mL, 14.5 mmol) were added and the mixture was heated to 60 °C and stirred overnight. The mixture was carefully diluted with water (25 mL) and extracted with DCM (3× 50 mL). The combined organic layers were washed with aq. 1 m HCl (3×25 mL) and brine, dried over Na2SO4, filtrated and concentrated. The crude product was purified by gradient column chromatography (pentane/EtOAc, 400:1 to 200:1), affording the title product as a white solid (1.44 g, 93 %). Analytical data were in accordance with those reported in literature.46
(4 aR ,6R,7S,8S,8 aR )‐2,2‐Di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)‐6‐(phenylthio)hexahydropyrano[3,2‐d][1,3,2]dioxasiline (11): The 4,6‐silydene compound was prepared from 10 28 (4.9 g, 18 mmol) as described for the preparation of 9 to afford the product (6.6 g, 89 %) as a white solid. 1H NMR (400 MHz, CDCl3): δ=7.52 −7.20 (m, 5 H), 5.53 (s, 1 H), 4.30 (d, J=3.1 Hz, 1 H), 4.24 (td, J=10.0, 5.0 Hz, 1 H), 4.15–4.08 (t, J=9.4 Hz, 1 H), 4.05 (dd, J=10.0, 5.0 Hz, 1 H), 3.96 (t, J=10.1 Hz, 1 H), 3.87 (dd, J=9.1, 3.3 Hz, 1 H), 2.67 (brs, 2xOH), 1.05 (s, 9 H), 1.03 ppm (s, 9 H). 13C NMR (101 MHz, CDCl3): δ=133.9, 131.5, 129.3, 127.7, 87.8, 75.0, 72.4, 72.1, 67.9, 66.2, 27.6, 27.2, 22.8, 20.2 ppm. IR (neat): =3384, 2932, 2858, 1474, 1064 cm−1. [α] (c 0.4, DCM): +227. HRMS (ESI) m/z: [M+Na]+ calcd for C20H32O5SSiNa 435.16319, found 435.16301. The title product was prepared from the 4,6‐silydene compound (6.6 g, 16 mmol) as described for the preparation of 9 to afford the product (9.3 g, 91 %) as a pale yellow oil which crystallized at −20 °C. 1H NMR (400 MHz, CDCl3): δ=7.48–7.25 (m, 5 H), 5.29 (d, J=1.5 Hz, 1 H), 4.28 (t, J=9.0 Hz, 1 H), 4.19 (m, 1 H), 4.17–4.11 (m, 1 H), 4.11–4.08 (m, 1 H), 3.96 (t, J=9.7 Hz, 1 H), 3.87 (dd, J=8.9, 2.5 Hz, 1 H), 1.09 (s, 9 H), 1.07 (s, 9 H), 0.99 (s, 9 H), 0.92 (s, 9 H), 0.21 (s, 3 H), 0.18 (s, 3 H), 0.14 (s, 3 H), 0.07 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=134.9, 131.3, 129.3, 127.4, 89.9, 75.0, 74.6, 73.0, 69.6, 67.1, 27.8, 27.3, 26.3, 25.8, 22.9, 20.2, 18.5, 18.2, −3.9, −4.1, −4.4, −4.4 ppm. IR (neat): =2931, 2857, 1471, 1250, 1096 cm−1. [α] (c 1.0, DCM): +91. HRMS (ESI) m/z: [M+H]+ calcd for C32H61O5SSi3 641.35420, found 641.36460.
(4 aR ,6S,7S,8R,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7R,8S,8 aR)‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐7‐yl benzoate (12 g): Compound 9 (2.00 g, 3.12 mmol) and compound 7 (1.58 g, 3.12 mmol) were combined and co‐evaporated with toluene (3×). The mixture was dissolved in dry CH2Cl2 (20 mL) and stirred with activated 4A MS for 30 minutes at room temperature. The reaction was cooled to −50 °C and NIS (842 mg, 3.74 mmol) and TMSOTf (68 μL, 0.37 mmol) were added. The reaction mixture was warmed to −40 °C, stirred for 1 hour and subsequently neutralized with NEt3 (2 mL). The mixture was diluted with CH2Cl2 (200 mL) and washed with saturated aqueous Na2SO3 (2×100 mL), H2O (100 mL) and subsequently dried over MgSO4. The solvents were removed under reduced pressure and the crude product was purified by gradient column chromatography (EtOAc/pentane, 1:50 to 1:40). The title product was obtained as a white foam (2.98 g, 92 %). 1H NMR (400 MHz, CDCl3): δ=8.08 (d, J=7.3 Hz, 2 H), 7.59 (t, J=7.4 Hz, 1 H), 7.47 (t, J=7.7 Hz, 2 H), 5.49 (s, 1 H), 5.16 (d, J=2.9 Hz, 1 H, H‐1 “donor”), 4.86 (s, 1 H, H‐1 “acceptor”), 4.44 (t, J=9.4 Hz, 1 H), 4.22–4.15 (m, 2 H, H‐3), 4.04 (t, J=10.2 Hz, 1 H), 3.95–3.79 (m, 3 H), 3.75–3.66 (m, 2 H), 3.61 (t, J=8.4 Hz, 1 H), 3.54 (dt, J=10.2, 5.7 Hz, 1 H), 3.49–3.41 (m, 2 H), 1.91 (dq, J=13.5, 6.9 Hz, 1 H), 1.13 (s, 9 H), 1.04 (s, 9 H), 1.03 (s, 9 H), 0.97 (s, 9 H), 0.91 (s, 9 H), 0.79 (s, 9 H), 0.17 (s, 3 H), 0.05 (s, 3 H), 0.03 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=165.4, 133.2, 129.9, 129.6, 128.5, 98.1, 97.9, 78.6, 75.2, 74.3, 73.4, 72.6, 71.1, 67.8, 67.8, 66.9, 66.6, 64.4, 48.1, 28.9, 27.5, 27.1, 27.0, 26.4, 26.2, 22.7, 22.7, 20.0, 20.0, 18.1, 18.0, −3.2, −3.5, −3.6, −4.4 ppm. 13C‐HMBC‐GATED NMR (101 MHz, CDCl3): δ=98.1 (J C1,H1=170.6 Hz, C1 “donor”), 97.9 (J C1,H1=172.1 Hz, C1 “acceptor”). IR (neat): =2966, 2859, 2093, 1732, 1472, 1260, 1096, 1069, 1045, 827 cm−1. [α] (c 0.1, DCM): +20. HRMS (ESI) m/z: [M+Na]+ calcd for C50H91N3O12Si4+Na 1060.55720, found 1060.55694.
(4 aR ,6S,7S,8R,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7S,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐7‐yl benzoate (12 m): This compound was prepared from 11 (378 mg, 0.59 mmol) and 7 (299 mg, 0.59 mmol) as described for the preparation of 12 g, to afford the title product (538 mg, 88 %) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ=8.02 (m, 2 H), 7.62–7.53 (m, 1 H), 7.45 (t, J=7.7 Hz, 2 H), 5.34 (dd, J=3.5, 1.6 Hz, 1 H), 4.96 (d, J=1.9 Hz, 1 H), 4.82 (d, J=1.4 Hz, 1 H), 4.28 (t, J=9.5 Hz, 1 H), 4.15 (m, 3 H), 4.08 (dd, J=9.4, 3.6 Hz, 1 H), 4.02–3.93 (t, J=10.3 Hz, 1 H), 3.90–3.77 (m, 4 H), 3.77–3.67 (m, 2 H), 3.57–3.37 (m, 3 H), 2.02–1.77 (m, 2 H), 1.09 (s, 9 H), 1.04–1.00 (s, 9 H), 1.00 (s, 9 H), 0.95–0.89 (m, 9 H), 0.87–0.82 (m, 9 H), 0.82–0.76 (m, 9 H), 0.07 (s, 3 H), 0.00 (s, 3 H), −0.13 (s, 3 H), −0.17 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=165.5, 133.4, 130.0, 129.7, 128.6, 103.4, 98.2, 75.2, 75.1, 74.0, 73.6, 72.4, 71.8, 69.5, 67.7, 67.5, 67.1, 64.8, 48.4, 29.0, 27.9, 27.7, 27.2, 27.2, 26.2, 25.8, 22.9, 22.9, 20.1, 19.9, 18.4, 18.2, −4.3, −4.4, −4.4, −4.7 ppm. 13C‐HMBC‐GATED NMR (101 MHz, CDCl3): δ=103.4 (J C1,H1=172.1 Hz, C1 “donor”), 98.2 ppm (J C1,H1=172.5 Hz, C1 “acceptor”). IR (neat): =2931, 2858, 20 998, 1729, 1472, 1226, 1096, 1068 cm−1. [α] (c 0.4, DCM): +1. HRMS (ESI) m/z: [M+H]+ calcd for C50H92N3O12Si4 1038.57526, found 1038.57587.
(4 aR ,6S,7S,8R,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7R,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐7‐ol (13 g): Compound 12 g (610 mg, 0.59 mmol) was co‐evaporated with toluene (3×) and dissolved in a mixture of DCM/MeOH (9 mL, 1:1). NaOMe (30 wt %, 560 μL) was added and the reaction mixture was stirred for 24 h. The reaction was neutralized with AcOH and the solvents were removed under reduced pressure. The crude product was purified by gradient column chromatography (EtOAc/pentane, 1:11 to 1:8). The title product was obtained as a white foam (519 mg, 95 %). 1H NMR (400 MHz, CDCl3): δ=5.34 (d, J=3.1 Hz, 1 H), 4.81 (d, J=0.7 Hz, 1 H), 4.29 (t, J=9.3 Hz, 1 H), 4.10–4.02 (m, 2 H), 3.98 (t, J=10.3 Hz, 1 H), 3.95 (s, 1 H), 3.88 (dd, J=9.2, 3.3 Hz, 1 H), 3.86–3.76 (m, 3 H), 3.76–3.66 (m, 3 H), 3.58 (dd, J=8.2, 3.1 Hz, 1 H), 3.54–3.47 (m, 1 H), 3.38 (td, J=6.5, 1.7 Hz, 2 H), 3.00 (s, 1 H, OH), 1.94–1.78 (m, 2 H), 1.05 (s, 9 H), 1.04 (s, 9 H), 1.00 (s, 9 H), 0.98 (s, 9 H), 0.93 (s, 9 H), 0.92 (s, 9 H), 0.14 (s, 3 H), 0.13 (s, 3 H), 0.11 (s, 3 H), 0.09 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=99.7, 97.4, 78.7, 75.1, 74.6, 74.5, 74.3, 71.1, 67.6, 67.4, 67.0, 66.4, 64.4, 48.4, 29.0, 27.6 (3×), 27.5 (3×), 27.2 (3×), 27.1 (3×), 26.4 (3×), 26.4 (3×), 22.9, 22.7, 20.1, 20.1, 18.3, 18.3, −3.1, −3.3, −3.4, −3.9 ppm. IR (neat): =2931, 2856, 2099, 1472, 1252, 1132, 1095, 1069, 1043, 868, 827, 772, 654 cm−1. [α] (c 0.1, DCM): +44. HRMS (ESI) m/z: [M+Na]+ calcd for C43H87N3O11Si4+Na 956.53099, found 956.53097.
(4 aR ,6S,7S,8R,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7S,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐7‐ol (13 m): This compound was prepared from 12 m (501 mg, 0.48 mmol) as described for the preparation of 13 g to afford the title product (386 mg, 86 %) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ=5.00 (d, J=1.9 Hz, 1 H), 4.79 (d, J=1.1 Hz, 1 H), 4.17 (t, J=9.2 Hz, 1 H), 4.11 (m, 3 H), 3.99–3.88 (m, 4 H), 3.88–3.82 (m, 2 H), 3.82–3.77 (m, 1 H), 3.77–3.62 (m, 2 H), 3.49 (m, 1 H), 3.40 (td, J=6.5, 3.1 Hz, 1 H), 2.37–2.03 (brs, OH), 1.97–1.77 (m, 2 H), 1.04 (m, 18 H), 0.99 (s, 9 H), 0.97 (s, 9 H), 0.93 (s, 9 H), 0.86 (s, 9 H), 0.12 (s, 3 H), 0.11 (s, 3 H), 0.10 (s, 3 H), 0.02 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=103.2, 99.6, 77.7, 74.5, 74.1, 73.3, 72.4, 71.5, 69.6, 67.4, 67.33, 66.8, 64.5, 48.4, 28.9, 27.8, 27.6, 27.2, 27.1, 26.3, 25.8, 22.9, 22.7, 20.1, 18.6, 18.2, −3.9, −4.1, −4.3, −4.6. ppm IR (neat): =2930, 2858, 2098, 1472, 1250, 1096, 1031 cm−1. [α] (c 0.4, DCM): +32. HRMS (ESI) m/z: [M+H]+ calcd for C43H88N3O11Si4 934.54904, found 934.54959.
(4 aR ,6S,8S,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7R,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydropyrano[3,2‐d][1,3,2]dioxasilin‐7(6H)‐one (14 g): Compound 13 g (2.20 g, 2.36 mmol) was co‐evaporated with dry toluene (3×) and dissolved in dry CH2Cl2 (65 mL). Dess–Martin periodinane (2.00 g, 4.71 mmol) was added and the mixture was stirred overnight. Celite was added and the solvents were removed under reduced pressure. The product was purified by gradient column chromatography (EtOAc/pentane, 1:70 to 1:4). The title product was obtained as a white foam (2.15 g, 98 %). 1H NMR (400 MHz, CDCl3): δ=5.18 (d, J=2.8 Hz, 1 H), 4.73–4.72 (m, 2 H), 4.24–4.02 (m, 5 H), 4.02–3.90 (m, 1 H), 3.90–3.73 (m, 3 H), 3.67 (t, J=8.6 Hz, 1 H), 3.62–3.54 (m, 2 H), 3.40 (t, J=6.6 Hz, 2 H), 1.94–1.81 (m, 2 H), 1.06 (s, 9 H), 1.04 (s, 9 H), 1.02 (s, 9 H), 0.98 (s, 9 H), 0.93 (s, 9 H), 0.92 (s, 9 H), 0.17 (s, 3 H), 0.12 (s, 3 H), 0.11 (s, 3 H), 0.08 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=196.2, 100.2, 98.2, 80.0, 78.9, 78.7, 74.9, 73.9, 67.49, 67.45, 67.0, 66.0, 65.3, 48.0, 27.5, 27.4 (3×), 27.1 (3×), 27.0 (3×), 26.5 (3×), 26.41 (3×), 22.7, 22.6, 20.0, 20.0, 18.3, 18.1, −3.0, −3.5, −3.7, −3.9 ppm. IR (neat): =2932, 2859, 2099, 1757, 1474, 1387, 1362, 1252, 1161, 1093, 1070, 1043, 866, 827, 775, 652 cm−1. [α] (c 0.1, DCM): +50. HRMS (ESI) m/z: [M+Na]+ calcd for C43H85N3O11Si4+Na 954.51534, found 954.51535.
(4 aR ,6S,8S,8 aR )‐6‐(3‐Azidopropoxy)‐2,2‐di‐tert‐butyl‐8‐(((4 aR ,6R,7S,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydropyrano[3,2‐d][1,3,2]dioxasilin‐7(6H)‐one (14 m): This compound was prepared from 13 m (355 mg, 0.38 mmol) as described for the preparation of 14 g to afford the title product (340 mg, 96 %) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ=4.81 (d, J=1.9 Hz, 1 H), 4.75 (s, 1 H), 4.45 (d, J=9.1 Hz, 1 H), 4.31–4.19 (m, 2 H), 4.18–4.12 (t, J=9.2 Hz, 1 H), 4.12–3.98 (m, 4 H), 3.97–3.88 (m, 2 H), 3.88–3.78 (m, 2 H), 3.61–3.52 (dt, J=9.9, 5.4 Hz, 1 H), 3.41 (t, J=6.5 Hz, 2 H), 2.00–1.76 (m, 2 H), 1.05 (s, 9 H), 1.05 (s, 9 H), 1.04 (s, 9 H), 1.00 (s, 9 H), 0.93 (s, 9 H), 0.86 (s, 9 H), 0.15 (s, 3 H), 0.12 (s, 3 H), 0.09 (s, 3 H), 0.01 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=195.8, 103.4, 100.3, 82.2, 79.5, 74.4, 73.3, 72.1, 68.9, 67.3, 67.1, 66.6, 65.3, 48.0, 28.8, 27.6, 27.4, 27.1, 27.0, 26.2, 25.7, 22.8, 22.7, 20.1, 20.0, 18.4, 18.1, −4.0, −4.2, −4.5, −4.7 ppm. IR (neat): =2933, 2858, 2087, 1755, 1471, 1254, 1155 cm−1. [α] (c 0.4, DCM): +47. HRMS (ESI) m/z: [M+H]+ calcd for C43H86N3O11Si4 932.53339, found 932.53363.
(2S,4 a′R ,6′S,8′S,8 a′R )‐6′‐(3‐Azidopropoxy)‐2′,2′‐di‐tert‐butyl‐8′‐(((4 aR ,6R,7R,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydro‐6′H‐spiro[oxirane‐2,7′‐pyrano[3,2‐d][1,3,2]dioxasiline] (15 g) and (2R,4 a′R ,6′S,8′S,8 a′R )‐6′‐(3‐azidopropoxy)‐2′,2′‐di‐tert‐butyl‐8′‐(((4 aR ,6R,7R,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydro‐6′H‐spiro[oxirane‐2,7′‐pyrano[3,2‐d][1,3,2]dioxasiline] (16 g): Method A: dimethyl sulfonium methylide: A 1 m solution of dimsyl‐sodium was prepared from sodium hydride (60 wt %, 200 mg, 5 mmol) in dry DMSO (2.5 mL) and heating this mixture to 70 °C for 1 h. The olive‐green solution was cooled to room temperature and diluted with dry THF (2.5 mL). A fraction (0.12 mL, 0.19 mmol) of this mixture was added to a dried flask and cooled on an ice‐salt bath. Then, a solution of trimethylsulfonium iodide (26.3 mg, 0.129 mmol) in dry DMSO (0.43 mL) and dry THF (0.4 mL) was added drop wise and the mixture was stirred for 5 minutes. Then, compound 14 g (100 mg, 0.11 mmol, co‐evaporated with toluene (3×) beforehand) in dry THF (0.64 mL) was added and the mixture was stirred for 30 minutes. The mixture was diluted with water (20 mL) and extracted with Et2O/pentane (2:1, 4x 15 mL). The combined organic layers were washed with water (20 mL), dried over Na2SO4, filtrated and concentrated. The crude product was purified by gradient column chromatography (pentane/EtOAc, 60:1 to 50:1) to afford solely product 15 g (51 mg, 50 %) as an oil.
Method B: dimethyl sulfoxonium methylide: Trimethylsulfoxonium iodide (37.8 mg, 0.172 mmol) was suspended in dry THF (2 mL) and cooled to 0 °C. nButyllithium (2 m in pentane, 80 μL, 0.16 mmol) was added and the mixture was heated to 60 °C. Compound 14 g (100 mg, 0.11 mmol) was co‐evaporated with toluene (3×), dissolved in dry THF (1 mL) and added drop wise to the ylide solution. After 10 minutes, the mixture was cooled to room temperature and quenched with MeOH (0.5 mL). The mixture was evaporated and the crude product was purified by gradient column chromatography (pentane/EtOAc, 60:1) to give a mixture of compounds 15 g and 16 g (90 mg, ratio 15 g:16 g 1:5, total yield 88 %) as a colorless oil.
Method C: diazomethane: To a glass tube were added aq. KOH (40 %, 5 mL) and Et2O (20 mL) and this mixture was cooled to 0 °C. Then, 1‐methyl‐3‐nitro‐1‐nitrosoguanidine (2.9 g, 10 mmol) was added in portions with swirling. A fraction (2 mL) of the bright yellow ether layer was added drop‐wise to a solution of compound 14 g (100 mg, 0.11 mmol) in EtOH (3 mL) at 0 °C. After stirring for 10 minutes, acetic acid (glacial) was added drop wise until the yellow mixture turned colorless. The mixture was concentrated and co‐evaporated with toluene (3×). The crude products were purified by column chromatography (pentane/acetone, 150:1), affording compound 15 g and 16 g (79 mg, ratio 15 g:16 g 1:1, total yield 78 %). Data for compound 15 g (equatorial methylene): 1H NMR (400 MHz, CDCl3): δ=5.33 (d, J=2.9 Hz, 1 H), 4.27–4.15 (m, 3 H), 4.18 (s, 1 H), 4.10–4.01 (m, 2 H), 4.00–3.95 (t, J=10.1 Hz, 1 H), 3.92–3.76 (m, 3 H), 3.75–3.59 (m, 4 H), 3.50–3.35 (m, 3 H), 3.15 (d, J=5.1 Hz, 1 H), 2.61 (d, J=5.1 Hz, 1 H), 1.86 (quintet, J=6.3 Hz, 2 H), 1.04 (s, 9 H), 1.04 (s, 9 H), 0.98 (s, 18 H), 0.91 (s, 9 H), 0.90 (s, 9 H), 0.13 (s, 3 H), 0.10 (s, 3 H), 0.09 (s, 3 H), 0.08 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=102.5, 97.4, 79.3, 78.5, 76.0, 74.8, 68.7, 67.6, 67.1, 66.3, 64.2, 59.4, 48.3, 46.4, 29.9, 29.1, 27.6, 27.5, 27.2, 27.1, 26.3, 22.9, 22.7, 20.1, 18.4, 18.3, −3.6, −3.6, −4.0 ppm. IR (neat): =2930, 2858, 2099, 1472, 1252, 1091, 1043 cm−1. [α] (c 0.1, DCM): +54. HRMS (ESI) m/z: [M+Na]+ calcd for C16H27N3O11Na 968.53099, found 968.53089.Data for compound 16 g (axial methylene) 1H NMR (400 MHz, CDCl3): δ=5.34 (d, J=3.0 Hz, 1 H), 4.40–4.22 (m, 2 H), 4.09 (dd, J=10.4, 5.1 Hz, 2 H), 4.04–3.93 (m, 3 H), 3.87–3.76 (m, 4 H), 3.66 (t, J=8.7 Hz, 1 H), 3.56–3.39 (m, 4 H), 3.23 (d, J=5.6 Hz, 1 H), 2.63 (d, J=5.6 Hz, 1 H), 1.89 (q, J=5.7 Hz, 2 H), 1.07 (s, 18 H), 1.05 (s, 9 H), 1.02 (s, 9 H), 0.96 (s, 9 H), 0.95 (s, 9 H), 0.16 (s, 3 H), 0.13 (s, 3 H), 0.11 (s, 3 H), 0.10 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=101.2, 96.9, 79.7, 78.2, 74.7, 73.8, 67.7, 67.4, 66.9, 66.8, 66.1, 64.4, 58.3, 48.1, 48.0, 29.0, 27.6 (3×), 27.3 (3×), 27.2 (3×), 27.0 (3×), 26.5 (3×), 26.4 (3×), 22.8, 22.5, 20.1, 20.0, 18.2, 18.1, −3.08, −3.38 (2×), −4.03 ppm. IR (neat): =2934, 2858, 2320, 2094, 1095, 1043, 827, 773 cm−1. [α] (c 0.1, DCM): +98 (c 0.1, DCM). HRMS (ESI) m/z: [M+H]+ calcd for C44H88N3O11Si4 946.54904, found 946.54953.
(2S,4 a′R ,6′S,8′S,8 a′R )‐6′‐(3‐Azidopropoxy)‐2′,2′‐di‐tert‐butyl‐8′‐(((4 aR ,6R,7S,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydro‐6′H‐spiro[oxirane‐2,7′‐pyrano[3,2‐d][1,3,2]dioxasiline] (15 m) and (2R,4 a′R ,6′S,8′S,8 a′R )‐6′‐(3‐azidopropoxy)‐2′,2′‐di‐tert‐butyl‐8′‐(((4 aR ,6R,7S,8S,8 aR )‐2,2‐di‐tert‐butyl‐7,8‐bis((tert‐butyldimethylsilyl)oxy)hexahydropyrano[3,2‐d][1,3,2]dioxasilin‐6‐yl)oxy)tetrahydro‐6′H‐spiro[oxirane‐2,7′‐pyrano[3,2‐d][1,3,2]dioxasiline] (16 m): Compounds 15 m and 16 m were prepared from 14 m as described for the preparation of 15 g and 16 g, and could be separated by careful column chromatography. Method A: Starting from compound 14 m (100 mg, 0.11 mmol), the product 16 m (54 mg, 53 %) was obtained as the single product. Method B: Starting from compound 14 m (150 mg, 0.16 mmol), product 15 m and 16 m were obtained as a mixture (129 mg, 85 %, ratio 15 m:16 m=1:2). Method C: Starting from compound 14 m (265 mg, 0.28 mmol), product 15 m and 16 m were obtained as a mixture (263 mg, 98 %, ratio 15 m:16 m=3:1). Data for 15 m (equatorial methylene): 1H NMR (400 MHz, CDCl3): δ=5.12 (d, J=2.0 Hz, 1 H), 4.28 (d, J=9.4 Hz, 1 H), 4.22–4.14 (m, 3 H), 4.14–4.05 (m, 2 H), 4.00–3.93 (t, J=10.3 Hz, 1 H), 3.93–3.83 (m, 3 H), 3.83–3.77 (m, 2 H), 3.55–3.43 (m, 2 H), 3.40 (m, 2 H), 3.08 (d, J=4.7 Hz, 1 H), 2.67 (d, J=4.7 Hz, 1 H), 1.94–1.82 (m, 2 H), 1.05 (s, 9 H), 1.04 (s, 9 H), 1.00 (s, 9 H), 0.99 (s, 9 H), 0.94–0.90 (m, 9 H), 0.84 (s 9 H), 0.14 (s, 3 H), 0.11 (s, 3 H), 0.10 (s, 3 H), 0.00 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=102.2, 101.9, 78.2, 74.4, 73.4, 72.1, 70.8, 69.7, 68.2, 67.3, 66.5, 64.2, 58.9, 48.2, 46.6, 28.8, 27.7, 27.5, 27.01, 26.2, 25.6, 22.7, 22.6, 20.0, 19.9, 18.4, 18.1, −4.0, −4.2, −4.2, −4.8 ppm. IR (neat): =2929, 2098, 1741, 1251, 1161, 1099 cm−1. [α] (c 0.05, DCM): +38. HRMS (ESI) m/z: [M+H]+ calcd for C44H88N3O11Si4 946.54904, found 946.54940. Data for compound 16 m (axial methylene): 1H NMR (400 MHz, CDCl3): δ=4.94 (d, J=2.0 Hz, 1 H), 4.28 (s, 1 H), 4.25–4.11 (m, 5 H), 3.98–3.85 (m, 6 H), 3.85–3.75 (m, 3 H), 3.48 (m, 3 H), 2.98 (d, J=5.3 Hz, 1 H), 2.64 (d, J=5.3 Hz, 1 H), 2.02–1.80 (m, 2 H), 1.08 (s, 9 H), 1.06 (s, 9 H), 1.03 (s, 9 H), 1.01 (s, 9 H), 0.96 (s, 9 H), 0.87 (s, 9 H), 0.15 (s, 3 H), 0.14 (s, 3 H), 0.11 (s, 3 H), 0.03 ppm (s, 3 H). 13C NMR (101 MHz, CDCl3): δ=102.1, 101.3, 79.0, 74.4, 73.5, 72.6, 68.7, 67.4, 67.2, 66.9, 64.5, 58.3, 48.2, 47.6, 28.9, 27.7, 27.7, 27.2, 27.1, 26.3, 25.7, 22.8, 22.8, 20.2, 20.1, 18.7, 18.2, −4.0, −4.1, −4.3, −4.6 ppm. IR (neat): =2931, 2098, 1741, 1251, 1159, 1097 cm−1. [α] (c 0.05, DCM): +44. HRMS (ESI) m/z: [M+H]+ calcd for C44H88N3O11Si4 946.54904, found 946.54933.
(2R,3R,4S,5S,6R)‐2‐(((3R,4S,6R,7R,8S)‐4‐(3‐Azidopropoxy)‐7‐hydroxy‐6‐(hydroxymethyl)‐1,5‐dioxaspiro[2.5]octan‐8‐yl)oxy)‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐3,4,5‐triol (17): Compound 16 g (145 mg, 0.153 mmol) was co‐evaporated with toluene (3×) and dissolved in dry THF (14.5 mL). TBAF (1 m in THF, 2.3 mL, 2.3 mmol) was added and the mixture was stirred 5 days at room temperature. The solution was eluted with THF over a small Dowex‐50WX4‐200‐Na+ packed column, concentrated and purified by gradient column chromatography (EtOAc/MeOH, 19:1 to 9:1). The product was dissolved in water and lyophilized to afford the title compound as a white solid (64.8 mg, 97 %). 1H NMR (400 MHz, D2O): δ=5.23 (d, J=3.8 Hz, 1 H), 4.50 (s, 1 H), 4.25 (d, J=9.0 Hz, 1 H), 3.95–3.68 (m, 8 H), 3.63–3.53 (m, 2 H), 3.53–3.43 (m, 3 H), 3.41–3.34 (t, J=8 Hz, 1 H), 3.17 (d, J=4.5 Hz, 1 H), 2.87 (d, J=4.6 Hz, 1 H), 1.97–1.85 ppm (m, 2 H). 13C NMR (101 MHz, D2O): δ=100.0, 99.2, 73.0, 72.9, 72.5, 71.7, 71.5, 70.91, 69.1, 64.8, 60.3, 60.2, 58.7, 48.4, 48.1, 27.8 ppm. IR (neat): =3369, 2927, 2108, 1521, 1026 cm−1. [α] (c 0.1, DCM): +174. HRMS (ESI) m/z: [M+NH4]+ calcd for C16H31N4O11 455.19838, found 455.19849.
(2R,3S,4S,5S,6R)‐2‐(((3R,4S,6R,7R,8S)‐4‐(3‐azidopropoxy)‐7‐hydroxy‐6‐(hydroxymethyl)‐1,5‐dioxaspiro[2.5]octan‐8‐yl)oxy)‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐3,4,5‐triol (18): This compound was prepared from 16 m (59 mg, 0.623 mmol) as described for the preparation of 17 to afford the title product (20 mg, 74 %) as a white solid. 1H NMR (400 MHz, D2O): δ=5.05 (d, J=1.6 Hz, 1 H), 4.45 (s, 1 H), 4.18 (d, J=9.3 Hz, 1 H), 3.95 (dd, J=3.2, 1.8 Hz, 1 H), 3.77 (m, 6 H), 3.64 (m, 3 H), 3.57–3.48 (m, 1 H), 3.41 (t, J=6.5 Hz, 2 H), 3.10 (d, J=4.5 Hz, 1 H), 2.81 (d, J=4.5 Hz, 1 H), 1.95–1.78 ppm (m, 2 H). 13C NMR (101 MHz, D2O): δ=101.0, 100.0, 73.3, 72.8, 72.5, 70.9, 70.4, 69.9, 66.3, 64.7, 60.8, 60.2, 58.7, 48.3, 48.0, 27.8 ppm. HRMS (ESI) m/z: [M+Na]+ calcd for C16H27N3O11 460.1538, found 460.1544.
1‐(6‐(((1‐(3‐(((3R,4S,6R,7R,8S)‐7‐Hydroxy‐6‐(hydroxymethyl)‐8‐(((2R,3R,4S,5S,6R)‐3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl)oxy)‐1,5‐dioxaspiro[2.5]octan‐4‐yl)oxy)propyl)‐1H‐1,2,3‐triazol‐4‐yl)methyl)amino)‐6‐oxohexyl)‐3,3‐dimethyl‐2‐((1E,3E)‐5‐((Z)‐1,3,3‐trimethylindolin‐2‐ylidene)penta‐1,3‐dien‐1‐yl)‐3H‐indol‐1‐ium (4): Compound 17 (4.83 mg, 11.0 μmol) was dissolved in DMF (0.5 mL) and placed under Argon. Then the Cy5‐alkyne47 (6.1 mg, 11.0 μmol), aq. CuSO4 (0.1 m, 44 μL, 4.4 μmol) and aq. sodium ascorbate (0.1 m, 44 μL, 4.4 μmol) were added and the mixture was stirred overnight at room temperature. The product was purified by HPLC (50 mm NH4CO3) to afford the title compound as a blue solid (3.54 mg, 32 %). 1H NMR (400 MHz, MeOD): δ=8.24 (t, J=13.0 Hz, 2 H), 7.89 (s, 1 H), 7.49 (d, J=7.4 Hz, 2 H), 7.44–7.38 (m, 2 H), 7.32–7.23 (m, 4 H), 6.62 (t, J=12.4 Hz, 1 H), 6.28 (d, J=13.7 Hz, 2 H), 5.14 (d, J=3.8 Hz, 1 H), 4.85 (s, 1 H), 4.53 (t, J=6.8 Hz, 2 H), 4.42 (s, 2 H), 4.32 (s, 1 H), 4.16 (d, J=9.1 Hz, 1 H), 4.10 (t, J=7.4 Hz, 2 H), 3.84–3.64 (m, 9 H), 3.63 (s, 3 H), 3.56 (t, J=9.3 Hz, 1 H), 3.40 (dd, J=9.7, 3.8 Hz, 1 H), 3.37–3.32 (1, 9 H), 3.07 (d, J=5.3 Hz, 1 H), 2.70 (d, J=5.4 Hz, 1 H), 2.25 (t, J=7.3 Hz, 2 H), 2.23–2.15 (m, 2 H), 1.88–1.76 (m, 2 H), 1.75–1.67 (m, 17 H), 1.51–1.44 ppm (m, 2 H). 13C NMR (101 MHz, MeOD): δ=180.3, 175.7, 175.4, 174.7, 155.5, 155.5, 146.1, 144.3, 143.6, 142.6, 142.5, 129.8, 129.7, 126.6, 126.3, 126.2, 124.7, 123.4, 123.3, 112.0, 111.9, 104.4, 104.3, 102.4, 101.8, 76.8, 75.1, 74.4, 74.0, 73.9, 73.0, 71.2, 65.3, 62.4, 62.4, 59.8, 50.6, 50.5, 48.4, 44.8, 36.5, 35.6, 31.5, 31.0, 28.1, 27.9, 27.8, 27.3, 26.4 ppm. HRMS (ESI) m/z: [M]+ calcd for C51H69N6O12 957.4968, found 957.5005.
1‐(6‐(((1‐(3‐(((3R,4S,6R,7R,8S)‐7‐Hydroxy‐6‐(hydroxymethyl)‐8‐(((2R,3S,4S,5S,6R)‐3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl)oxy)‐1,5‐dioxaspiro[2.5]octan‐4‐yl)oxy)propyl)‐1H‐1,2,3‐triazol‐4‐yl)methyl)amino)‐6‐oxohexyl)‐3,3‐dimethyl‐2‐((1E,3E)‐5‐((Z)‐1,3,3‐trimethylindolin‐2‐ylidene)penta‐1,3‐dien‐1‐yl)‐3H‐indol‐1‐ium (5): This compound was prepared from 18 (3.72 mg, 8.5 μmol) as described for the preparation of 4 to afford the product (2.9 mg, 34 %) as a blue solid. 1H NMR (600 MHz, MeOD): δ=8.24 (t, J=13.0 Hz, 2 H), 7.90 (s, 1 H), 7.49 (d, J=7.4 Hz, 2 H), 7.44–7.39 (m, 2 H), 7.28 (dt, J=16.4, 7.6 Hz, 4 H), 6.62 (t, J=12.4 Hz, 1 H), 6.28 (d, J=13.7 Hz, 2 H), 5.19 (d, J=1.3 Hz, 1 H), 4.85 (s, 1 H), 4.54 (t, J=6.8 Hz, 2 H), 4.42 (s, 2 H), 4.29 (s, 1 H), 4.23 (d, J=9.2 Hz, 1 H), 4.10 (t, J=7.4 Hz, 3 H), 3.90 (dd, J=3.2, 1.7 Hz, 1 H), 3.85–3.68 (m, 9 H), 3.66 (d, J=9.5 Hz, 1 H), 3.63 (s, 3 H), 3.60 (dd, J=9.5, 3.3 Hz, 1 H), 3.57–3.52 (m, 1 H), 3.36–3.32 (m, 1 H), 3.01 (d, J=5.3 Hz, 1 H), 2.68 (d, J=5.4 Hz, 1 H), 2.26 (t, J=7.3 Hz, 2 H), 2.20 (dq, J=13.1, 6.7 Hz, 2 H), 1.83 (m, 2 H), 1.73 (s, 17 H), 1.47 ppm (m, 2 H). 13C NMR (150 MHz, MeOD): δ=180.3, 175.7, 175.4, 174.7, 155.5, 155.5, 146.1, 144.3, 143.6, 142.6, 142.5, 129.8, 129.7, 126.6, 126.3, 126.2, 124.8, 123.4, 123.3, 112.1, 111.8, 104.4, 104.3, 102.6, 102.4, 74.7, 74.3, 74.1, 73.3, 72.7, 72.1, 68.2, 65.2, 62.7, 62.3, 59.9, 50.6, 50.5, 48.3, 44.8, 36.5, 35.7, 31.5, 31.1, 31.0, 28.1, 28.0, 27.8, 27.3, 26.4 ppm. HRMS (ESI) m/z: [M]+ calcd for C51H69N6O12 957.4968, found 957.4995.
Labeling of BtGH99 and BxGH99 enzymes
To determine the detection limit, 400 ng recombinant B. thetaiotaomicron (Bt) and B. xylanisolvens (Bx) GH99 enzymes were labeled in 150 mm McIlvaine buffer, pH 7.0 (citric acid‐Na2HPO4) with 0.0001–10 μm spiro‐epoxyglycoside 4 or 5 for 1 h at 37 °C. The samples were then denatured with 5× Laemmli buffer (50 % (v/v) 1 m Tris‐HCl, pH 6.8, 50 % (v/v) 100 % (v/v) glycerol, 10 % (w/v) DTT, 10 % (w/v) SDS, 0.01 % (w/v) bromophenol blue), boiled for 4 min at 100 °C, and separated by electrophoresis on 10 % (w/v) SDS‐PAGE gel running continuously at 90 V.43 Wet slab‐gels were scanned on fluorescence using a Typhoon FLA 9500 (GE Healthcare at λ EX 532 nm and λ EM 575 nm for ABP TB340; and at λ EX 635 nm and λ EM 665 nm for 4 and 5. The pH optimum was analyzed using 4 ng enzyme incubated with 1 μm 4 or 5 dissolved in McIlvaine buffer, pH 3–8, for 30 min at 37 °C. Time‐dependent labeling of BxGH99 wild‐type, E333Q and E336Q enzymes was assessed by incubating 400 ng for 5, 15 or 30 min with 1 μm 4 or 5 dissolved in McIlvaine buffer, pH 7.0. The effect of denaturation was assessed on 4 ng wild‐type BtGH99 and BxGH99 by boiling for 4 min at 100 °C prior to incubating with 1 μm 4 and 5 for 30 min at 37 °C. Competitive ABPP assays utilized 4 ng BtGH99 and BxGH99 enzyme that was pre‐incubated with 10–1000 μm 17, 18 or ManIFG, or 0.3–30 μg μL−1 yeast mannan (S. cerevisiae), at pH 7.0 for 30 min at 37 °C, followed by labeling with 1 μm 4 and 5 for 30 min at 37 °C.
Functional GLA assay
Recombinant α‐galactosidase GLA was diluted 1:2 in 50 mm McIlvaine buffer, pH 4.6, and pre‐labeled with 2 μm TB340 for 1 h at 37 °C. Subsequently, the mixture was diluted to 1:500 in 150 mm McIlvaine buffer, pH 7.0. In parallel, 400 ng BxGH99 wild‐type, E333Q and E336Q were incubated in the presence or absence of 10 μm 4 or 5, dissolved in 150 mm McIlvaine buffer, pH 7.0, for 1 h at 37 °C. Subsequently, the BxGH99 mixture (10 μL) was incubated with 10 μL TB340‐labeled GLA for 8 h at 37 °C. Hereafter, samples were denatured, separated on SDS‐PAGE gel and visualized by fluorescence scanning, as described above (vide supra). As control, 10 μL TB340‐labeled GLA was treated by either Endo‐H or PNGase‐F, following the manufacturer's instructions (New England Biolabs).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Netherlands Organization for Scientific Research (NWO‐CW ChemThem grant to JMFGA and HSO), and the European Research Council (ERC‐2011‐AdG‐290836 “Chembiosphing” to HSO, and ERC‐2012‐AdG‐32294 “Glycopoise” to GJD). GJD thanks the Royal Society for the Ken Murray Research Professorship.
S. P. Schröder, W. W. Kallemeijn, M. F. Debets, T. Hansen, L. F. Sobala, Z. Hakki, S. J. Williams, T. J. M. Beenakker, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée, G. J. Davies, H. S. Overkleeft, Chem. Eur. J. 2018, 24, 9983.
Contributor Information
Prof. Dr. Gideon J. Davies, Email: gideon.davies@york.ac.uk.
Prof. Dr. Herman S. Overkleeft, Email: h.s.overkleeft@chem.leidenuniv.nl.
References
- 1. Helenius A., Aebi M., Science 2001, 291, 2364. [DOI] [PubMed] [Google Scholar]
- 2. Molinari M., Nat. Chem. Biol. 2007, 3, 313. [DOI] [PubMed] [Google Scholar]
- 3. Feizi T., Larkin M., Glycobiology 1990, 1, 17. [DOI] [PubMed] [Google Scholar]
- 4. Zhao Y.-Y., Takahashi M., Gu J.-G., Miyoshi E., Matsumoto A., Kitazume S., Taniguchi N., Cancer Sci. 2008, 99, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akasaka-Manya K., Manya H., Sakurai Y., Wojczyk B. S., Kozutsumi Y., Saito Y., Taniguchi N., Murayama S., Spitalnik S. L., Endo T., Glycobiology 2010, 20, 99. [DOI] [PubMed] [Google Scholar]
- 6. Herscovics A., Biochim. Biophys. Acta Gen. Subj. 1999, 1473, 96. [DOI] [PubMed] [Google Scholar]
- 7. Pan Y. T., Hori H., Saul R., Sanford B. A., Molyneux R. J., Elbein A. D., Biochemistry 1983, 22, 3975. [DOI] [PubMed] [Google Scholar]
- 8. Sasak V. W., Ordovas J. M., Elbein A. D., Berninger R. W., Biochem. J. 1985, 232, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foddy L., Hughes R. C., Eur. J. Biochem. 1988, 175, 291. [DOI] [PubMed] [Google Scholar]
- 10. Spiro R. G., J. Biol. Chem. 2000, 275, 35657. [DOI] [PubMed] [Google Scholar]
- 11. Moore S. E. H., Spiro R. G., J. Biol. Chem. 1990, 265, 13104. [PubMed] [Google Scholar]
- 12. Fujimoto K., Kornfeld R., J. Biol. Chem. 1991, 266, 3571. [PubMed] [Google Scholar]
- 13. Völker C., De Praeter C. M., Hardt B., Breuer W., Kalz-Füller B., Van Coster R. N., Bause E., Glycobiology 2002, 12, 473. [DOI] [PubMed] [Google Scholar]
- 14. Lubas W. A., Spiro R. G., J. Biol. Chem. 1987, 262, 3775. [PubMed] [Google Scholar]
- 15. Lubas W. A., Spiro R. G., J. Biol. Chem. 1988, 263, 3990. [PubMed] [Google Scholar]
- 16.
