

SUMMARY
Background
Transport protein particle (TRAPP) is a multisubunit complex that regulates membrane trafficking through the Golgi apparatus. The clinical phenotype associated with mutations in various TRAPP subunits has allowed elucidation of their functions in specific tissues. The role of some subunits in human disease, however, has not been fully established, and their functions remain uncertain.
Objective
We aimed to expand the range of neurodevelopmental disorders associated with mutations in TRAPP subunits by exome sequencing of consanguineous families.
Methods
Linkage and homozygosity mapping and candidate gene analysis were used to identify homozygous mutations in families. Patient fibroblasts were used to study splicing defect and zebrafish to model the disease.
Results
We identified six individuals from three unrelated families with a founder homozygous splice mutation in TRAPPC6B, encoding a core subunit of the complex TRAPP I. Patients manifested a neurodevelopmental disorder characterised by microcephaly, epilepsy and autistic features, and showed splicing defect. Zebrafish trappc6b morphants replicated the human phenotype, displaying decreased head size and neuronal hyperexcitability, leading to a lower seizure threshold.
Conclusion
This study provides clinical and functional evidence of the role of TRAPPC6B in brain development and function.
INTRODUCTION
Transport protein particle (TRAPP) embodies a series of protein complexes that regulate membrane trafficking from the endoplasmic reticulum (ER) to the Golgi and plasma membrane. There are three TRAPP complexes that share a core of seven subunits and contain additional subunits that give their specific functions (figure 1A): 1) TRAPP I regulates vesicle trafficking from the ER to early Golgi; 2) TRAPP II controls intra-Golgi trafficking and 3) TRAPP III is required for autophagy.1 Mutations in TRAPP subunits have been associated with several human diseases, ranging from spondyloepiphyseal dysplasia tarda (TrappC2 (MIM 300202); adaptor),2 limb girdle muscular dystrophy (TrappC11 (MIM 614138); TRAPP III)3 and intellectual disability with microcephaly (TrappC9 (MIM 611966); TRAPP II).4 However, how mutations in different TRAPP subunits lead to such diverse clinical phenotypes remains unclear. Efforts are required to uncover the full spectrum of diseases associated with individual TRAPP subunits in order to elucidate their cell or tissue-specific functions and their roles within and outside their respective complexes. In this report, we demonstrate the role of TrappC6B in human brain development by describing six children with microcephaly, epilepsy and autistic features who carried a homozygous splice site mutation in TRAPPC6B (trafficking protein particle complex, subunit 6B, (MIM 610397)).
Figure 1.
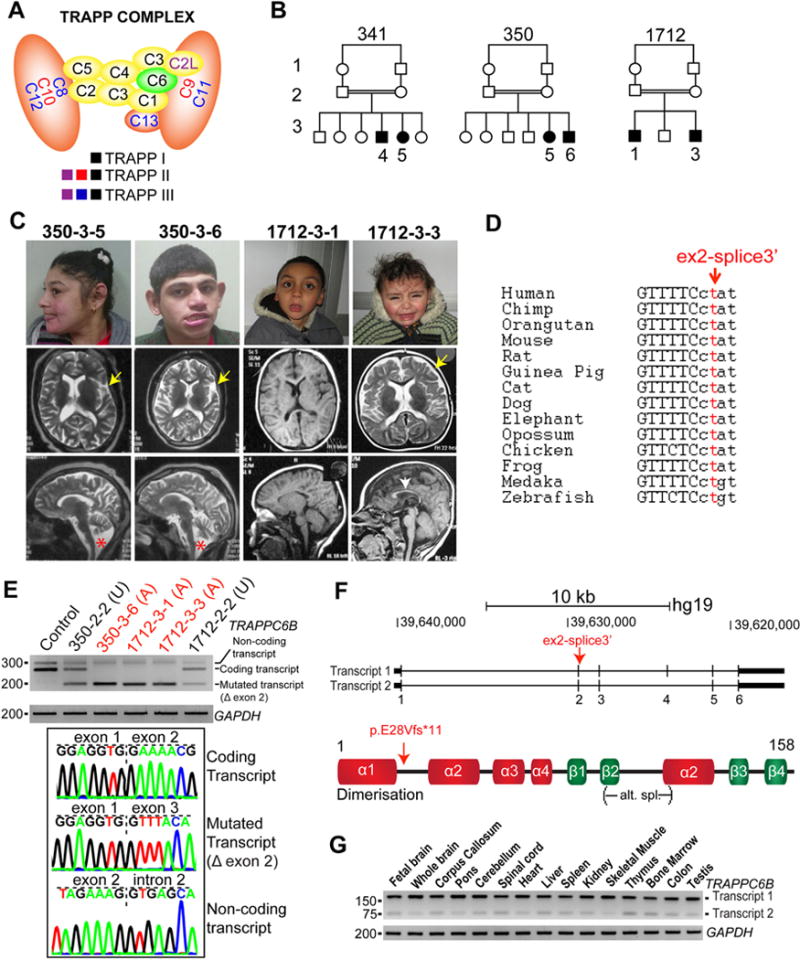
A homozygous mutation in TRAPPC6B leads to a distinctive clinical phenotype. (A) Subunit composition of the different transport protein particle (TRAPP) complexes.1 The subunit mutated in our patients is highlighted in green. (B) Pedigrees of families 341, 350 and 1712 show consanguinity (first cousins). (C) Patient faces and their available sagittal and axial brain MRIs. All individuals were microcephalic without other facial or cranial dysmorphisms. Abnormal brain structural findings included cortical atrophy (350-3-5 (23 years of age), 350-3-6 (21 years of age) and 1712-3-3 (2 years of age), yellow arrow), enlarged lateral ventricles (350-3-5, 350-3-6 and 1712-3-3), hypoplasia of corpus callosum (all patients (1712-3-1, at 3 years of age), white arrow) and cerebellar and brainstem atrophy (350-3-5 and 350-3-6, red star). (D) DNA sequence and splice site conservation of TRAPPC6B in vertebrates. (E) Expression of TRAPPC6B transcript in dermal fibroblasts from control, unaffected (U) and affected (A) individuals by RT-PCR flanking internal regions of exon 1 and exon 3 (primers sequences are available on request). Note decreased and absent coding transcript in unaffected and affected individuals, respectively, compared with control, and the presence of the mutated transcript in both unaffected and affected individuals. Sequence of PCR bands demonstrate skipping of exon 2 in the mutated transcript. The top band represents a non-coding transcript that includes part of intron 2. (F) Scheme of the two transcripts of human TRAPPC6B (transcript 1: NM_001079537.1, transcript 2: NM_177452.3) and of the human protein. The splice site mutation is predicted to shift the reading frame in both transcripts, resulting in a truncated protein. (G) RT-PCR covering internal regions of exons 3–5 shows that both TRAPPC6B transcripts are expressed ubiquitously in human tissues, transcript 1 being the most predominant.
RESULTS
A total of 5000 families were recruited based on the presentation of at least one child with a neurological disorder of likely genetic origin. Recruitment was focused in the major population centres of the Middle East including Morocco, Libya, Egypt, Saudi Arabia, Kuwait, UAE, Oman, Jordan, Pakistan, Turkey and Iran, with consanguinity rates (ie, rate of marriage between first or second cousins) of approximately 50% compared with <1% in the USA and Western Europe. Whole exome sequencing (WES) was performed on at least one affected member of each family as we reported previously.5 We focused on homozygous, rare (<0.1% allele frequency in our exome database) and potentially deleterious variants (genomic evolutionary rate profile score >4 or phastCons>0.9). We identified a disease-causative mutation in the gene TRAPPC6B in six affected individuals from three unrelated consanguineous families (family 341, 350, 1712), who originated from different Egyptian governorates (figure 1B; table 1). A local paediatric neurologist and geneticist clinically evaluated all patients. Samples including blood, saliva and skin biopsy were collected after informed consent from patients and family members. The study was consistent with Institutional Review Board guidelines and approved by the ethical committees of UC San Diego and other participating institutions.
Table 1.
Comprehensive description of clinical findings of patients with TRAPPC6B mutations
| 341-3-4 | 341-3-5 | 350-3-5 | 350-3-6 | 1712-3-1 | 1712-3-3 | |
|---|---|---|---|---|---|---|
| Mutation (genomic DNA) | hg19:14:g.39628756T> C:TRAPPC6B:NM_177452 |
hg19:14:g.39628756T> C:TRAPPC6B:NM_177452 |
hg19:14:g.39628756T> C:TRAPPC6B:NM 177452 |
hg19:14:g.39628756T> C:TRAPPC6B:NM_177452 |
hg19:14:g.39628756T> C:TRAPPC6B:NMJ 77452 |
hg19:14:g.39628756T> C:TRAPPC6B:NM_177452 |
| Mutation (cDNA) | c.82-2A>G | c.82-2A>G | c.82-2A>G | c.82-2A>G | c.82-2A>G | c.82-2A>G |
| Mutation (protein) | p.E28Vfs*11 | p.E28Vfs*11 | p.E28Vfs*11 | p.E28Vfs*11 | p.E28Vfs*11 | p.E28Vfs*11 |
| Origin | Beni Suef (Upper Egypt) |
Beni Suef (Upper Egypt) |
Qalyubia (Lower Egypt) |
Qalyubia (Lower Egypt) |
Sharqia (Lower Egypt) |
Sharqia (Lower Egypt) |
| Gestational age | Full term | Full term | Full term | Full term | Full term | Full term |
| Weight at birth (kg) | 3 | 2.5 | 3 | 3.2 | 3.5 | 3.4 |
| Length at birth (cm) | 49 | 48 | N/A | N/A | 50 | 49 |
| HC at birth (cm, SD) | 32 (−1.9) | 32 (−1.9) | 32 (−1.4) | 32.5 (−1.7) | 32.4 (−1.7) | 33 (−1.5) |
| Postnatal growth | ||||||
| Age at examination (years) | 8 | 3 | 12/23 | 10/21 | 10 | 2 |
| HC (cm, SD) | 47 (−4) | 12.6 (−4) | 46 (−4.5)/46 (−7) | 46 (−4)/48 (−5) | 46 (−4) | 43.2 (−3.9) |
| Height (cm, SD) | 109 (−3) | 84 (−2.5) | 138 (−2)/147 (−2.6) | 122 (−2.2)/156 (−2.5) | 118 (−3) | 80 (−2) |
| Weight (kg, SD) | 20 (−1.6) | 12.6 (−1.5) | 48 (+1)/55 (0) | 36 (+0.7)/50 (−1.5) | 23 (−1) | 14 (+1) |
| Psychomotor development | ||||||
| Gross motor | Delayed, walked at 5 years | Delayed, walks supported | Delayed, walked at 4 years | Delayed, walked at 3 years | Delayed walked at 5 years | Unable to walk, able to sit |
| Fine motor | Severely delayed | Delayed | Severely delayed | Severely delayed | Severely delayed | Delayed |
| Language | No language | No language | Few double syllable words | