

Abstract
Introduction
Sustained proliferative signaling and de-regulated cellular bioenergetics are two of the chief hallmarks of cancer. Alterations in the Ras pathway and its downstream effectors are among the major drivers for uncontrolled cell growth in many cancers. The GTPases are one of the signaling molecules that activate crucial signal transducing pathways downstream of Ras through several effector proteins. The GTPases (GTP bound) interact with several effectors and modulate a number of different biological pathways including those that regulate cytoskeleton, cellular motility, cytokinesis, proliferation, apoptosis, transcription and nuclear signaling. Similarly, the altered glycolytic pathway, the so-called “Warburg effect”, rewires tumor cell metabolism to support the biosynthetic requirements of uncontrolled proliferation. There exists strong evidence for the critical role of the glycolytic pathway’s rate limiting enzymes in promoting immunosuppression.
Areas covered
We review the emerging roles of GTPase effector proteins particularly the p21 activated kinase 4 (PAK4) and nicotinamide biosynthetic pathway enzyme nicotinamide phosphoribosyltransferase (NAMPT) as signaling molecules in immune surveillance and the immune response.
Expert Opinion
In this expert opinion article we highlight the recent information on the role of GTPases and the metabolic enzymes on the immune microenvironment and propose some unique immune therapeutic opportunities.
Keywords: P21 activated Kinases, PAK4, NAD, NAMPT, NAPRT, Immune checkpoint, PD-1, PD-L1, Ras, GTPase, PAK4 inhibitor, NAMPT inhibitor
1) Introduction
A majority of solid tumors are heterogeneous and thrive in a milieu of multiple cell types with different metabolic profiles [1]. Such heterogeneity includes a significant number of immune surveillance cells, some of which generate a suppressive tumor microenvironment (TME), contributing to the lack of an optimal immunogenic response post-treatment with chemo or targeted therapies [2]. These observations prompted developing immunotherapy strategies that specifically target immune suppression and surveillance molecules. In particular, the immune checkpoint inhibitors (ICIs) are being widely evaluated in the clinic in combination therapies for various tumor indications. Nevertheless, despite some positive responses, ICIs alone have failed to demonstrate robust activity in most cancer patients [3]. Therefore, novel combination strategies that can enhance the efficacy of ICIs with minimal additional toxicities are urgently needed.
Sustained proliferation and disturbed cellular bioenergetics are critical to tumor survival. The uncontrolled proliferation has been attributed to the alterations in Kras signaling which occurs in many tumors (http://cancer.sanger.ac.uk/cosmic). Simultaneously, the disturbances in the glycolytic signaling (“Warburg Effect”) and/or cellular energy generating pathways are considered as the major driver of excessive cellular growth in most solid tumors [4]. As presented in this article, both these cancer hallmarks influence immune signaling in the tumor microenvironment. Nevertheless, Kras has remained by far undruggable and strategies to target the unique tumor bioenergetics are yet to yield a clinically beneficial outcome. There is a need to identify unique druggable avenues within these critical pathways for a successful therapeutic outcome.
The Rho GTPases and their effectors hold special position downstream of mutant Kras exerting proliferative signaling, cellular differentiation and plasticity. It is not unusual to note that the majority of Rho GTPase effectors are found to be activated in a large proportion of cancers [5]. More significantly, their role in immune surveillance is slowly being recognized. In parallel, the increased tumor glucose consumption acts as a continuous source of carbon that is needed for anabolic processes for rapidly proliferating cells [6]. Such continuous generation of carbon is used as building blocks for several macromolecules such as proteins, lipids and nucleotides. Highly proliferative cells acutely depend on the reducing equivalents particularly the nicotinamide adenine dinucleotide phosphate (NADPH) [7]. Enhancement in glucose consumption forces higher production of the reducing equivalents in the oxidative branch of the pentose phosphate pathway. These reducing equivalents are then utilized in de novo lipid synthesis pathway (reductive biosynthesis) [8]. Nicotinamide phosphoribosyltransferase (NAMPT) controls the level of intracellular nicotinamide adenine dinucleotide (NAD) thereby controlling the activity of NAD-dependent enzymes [9]. Together with several glycolytic pathway enzymes, NAMPT has also been shown to play a major role in the maturation of several types of cells including those belonging to the immunological niche [10]. There are strong indications to the presence of unique therapeutic opportunities to re-ignite the tumoricidal immune response in cancers that are sustained on Rho GTPase effectors and hyperactivation of cellular energy pathway enzymes.
2) Immune suppression in the solid tumor microenvironment
2.1) Immunosuppressive Cells
Many cells in the tumor microenvironment (TME) have been found to promote a local immunosuppressive state that renders solid tumors unresponsive to immunotherapy. Cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) that are major constituents of stroma promote this TME immunosuppression (Figure 1)[11]. MDSCs were shown to produce reactive nitrogen species that cause nitration of CCL2, forming N-CCL2, which results in T cell apoptosis and trapping in the extracellular matrix (ECM) surrounding numerous tumor types including colon and prostate cancers. MDSCs also secrete arginase which catabolizes L-arginine, a crucial amino acid for T cells, and down regulate L-selectin leading to decreased T cell motility [12]. TAMs resemble M2 macrophages and produce cytokines, like IL-10, that inhibit IL-12 production by intratumoral CD103+ dendritic cells (DCs), which is crucial to T cell proliferation [11]. The increase in lactate production caused by aerobic glycolysis in tumor cells, increases M2 TAMs in the TME. CAFs expressing fibroblast activation protein-α (FAP) trap T cells in the ECM that they produce, and secrete the chemokine CXCL12, which has been found to bind tumor cells, causing T cell exclusion in pancreatic ductal adenocarcinoma, colorectal cancer, and ovarian cancer [13–16]. Another very important immunosuppressive cell type in the TME is the regulatory T cell (Treg). Treg cells (CD4+, CD25+, FOXP3+, CTLA4+, GITR+) are part of the natural immune modulating mechanism, but their presence within tumors is correlated with poor prognosis [17]. Treg cells recognize tumor associated antigens and induce a tolerant microenvironment, through the production of cytokines like IL-10, and suppress effector T cell activation and proliferation [12]. Cytotoxic lymphocyte-associated antigen-4 (CTLA-4), an immune checkpoint protein expressed on Treg cells and recently activated T cells, competes for APC’s CD80 and CD86 (B7 ligands which normally provide co-stimulatory signals to T cells via CD28) causing an increase in the T cell activation signaling threshold [12, 18]. Treg cells also express high levels of the transcription factor FoxP3 that produces TGF-β, preventing the production of pro-inflammatory cytokines [12]. CD8+ T cell mediated inflammation in early tumorigenesis promotes Treg cell production. Indole 2,3-dioxygenase (IDO) expressed by DCs, MDSCs and tumor cells also promotes the formation of Treg cells, as well as metabolizes tryptophan into kynurenine which inhibits T cell proliferation [11]. Tumor and stromal cells produce C-C motif chemokine ligand 28 (CCL28) which attracts Treg cells to the TME [17]. The vasculature of some tumors have increased expression of the apoptosis inducer Fas ligand (FasL) [11]. Treg cells have a high expression of the apoptosis inhibitor c-FLIP and so escape FasL-mediated killing, while CD8+ T cells undergo apoptosis, promoting a Treg dominated immunosuppressed TME.
Figure 1. GTPase and NAD signaling mediated immune suppression mechanisms.
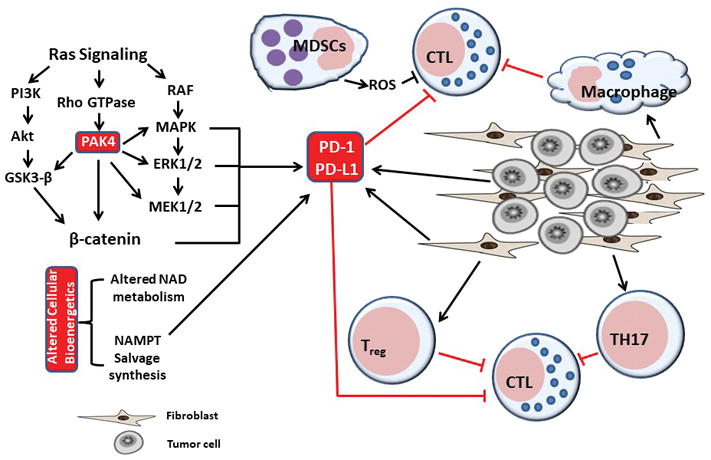
Tumors thrive in a milieu of different types of cells including fibroblasts and collagen. Tumor stromal cells regulate the function of several types of immune cells such as Tregs, TH17, macrophages and MDSCs. Mutant ras promotes canonical RAF-MAPK-ERK-MEK pathway and also activates the Rho GTPase effector protein PAK4. RAF and PAK4 regulate the expression of pro-pro-survival signaling molecules that directly promote immune checkpoint protein PD-1 activation. PD-1 in turn block proper activation of immune surveillance molecules. NAMPT, a rate limiting enzyme in NADPH turnover, can directly activate PD-1 expression.
2.2) Immune Checkpoint
The mechanisms of T-cell activation are very important when discussing immunosuppression in the TME. Antigen presenting cells (APCs) activate T cells by providing 3 signals [19]. The first signal involves binding of the APC’s antigen peptide-MHC complex to the T-cell receptor (TCR), activating the TCR signaling cascade. The second signal involves the co-stimulatory interaction of APCs’ CD80 and T cells’ CD28, activating PI3K-PIP3-AKT and PKCθ pathways necessary for T-cell effector functions and proliferation, as well as preventing T cells apoptosis. The second signal can also be inhibitory through the interaction of the aforementioned APC’s CD80 and T cell’s CTLA4. Another inhibitory signal is through the interaction of programmed death receptor 1 (PD-1) on T cells, with programmed death ligand 1 (PD-L1) expressed on many cells including APCs and cancer cells, which causes strong countering of both TCR signaling and CD28 co-stimulation. The expression of PD-L1 on cancer cells occurs through innate and adaptive immune resistance processes. The innate immune resistance of cancer cells occurs when PI3K/AKT and IL-6/STAT3 signaling cause an increase in PD-L1 expression, while adaptive immune resistance is when PD-L1 is expressed in response to inflammatory cytokines, like INF-γ, released by T cells recognizing antigen on MHC [12, 20]. Hypoxia within the TME causes HIF-1α to bind to a hypoxia-responsive region in the PD-L1 promotor of DCs, thereby increasing PD-L1 expression on their surface [11]*. Lymphocyte Activation Gene-3 (LAG-3), a transmembrane protein expressed on activated immune cells, can also provide an inhibitory signal by binding to MHC2, therefore blocking downstream TCR signaling and inhibiting T-cell proliferation [12]. These inhibitory signals are known as immune checkpoints. Cytokines regulating T-cell differentiation constitute the third signal of T-cell activation [19].
The PD-1/PD-L1 immune checkpoint complex provides a direct cause of immunosuppression within the TME and therefore will be examined in further detail. PD-1 on T cells, when associated with its ligand, PD-L1, directly inhibits the TCR by recruiting SHP1 and SHP2 phosphatases which inhibit ZAP70 and PI3k phosphorylation [19]. This inhibition of phosphorylation has multiple effects like T cells shifting from the glycolysis needed for T-cell activation to fatty acid β-oxidation, as well as T-cell exhaustion and IL-10 production within the tumor. PD-L1 bound PD-1 can also indirectly inhibit TCR signaling by reducing CK2 expression, yielding an active PTEN that converts PIP3 to PIP2, therefore inhibiting the PI3K-AKT-PKCθ pathway and causing T-cell arrest. Also, E3-ubiquitin ligases CBL-B, c-CBL and ITCH are upregulated by engaged PD-1 leading to removal of the TCR from the T-cell surface by endocytosis. PD-1 expressed on Treg cells increases the immune resistance of the TME by upregulating the conversion of naïve T cells to Treg cells in the presence of CD3 and TGF-β [20]. Some melanoma cells have also been shown to express PD-1 that promoted tumor growth through activation of survival pathways [19]. PD-1/PD-L1 blockade was observed to prevent T cell exhaustion and delay tumor growth.
There are about 10 PD1 or PD-L1 directed antibodies under clinical evaluation. Five of those have been approved by FDA (Table 1). Pembrolizumab (pembro) is a monoclonal antibody directed against PD-1 on T cells, disrupting the interaction between PD-1 and its ligand, PD-L1, promotes T cell activation and tumor killing. Darvalumumab and Ipilimumab are monoclonal antibodies against another immune checkpoint, CTLA-4/B7 pair. Evidence suggests that the patients most likely to respond to immunotherapies are those who have a baseline CD8+ T cell infiltration within the TME. In contrast, patients who lack a T cell infiltrate have poor responses to checkpoint inhibitors [21, 22].
Table 1.
FDA approved PD-1/PD-L1 inhibitors
| Drug Name | Type | Indication |
|---|---|---|
| Imfinzi (durvalumab) | Anti-PD-L1 checkpoint inhibitor |
|
| Tecentriq (atezolizumab) | Anti-PD-L1 checkpoint inhibitor |
|
| Bavencio (avelumab) | Anti-PD-L1 checkpoint inhibitor |
|
| Keytruda (pembrolizumab) | Anti-PD-1 checkpoint inhibitor |
|
| Opdivo (nivolumab) | Anti-PD-1 checkpoint inhibitor |
|
Column 3 indications information obtained from Clinicaltrials.gov
2.3) WNT/β-catenin and immune suppression
The Wnt/β-catenin pathway (which is crucial in embryonic development, hematopoiesis, cell migration and wound repair) has been shown to promote cancer growth [23]. WNTs (cysteine-rich glycoproteins) bind to a frizzled receptor and the LRP co-receptor, inhibiting glycogen synthase kinase-3β (GSK-3β) and ultimately increasing un-phosphorylated active β-catenin cytoplasmic levels [24]. Without WNT ligand stimulation, the β-catenin destruction complex (made up of axin, APC, CK1α, and GSK-3β) phosphorylates β-catenin, causing its proteasomal degradation. The function of β-catenin is dependent on its location. At the cell membrane, β-catenin is an important junctional component that links E-cadherins to the actin cytoskeleton, mediating cell to cell adhesion [23, 25]. In the nucleus β-catenin activates T-cell factor/lymphoid enhancer factor-1 (TCF/Lef1) transcriptional complex, activating the expression of several cell proliferation genes such as c-myc and cyclin D1 [26]. Higher β-catenin expression levels are associated with lower CD8+ T cell infiltration, through expression of ATF3 which suppresses CCL4 transcription leading to defective CD103+/CD8α DC recruitment, and higher Treg infiltration and activity, due to the disruption of Foxp3 transcriptional activity [24]. In melanoma, β-catenin signaling increases the expression of IL-10, which hinders the ability of DCs to induce a CD8+ T cell response [27]. In one study, melanoma tumors with active β-catenin were shown to be nearly devoid of T cells [28]. In another study in breast cancer WNT/β-catenin signaling was shown to regulate cancer stem cell (CSC) self-renewal and migration [24]. Also, β-catenin within T cells seems to inhibit their activation; a possible mechanism of immunotherapy resistance [28]. The end result is T-cell exclusion from the TME and unresponsiveness to ICIs.
A correlation was reported between activated β-catenin, a component of the Wnt pathway, and T cell exclusion from the TME in melanoma patients [28–32] as well as in other experimental cancer models including bladder cancer [33], breast cancer [34], and rectal cancer, non-small cell lung cancer and renal cell carcinoma [35]. The mechanism by which active β-catenin reduces T-cell recruitment is not certain, but appears to involve an early step in T-cell priming, one that involves a reduction of antigen-presenting cells/DCs [28]. Specifically, β-catenin is thought to suppress the expression levels of the chemokine CCL4, via its induction of the transcriptional repressor ATF3, which consequently leads to reduced CD103/CD8α DCs [28]. The correlation between active β-catenin and a reduction in T-cell recruitment to the TME is an important concept for the development of combination therapies. These data indicate that combination therapies, in which the Wnt/β-catenin pathway is inhibited, while simultaneously targeting immune checkpoints, may be beneficial in patients who are otherwise resistant to ICI treatment. Nuclear localization of β-catenin is important for the production of its oncogenic transcriptional products [23]. The p21 activated kinase 4 (PAK4), a Rho GTPase (CDC42) effector is a well-recognized regulator for β-catenin and was shown to modulate WNT signaling [26]. In the following section we describe the role of Rho GTPase effectors in immunosuppression and also highlight therapeutic strategies that can be used to re-ignite the immune microenvironment from cold to hot through small molecule strategies.
2.4) Rho GTPase effectors in immune suppression
Members of the Ras family of GTPases, particularly, Ras and Rap1A, and the Rho family GTPases, Cdc42, Rac and RhoA, hold critical positions as signal transducers. Presence of various Ras mutation has been directly linked to the observed enhanced expression of PD-L1 [36–38]. Ras downstream signaling (both canonical and non-canonical) can influence several immune related pathways.
Ras-MAPK pathway activation has been shown to promote tumor immune-evasion. Mitogen activated kinases (MAPK) and epidermal growth factor receptor (EGFR) signaling in immune checkpoint regulation has been clearly demonstrated for solid tumors including enhanced PD-1 expression in pancreatic cancer [39]. Similarly, Ras-MAPK signals are required for PD-L1 expression in lung tumors [40]. MAPK is in turn activated by a variety of mechanisms, some of which are Rho, Rac and CDC42, which are downstream of mutant ras. These GTPases bind to a wide range of effectors that in turn play important roles in lymphocyte biology [41]. The RhoGTPase effector protein has important functions in normal cellular functions such as cell-cell adhesion, regulating cellular polarity, cell motility, cell invasion, cell cycle progression, cell survival, cellular innate immunity and inflammation [42]. RhoA was shown to trigger adhesion through leukocyte integrins thereby regulating immune cell-cell interactions and trafficking [43]. Rac has been demonstrated to regulate integrin-mediated spreading and increased adhesion of T lymphocytes [44]. Various mutations in GTPases are highly relevant to immune suppression, for example Rac1 mutation is associated with increased expression of PD-L1 in mutated Rac1P29S melanoma patients compared to Rac1 wild type or other Rac1 mutants [45] and this has been proposed to cause cancer cells to evade the host immune system. Earlier, Marques and colleagues had identified GTPase Cdc42 as the determinator of cancer cell susceptibility to antigen-specific CTLs in vitro and adoptively transferred immune effectors in vivo [46]. Results from their studies showed that Cdc42 prevents CTL-induced apoptosis via MAPK signaling and post-transcriptional stabilization of B cell lymphoma-2 (Bcl-2). More significantly, pharmacologic inhibition of MEK and/or ERK, components of MAPK signaling cascade was shown to overcome Cdc42-mediated immuno-resistance and activation of Bcl-2 in vivo. These studies clearly demonstrated the role of Cdc42 signaling in mediating immune escape of cancer cells and leading to the idea that targeting Cdc42 may improve the efficacy of cancer immunotherapies. Deregulation of Rho GTPase effectors, particularly the p21-activated kinases [PAKs (Group I PAK1, 2 and 3 and Group II PAK4, 5 and 6)] is linked to oncogenic transformation through Wnt/β-catenin activation, cell survival, altered tumor metabolisms, as well as metastasis.
As discussed above, β-catenin mediated regulation of immune cell function is well recognized, and this protein happens to be within the Rho GTPase network. PAK4 phosphorylates β-catenin on Ser675 in the cytoplasm inhibiting its degradation and increasing its transcriptional activity. At the cell membrane, PAK4 co-localizes areas of cellular junction along with β-catenin, phosphorylating it, thereby contributing to the altered cell polarity seen in many cancers [47]. PAK4 has two nuclear localization signal (NLS) motifs, NLS1 and NLS3, which interact with importin α5, allowing it to enter the nucleus [26]. It is possible that PAK4 not only stabilizes β-catenin (which lacks a NLS motif) in the cytoplasm but also helps shuttle it across the nuclear envelope, as nuclear accumulation of PAK4 accompanies increased β-catenin nuclear levels and increased TCF/LEF transcriptional activity. The association of PAK4 in the nucleus with the β-catenin TCF/LEF complex increases expression of transcribed gene products such as c-myc and cyclin D1, which increase cell proliferation. Silencing of PAK4 decreased cytosolic and nuclear β-catenin, reduction in TCF/LEF transcriptional activity, as well as cyclin D1 expression [25]. It is interesting to note that PAK4, which is oncogenic when overexpressed [47], was also shown to increase cyclin D1 and cell proliferation through an alternate PAK4/c-Src/EGFR pathway, and so inhibiting it would quell multiple proliferative pathways [26].
The above presented studies clearly show the impact of Rho GTPase on immune cell function, and immune suppression. More importantly, the Rho GTPases are intertwined with the cellular energy pathways that are often altered in tumors. Cellular bioenergetics can significantly impact immune cell development and can also alter the immune response. In the following sections we will highlight the role of glycolytic and NAD biosynthetic pathway enzymes in immune cell regulation with a focus on NAMPT.
2.5) Altered tumor metabolism and immune suppression
Many tumors’ increased dependence on aerobic glycolysis, or the “Warburg Effect”, has been known for quite some time. The impact of such altered metabolic state on immune cell function is also emerging. Studies have shown that activated T cells upregulates the Glut1 glucose transporters through TCR and CD28 induced Akt activation, thereby increasing their glucose uptake eighteen times more than naïve T cells [48]. This allows cells to undergo aerobic glycolysis, and without this change T cells become hyporesponsive where even full antigenic stimulation cannot induce a response. Given that, activated T cells engage with aerobic glycolysis and anabolic metabolic pathways for their subsistence, propagation, and functionality, a glucose deficient environment is anticipated to prevent their maturation. Interestingly, competition between tumor cells and T cells for the glucose pool in the aerobic microenvironment was shown to be linked to suppressed T-cell effector functions [49]. Such nutrient competition has been demonstrated to result in loss of tumoricidal action of the T-cells. In a low glucose TME, T cells decrease their Akt activity to lower their glycolytic potential, rendering them hyporesponsive. T cells also activate apoptosis inducing members of the Bcl-2 family [48]. A decrease in methyltransferase enhancer of zeste homolog 2 (EZH2) in T cells of a low glucose TME of ovarian cancer has also been noted, which decreases T cells’ glycolytic potential and therefore function. It is interesting to note that tumor specific checkpoint blockade was shown to reduce tumor glucose uptake and increasing glucose available to T cells. Bone marrow transplant patients receiving allogenic PD-L1−/− T cells had higher levels of GLUT1 and lactate production, hinting that PD-1 signaling possibly decreases T cells’ glucose metabolism [50].
The TME is low in other nutrients essential to T-cell function. TMEs are known to be hypoxic due to decreased blood supply and increased cancer metabolism. Hypoxic conditions in the TME decrease T-cell function and proliferation [48]. In mice, decreasing hypoxia through respiratory hyperoxia increased T-cell infiltration and tumor suppression. Hypoxia also stabilizes hypoxia-inducible factor-1α (HIF-1α) which increases the expression of PD-L1 on tumor cells [50]. Through PD-1/PD-L1 interactions, T cells’ Akt-mTOR pathway is inhibited, decreasing T-cell glycolysis and functions. The TME also has low levels of two amino acids, glutamine and arginine, which are necessary for T-cell activation, differentiation and proliferation, and lack thereof is inhibitory to T cells [48]. The TME demonstrates high levels of immunosuppressive metabolic byproducts, lactic acid, which is produced in large amounts by tumor cells undergoing glycolysis and inhibits T-cell function and cytokine production by pH-dependent alterations and loss of cytosolic NAD+ regeneration. Dying cells in the TME release ATP and NAD which are metabolized by the ectoenzymes CD39, CD73, and the NADase CD38 which are upregulated in some cancer cells, to adenosine [51]. Adenosine binds to the T-cell adenosine A2R receptor inhibiting effector T-cell functions and stimulating regulatory T cells.
Another way PD-1 signaling affects T-cell metabolism is by decreasing expression of the transcription co-activator, peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α), which is a master regulator of mitochondrial biogenesis genes [51]. PGC-1α normally increases glucose uptake and mitochondrial membrane potential in T cells enhancing their activities and prolonging their survival. PD-1 signaling negatively regulates PGC-1α, causing a failure of mitochondrial quality control in T cells, an accumulation of exhausted T cells with high levels of reactive oxygen species, and a possible inhibition of T-cell function.
Aside from T-cells, NAD pathway enzymes and metabolites were shown to affect other immune-cell functions as well. The MDSCs have been well studied for their immunosuppressive role in the tumor microenvironment. MDSCs also deplete the TME of arginine, impairing T-cell functions [52]. Goffaux et al., conducted a modeling study on the MDSCs central carbon metabolism and bioenergetics dynamic using as an in vitro model where the MDSCs were derived from matured mouse bone marrow cells. MDSCs maturation is demonstrated to directly link to high glycolytic flux with minimal participation of the pentose phosphate pathway and oxidative phosphorylation to ensure NADPH production and anabolic metabolite generation [53]. Collectively, these studies clearly illustrate that the tumor metabolism associated microenvironment and Warburg effect play key roles in immunosuppression (Figure 1). Further understanding of the critical components within the tumor microenvironment and their specific roles in immune regulation will help design rational metabolism targeted therapeutic strategies for better treatment outcomes of immunotherapy regimens in cancer.
2.6) Role of NADPH pathway enzyme NAMPT in immune suppression
Tumor cells undertake a metabolic shift and have pronounced increase in glycolytic, pentose and fatty acid synthesis pathways. Such metabolic shift leads to increased tumor cell survival, proliferation and metastasis. The nicotinamide adenine dinucleotide (NAD) is a substrate for several biological enzymes. Many types of cancer cells have a pronounced NAD turnover rate compared to their normal counterparts, due in part to their genomic instability. Nicotinamide, nicotine and tryptophan are three major sources of NAD biosynthesis in mammalian cells. Since NAD is central for these metabolic changes, as the levels of NAD is to be balanced to accommodate these processes. NAD levels are regulated by an equilibrium between its synthesis and degradation which is mediated by enzymes such as CD38, PAPRs and SAMR1 [54]. All these enzymes appear to be involved in immune function and cancer biology. Synthesis of NAD is mainly mediated by Nicotinamide phosphoribosyltransferase (NAMPT) controls the rate-limiting reaction in the NAD biosynthesis pathway [55]. Quite interestingly, NAD precursors generated by NAMPT such as NMN and another one named NR (nicotinamide riboside) can be further metabolized by surface molecules such as CD38 and CD73 located in the tumor environment. These indicate that approaching NAD metabolism can be achieved by manipulation of both the synthetic and degradation pathways [56].
Many tumor cell types are recognized to have higher expression of NAMPT and this increase has been linked to immune suppression. Often considered as a chemokine, extracellular NAMPT has been shown to regulate signaling in inflammatory cells, such as human monocytes, mouse peritoneal macrophages and bone marrow-derived macrophages [57, 58]. Among the earlier studies, in non-cancer models, NAMPT was shown to link NAD metabolism to inflammatory cytokine secretion by leukocytes [59]. These findings led to the hypothesis that NAMPT inhibition may have therapeutic efficacy in immune-mediated inflammatory disorders. Stimulation of resting monocytes isolated from chronic lymphocytic leukemia with NAMPT polarizes them towards tumor-supporting M2-macrophages [60]. Moreover, NAMPT increases expression of immunosuppressive (IL-10) and tumor promoting (IL-6 and IL-8) cytokines. These data suggest an immunosuppressive role of NAMPT in cancer-related inflammation. The enzyme activity independent immune suppression of NAMPT has also been described. In chronic lymphocytic leukemia (CLL) patients with intra- and extra-cellular plasma over-expression of NAMPT promoted the differentiation of resting monocytes causing their polarizing towards tumor-supporting M2 macrophages [60]. These differentiated monocytes have elevated expression of CD163, CD206, and indoleamine 2,3-dioxygenase and secrete immunosuppressive (interleukin [IL] 10, CC chemokine ligand 18) and tumor-promoting (IL-6, IL-8) cytokines.
Earlier studies using conditional knockout mouse models, site directed mutagenesis and NAMPT selective small molecule inhibitor, FK866, showed that inhibition of NAMPT via conditional knockout or through mutation inactivation or chemical inhibition drastically affects development of both T and B lymphocytes leading to tumor immune suppression [61]. Similarly, NAMPT inhibitor APO866 decreased neutrophil NAD(P)/H levels in neutrophils in a dose- and time-dependent manner [62]. Such NAMPT inhibition was shown to alter the expression of several cell-surface receptors. NAMPT inhibitors were also shown to synergize with other drugs that target immune suppression promoting enzymes. For example, APO866 synergizes with L-1-methyl-tryptophan (L-1MT), the inhibitor of negative immune regulator enzyme Indoleamine 2,3-dioxygenase (IDO). The IDO-specific inhibitor also activates the immune responses and reduces tumor volume in mice tumor xenograft. It is anticipated that the combination of APO866 and L-1MT may yield better therapeutic outcome than single agent. Nevertheless, these NAMPT inhibitors were discontinued from clinical studies due to the lack of objective response in patients. Therefore, better agents to target PAK4 and NAMPT are yet to be developed.
3) PAK4-NAMPT dual inhibition
Based upon pre-clinical data and a recently initiated Phase 1 study, targeted inhibition of PAK4 or NAMPT has been demonstrated to be a feasible anti-cancer strategy [63]. However, earlier attempts to develop specific small molecule inhibitors have been challenging. For example, PF-3758309 is a pan PAK inhibitor, which inhibits both the group A PAKs (PAKs 1, 2, 3) and the group B PAKs (PAKs 4, 5, 6) [64], and FK866 was shown to be a highly selective NAMPT inhibitor [65]. Despite strong pre-clinical data from both compounds, but due to lack of objective response in Phase I studies, both inhibitors were discontinued. In the case of PF-3758309 this was due in part to undesirable PK characteristics [66, 67]. To address the unmet therapeutic need for effective PAK4 and NAMPT inhibitors [68], we and others collaborated with Karyopharm Therapeutics, to develop the PAK4-NAMPT dual inhibitor, KPT-9274. In preclinical studies, KPT-9274 demonstrates potent activity against a spectrum of solid tumor and hematological malignancies in both in vitro and in vivo settings [69–73]. KPT-9274 is currently undergoing Phase I clinical evaluation and remains the only compound targeting both PAK4 and NAMPT. Our previous work showed that KPT-9274 could reduce growth of several tumor cell lines and xenograft models, while modulating Wnt/β-catenin signaling [69, 71]. In our previous study, KPT-9274 was effective in inducing dose-dependent apoptosis in non-Hodgkin lymphoma (NHL) cell lines [74]. Also, when used in R-CHOP combination studies, enhanced viability suppression and increased apoptosis was noted. KPT-9274 was well tolerated and showed remarkable anti-tumor activity in sub-cutaneous NHL xenograft in mice. While KPT-9274 has strong anti-tumor activity in immunodeficient nude mice, our preliminary studies now show that KPT-9274 and anti-PD-1 antibody reduce tumor growth even more effectively than either agent alone, in two tumor allograft models in immunocompetent host. Based on published evidence for the role of PAK4-NAMPT in tumor immune suppression (Figure 2), along with emerging data, it would be worthwhile to characterize the cooperative interactions between KPT-9274 and ICIs in therapy resistant cancers. Success of such pre-clinical studies would strengthen the case for KPT-9274-ICI combination as a rational designed strategy for improved treatment outcomes in ICI-resistant patients.
Figure 2. Dual inhibition of PAK4 and NAMPT as a feasible strategy to re-ignite a compromised tumor immune micro-environment.
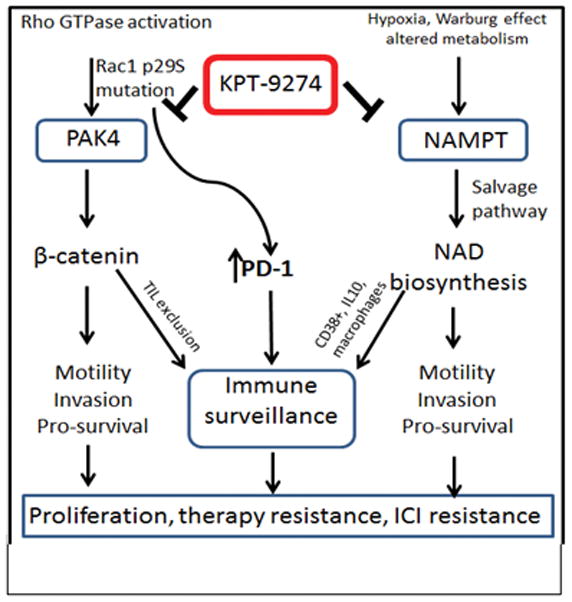
PAK4 signaling is active during cancer, in response to oncogenes such as Ras and mediated by Rho GTPases including Rac. Hypoxia/Warburg effect promotes the salvage biosynthesis pathway. This in turn activates immune surveillance molecules, thereby suppressing ICI activity
4) Expert opinion
The complex interaction between tumor and immune cells in the tumor microenvironment is not fully understood. It is clear that greater understanding of these interactions will be necessary for immune therapy to be successful. Recently, there has been a substantial increase in tumor immune microenvironment research. These efforts have led to the identification of immune suppressive molecules that are now being targeted in the clinics for the treatment of cancers. Among the major immune molecules, the immune checkpoint PD-1 and PD-L1 targeted strategies are being aggressively pursued by many clinics. Nevertheless, only a fraction of patients have benefitted from immune checkpoint blockade strategies. Lack of optimal response indicates the need for deciphering actionable targets that could enhance the efficacy of immunotherapy, particularly the immune checkpoint inhibitor therapies.
The Ras signaling is altered in many cancers and this problem is exacerbated further in some of the difficult to treat malignancies (>90% mutations in pancreatic cancer). Mutant Ras promotes several key proteins that are responsible for uncontrolled tumor cell survival, differentiation and division. Studies show that Ras pathway proteins can directly promote the expression of PD-1 and PD-L1. This signal is mediated through Ras activated MAPK/ERK1/2 or through the Rho GTPases effector PAKs. PAKs, particularly PAK4 can also enhance the expression of PD-1 through β-catenin. On the other hand, the cellular energy generating mechanisms are also found to be de-regulated in most tumors. Switching to glycolytic pathway provides a continuous source of energy and macromolecules for fast growing tumor cells. High NAD turnover rate is a common feature of rapidly dividing tumor cells. NAMPT, which controls the rate-limiting reaction in the NAD biosynthesis pathway, is also a promoter of an immune suppressive microenvironment. In our expert opinion drugging the Rho GTPase effector or NAD pathway enzymes can in principle become an effective strategy to tame immune suppressive molecules particularly the immune checkpoint regulators. The PAK4-NAMPT dual inhibitor KPT-9274 has recently been introduced in Phase I clinical trials for solid tumor and hematological malignancies. The results from these Phase I studies are encouraging and attest to the feasibility of such dual inhibition strategies. Nevertheless, more pre-clinical work especially in humanized patient derived xenograft (PDX) mouse models is needed to justify future Phase II studies combining KPT-9274 with immune checkpoint inhibitors for the treatment of therapy resistance cancers. Such combined therapy is anticipated to lead to unleashing the true potential of immune checkpoint inhibitors for optimal therapeutic outcome of immunotherapy strategies in cancer patients.
Acknowledgments
Work in the lab of ASA is supported by NIH grant 1R21CA188818-01A1. The authors acknowledge the generous support of SKY foundation Inc. for pancreas cancer research.
References
- 1.Lv J, Wang J, Chang S, et al. The greedy nature of mutant RAS: a boon for drug discovery targeting cancer metabolism? Acta Biochim Biophys Sin (Shanghai) 2016;48:17–26. doi: 10.1093/abbs/gmv102. [DOI] [PubMed] [Google Scholar]
- 2.Tufail S, Badrealam KF, Sherwani A, et al. Tissue specific heterogeneity in effector immune cell response. Front Immunol. 2013;4:254. doi: 10.3389/fimmu.2013.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato S, Goodman A, Walavalkar V, et al. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potter M, Newport E, Morten KJ. The Warburg effect: 80 years on. Biochem Soc Trans. 2016;44:1499–505. doi: 10.1042/BST20160094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansen S, Gosens R, Wieland T, et al. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol Ther. 2017 doi: 10.1016/j.pharmthera.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Gillies RJ, Gatenby RA. Metabolism and its sequelae in cancer evolution and therapy. Cancer J. 2015;21:88–96. doi: 10.1097/PPO.0000000000000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41:211–8. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imai S. Nicotinamide phosphoribosyltransferase (Nampt): a link between NAD biology, metabolism, and diseases. Curr Pharm Des. 2009;15:20–8. doi: 10.2174/138161209787185814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grahnert A, Grahnert A, Klein C, et al. Review: NAD +: a modulator of immune functions. Innate Immun. 2011;17:212–33. doi: 10.1177/1753425910361989. [DOI] [PubMed] [Google Scholar]
- 11.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 12.Skelton RA, Javed A, Zheng L, et al. Overcoming the resistance of pancreatic cancer to immune checkpoint inhibitors. J Surg Oncol. 2017;116:55–62. doi: 10.1002/jso.24642. [DOI] [PubMed] [Google Scholar]
- 13.Scotton CJ, Wilson JL, Scott K, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62:5930–8. [PubMed] [Google Scholar]
- 14.Liang JJ, Zhu S, Bruggeman R, et al. High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2010;19:2598–604. doi: 10.1158/1055-9965.EPI-10-0405. [DOI] [PubMed] [Google Scholar]
- 15.Akishima-Fukasawa Y, Nakanishi Y, Ino Y, et al. Prognostic significance of CXCL12 expression in patients with colorectal carcinoma. Am J Clin Pathol. 2009;132:202–10. doi: 10.1309/AJCPK35VZJEWCUTL. quiz 307. [DOI] [PubMed] [Google Scholar]
- 16.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sukari A, Nagasaka M, Al-Hadidi A, et al. Cancer Immunology and Immunotherapy. Anticancer Res. 2016;36:5593–606. doi: 10.21873/anticanres.11144. [DOI] [PubMed] [Google Scholar]
- 18.Bengsch F, Knoblock DM, Liu A, et al. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother. 2017 doi: 10.1007/s00262-017-2053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arasanz H, Gato-Canas M, Zuazo M, et al. PD1 signal transduction pathways in T cells. Oncotarget. 2017;8:51936–45. doi: 10.18632/oncotarget.17232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji RR, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–31. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RG, Ridsdale J, Tonks A, et al. Factors affecting the nuclear localization of beta-catenin in normal and malignant tissue. J Cell Biochem. 2014;115:1351–61. doi: 10.1002/jcb.24803. [DOI] [PubMed] [Google Scholar]
- 24.Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:101. doi: 10.1186/s13045-017-0471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin R, Liu W, Menezes S, et al. The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of beta-catenin through mechanisms involving FRAT1 and PAK4. J Cell Sci. 2014;127:3116–30. doi: 10.1242/jcs.147835. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Shao Y, Tong Y, et al. Nucleo-cytoplasmic shuttling of PAK4 modulates beta-catenin intracellular translocation and signaling. Biochim Biophys Acta. 2012;1823:465–75. doi: 10.1016/j.bbamcr.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Szczepaniak Sloane RA, Gopalakrishnan V, Reddy SM, et al. Interaction of molecular alterations with immune response in melanoma. Cancer. 2017;123:2130–42. doi: 10.1002/cncr.30681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 29.Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43. doi: 10.1186/s40425-015-0089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spranger S, Gajewski TF. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology. 2016;5:e1086862. doi: 10.1080/2162402X.2015.1086862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. 2010;16:399–403. doi: 10.1097/PPO.0b013e3181eacbd8. [DOI] [PubMed] [Google Scholar]
- 32.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sweis RF, Spranger S, Bao R, et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res. 2016;4:563–8. doi: 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajagopalan A, Berezhnoy A, Schrand B, et al. Aptamer-Targeted Attenuation of IL-2 Signaling in CD8+ T Cells Enhances Antitumor Immunity. Mol Ther. 2017;25:54–61. doi: 10.1016/j.ymthe.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luke JJ, Riyue B, Spranger S, et al. Correlation of WNT/beta-catenin pathway activation with immune exclusion across most human cancers. American Society of Clinical Oncology (ASCO) 2016 Abstract 3004. [Google Scholar]
- 36.Yang H, Chen H, Luo S, et al. The correlation between programmed death-ligand 1 expression and driver gene mutations in NSCLC. Oncotarget. 2017;8:23517–28. doi: 10.18632/oncotarget.15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Zhu X, Wang H, et al. Association between PD-L1 expression and driven gene status in NSCLC: A meta-analysis. Eur J Surg Oncol. 2017;43:1372–9. doi: 10.1016/j.ejso.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Koh J, Kwon D, et al. Comparative analysis of PD-L1 expression between primary and metastatic pulmonary adenocarcinomas. Eur J Cancer. 2017;75:141–9. doi: 10.1016/j.ejca.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res. 2016;22:1499–509. doi: 10.1158/1078-0432.CCR-15-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sumimoto H, Takano A, Teramoto K, et al. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS One. 2016;11:e0166626. doi: 10.1371/journal.pone.0166626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henning S, Cleverley S. Small GTPases in lymphocyte biology: Rho proteins take center stage. Immunol Res. 1999;20:29–42. doi: 10.1007/BF02786505. [DOI] [PubMed] [Google Scholar]
- 42.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during development and progression of cancer. Nature Reviews. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laudanna C, Campbell JJ, Butcher EC. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 1996;271:981–3. doi: 10.1126/science.271.5251.981. [DOI] [PubMed] [Google Scholar]
- 44.D’Souza-Schorey C, Boettner B, Van Aelst L. Rac regulates integrin-mediated spreading and increased adhesion of T lymphocytes. Mol Cell Biol. 1998;18:3936–46. doi: 10.1128/mcb.18.7.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vu HL, Rosenbaum S, Purwin TJ, et al. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015;28:590–8. doi: 10.1111/pcmr.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marques CA, Hahnel PS, Wolfel C, et al. An immune escape screen reveals Cdc42 as regulator of cancer susceptibility to lymphocyte-mediated tumor suppression. Blood. 2008;111:1413–9. doi: 10.1182/blood-2007-05-089458. [DOI] [PubMed] [Google Scholar]
- 47.Selamat W, Tay PL, Baskaran Y, et al. The Cdc42 Effector Kinase PAK4 Localizes to Cell-Cell Junctions and Contributes to Establishing Cell Polarity. PLoS One. 2015;10:e0129634. doi: 10.1371/journal.pone.0129634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scharping NE, Delgoffe GM. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines (Basel) 2016:4. doi: 10.3390/vaccines4040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho PC, Bihuniak JD, Macintyre AN, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–28. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kouidhi S, Elgaaied AB, Chouaib S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front Immunol. 2017;8:270. doi: 10.3389/fimmu.2017.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017;35:7–14. doi: 10.1016/j.cytogfr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Renner K, Singer K, Koehl GE, et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol. 2017;8:248. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goffaux G, Hammami I, Jolicoeur M. A Dynamic Metabolic Flux Analysis of Myeloid-Derived Suppressor Cells Confirms Immunosuppression-Related Metabolic Plasticity. Sci Rep. 2017;7:9850. doi: 10.1038/s41598-017-10464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chini CCS, Tarrago MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T, Zhang X, Bheda P, et al. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–2. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- 56.Chini CC, Guerrico AM, Nin V, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20:120–30. doi: 10.1158/1078-0432.CCR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Zhang Y, Dorweiler B, et al. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J Biol Chem. 2008;283:34833–43. doi: 10.1074/jbc.M805866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang TS, Korber DR, Tanaka T. Glycerol and environmental factors: effects on 1,3-propanediol production and NAD(+) regeneration in Lactobacillus panis PM1. J Appl Microbiol. 2013;115:1003–11. doi: 10.1111/jam.12291. [DOI] [PubMed] [Google Scholar]
- 59.Busso N, Karababa M, Nobile M, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One. 2008;3:e2267. doi: 10.1371/journal.pone.0002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Audrito V, Serra S, Brusa D, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. 2015;125:111–23. doi: 10.1182/blood-2014-07-589069. [DOI] [PubMed] [Google Scholar]
- 61.Rongvaux A, Galli M, Denanglaire S, et al. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol. 2008;181:4685–95. doi: 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- 62.Roberts KJ, Cross A, Vasieva O, et al. Inhibition of pre-B cell colony-enhancing factor (PBEF/NAMPT/visfatin) decreases the ability of human neutrophils to generate reactive oxidants but does not impair bacterial killing. J Leukoc Biol. 2013;94:481–92. doi: 10.1189/jlb.1012527. [DOI] [PubMed] [Google Scholar]
- 63.Naing A, Leong S, Pishvaian MJ, et al. 374PD - A First in Human Phase 1 Study of KPT-9274, a First in Class Dual Inhibitor of PAK4 and NAMPT, in Patients with Advanced Solid Malignancies or NHL. Annals of Oncology. 2017:28. [Google Scholar]
- 64.Murray BW, Guo C, Piraino J, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–51. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–42. [PubMed] [Google Scholar]
- 66.Rosen LS, Blumenkopf TA, Breazna A, Darang S, Gallo JD, Goldman J, Want D, Mileshkin L, Eckhardt SG. Phase 1, dose-escalateion, safety, pharmacokinetic and pharmacodynamic study of single agent PF-03758309, an oral PAK inhibitor, in patients with advanced solid tumors. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2011. ABSTRACT A177. [Google Scholar]
- 67.Crawford JJ, Hoeflich KP, Rudolph J. p21-Activated kinase inhibitors: a patent review. Expert Opin Ther Pat. 2012;22:293–310. doi: 10.1517/13543776.2012.668758. [DOI] [PubMed] [Google Scholar]
- 68.Zhao ZS, Manser E. Do PAKs make good drug targets? F1000 Biol Rep. 2010;2:70. doi: 10.3410/B2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abu Aboud O, Chen CH, Senapedis W, et al. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol Cancer Ther. 2016;15:2119–29. doi: 10.1158/1535-7163.MCT-16-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aboukameel A, Muqbil I, Senapedis W, et al. Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther. 2017;16:76–87. doi: 10.1158/1535-7163.MCT-16-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rane C, Senapedis W, Baloglu E, et al. A novel orally bioavailable compound KPT-9274 inhibits PAK4, and blocks triple negative breast cancer tumor growth. Sci Rep. 2017;7:42555. doi: 10.1038/srep42555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fulciniti M, Martinez-Lopez J, Senapedis W, et al. Functional role and therapeutic targeting of p21-associated kinase 4 (PAK4) in multiple myeloma. Blood. 2017 doi: 10.1182/blood-2016-06-724831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang YY, Lin DC, Mayakonda A, et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2016 doi: 10.1136/gutjnl-2016-311818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Azmi S, Aboukameel A, Muqbil AI, et al. Abstract 1358: p21 activated kinase 4 (pak4) as a novel therapeutic target for non-hodgkin’s lymphoma. Cancer Research. 2017:77. [Google Scholar]