- 16a. Lombard V., Ramulu H. G., Drula E., Coutinho P. M., Henrissat B., Nucleic Acids Res. 2014, 42, D490–D495; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.The Cazypedia Consortium, Glycobiology 2017, 28, 3. [DOI] [PubMed]
- 17. Cuskin F., Lowe E. C., Temple M. J., Zhu Y., Cameron E. A., Pudlo N. A., Porter N. T., Urs K., Thompson A. J., Cartmell A., Rogowski A., Hamilton B. S., Chen R., Tolbert T. J., Piens K., Bracke D., Vervecken W., Hakki Z., Speciale G., Munōz-Munōz J. L., Day A., Peña M. J., McLean R., Suits M. D., Boraston A. B., Atherly T., Ziemer C. J., Williams S. J., Davies G. J., Abbott D. W., Martens E. C., Gilbert H. J., Nature 2015, 517, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hakki Z., Thompson A. J., Bellmaine S., Speciale G., Davies G. J., Williams S. J., Chem. Eur. J. 2015, 21, 1966. [DOI] [PubMed] [Google Scholar]
- 19. Petricevic M., Sobala L. F., Fernandes P. Z., Raich L., Thompson A. J., Bernardo-Seisdedos G., Millet O., Zhu S., Sollogoub M., Jiménez-Barbero J., Rovira C., Davies G. J., Williams S. J., J. Am. Chem. Soc. 2017, 139, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iwamoto S., Kasahara Y., Kamei K. I., Seko A., Takeda Y., Ito Y., Matsuo I., Biosci. Biotechnol. Biochem. 2014, 78, 927. [DOI] [PubMed] [Google Scholar]
- 21. Vogel C., Pohlentz G., J. Carbohydr. Chem. 2000, 19, 1247. [Google Scholar]
- 22. Koshland D. E., Biol. Rev. 1953, 28, 416. [Google Scholar]
- 23. Thompson A. J., Williams R. J., Hakki Z., Alonzi D. S., Wennekes T., Gloster T. M., Songsrirote K., Thomas-Oates J. E., Wrodnigg T. M., Spreitz J., Stutz A. E., Butters T. D., Williams S. J., Davies G. J., Proc. Natl. Acad. Sci. USA 2012, 109, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cravatt B. F., Wright A. T., Kozarich J. W., Annu. Rev. Biochem. 2008, 77, 383. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y., Chen C., Jin L., Tan H., Wang F., Cao H., Carbohydr. Res. 2015, 401, 109. [DOI] [PubMed] [Google Scholar]
- 26. Despras G., Robert R., Sendid B., MacHez E., Poulain D., Mallet J. M., Bioorg. Med. Chem. 2012, 20, 1817. [DOI] [PubMed] [Google Scholar]
- 27. Motawia M. S., Olsen C. E., Enevoldsen K., Marcussen J., Møller B. L., Carbohydr. Res. 1995, 277, 109. [DOI] [PubMed] [Google Scholar]
- 28. Martín-Lomas M., Khiar N., García S., Koessler J. L., Nieto P. M., Rademacher T. W., Chem. Eur. J. 2000, 6, 3608. [DOI] [PubMed] [Google Scholar]
- 29. Heuckendorff M., Bendix J., Pedersen C. M., Bols M., Org. Lett. 2014, 16, 1116. [DOI] [PubMed] [Google Scholar]
- 30. Crich D., Dudkin V., Tetrahedron Lett. 2000, 41, 5643. [Google Scholar]
- 31. van der Vorm S., Hansen T., Overkleeft H. S., van der Marel G. A., Codée J. D. C., Chem. Sci. 2017, 8, 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van der Vorm S., Overkleeft H. S., Van der Marel G. A., Codée J. D. C., J. Org. Chem. 2017, 82, 4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sato K., Yoshimura J., Carbohydr. Res. 1979, 73, 75. [Google Scholar]
- 34. Corey E. J., Chaykovsky M., J. Am. Chem. Soc. 1965, 87, 1353. [Google Scholar]
- 35. Garman S. C., Garboczi D. N., J. Mol. Biol. 2004, 337, 319. [DOI] [PubMed] [Google Scholar]
- 36. Sohn Y., Lee J. M., Park H. R., Jung S. C., Park T. H., Oh D. B., BMB Rep. 2013, 46, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang J., Beenakker T. J. M., Kallemeijn W. W., van der Marel G. A., van den Elst H., Codée J. D. C., Aerts J. M. F. G., Overkleeft H. S., Chem. Eur. J. 2015, 21, 10861. [DOI] [PubMed] [Google Scholar]
- 38. Nakajima T., Ballou C. E., J. Biol. Chem. 1974, 249, 7685. [PubMed] [Google Scholar]
- 39. Legler G., Bause E., Carbohydr. Res. 1973, 28, 45. [DOI] [PubMed] [Google Scholar]
- 40. Havukainen R., Törrönen A., Laitinen T., Rouvinen J., Biochemistry 1996, 35, 9617. [DOI] [PubMed] [Google Scholar]
- 41. Gebler J. C., Aebersold R., Withers S. G., J. Biol. Chem. 1992, 267, 11126. [PubMed] [Google Scholar]
- 42. Willems L. I., Jiang J., Li K. Y., Witte M. D., Kallemeijn W. W., Beenakker T. J. N., Schröder S. P., Aerts J. M. F. G., van der Marel G. A., Codée J. D. C., Overkleeft H. S., Chem. Eur. J. 2014, 20, 10864. [DOI] [PubMed] [Google Scholar]
- 43. Witte M. D., Kallemeijn W. W., Aten J., Li K.-Y., Strijland A., Donker-Koopman W. E., van den Nieuwendijk A. M. C. H., Bleijlevens B., Kramer G., Florea B. I., Hooibrink B., Hollak C. E. M., Ottenhoff R., Boot R. G., van der Marel G. A., Overkleeft H. S., Aerts J. M. F. G., Nat. Chem. Biol. 2010, 6, 907. [DOI] [PubMed] [Google Scholar]
- 44. Kallemeijn W. W., Li K. Y., Witte M. D., Marques A. R. A., Aten J., Scheij S., Jiang J., Willems L. I., Voorn-Brouwer T. M., van Roomen C. P. A. A., Ottenhoff R., Boot R. G., van den Elst H., Walvoort M. T. C., Florea B. I., Codée J. D. C., van der Marel G. A., Aerts J. M. F. G., Overkleeft H. S., Angew. Chem. Int. Ed. 2012, 51, 12529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12697. [Google Scholar]
- 45. Ladmiral V., Mantovani G., Clarkson G. J., Cauet S., Irwin J. L., Haddleton D. M., J. Am. Chem. Soc. 2006, 128, 4823. [DOI] [PubMed] [Google Scholar]
- 46. Pedersen C. M., Nordstrøm L. U., Bols M., J. Am. Chem. Soc. 2007, 129, 9222. [DOI] [PubMed] [Google Scholar]
- 47. Kaczmarek O., Scheidt H. A., Bunge A., Föse D., Karsten S., Arbuzova A., Huster D., Liebscher J., Eur. J. Org. Chem. 2010, 1579. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary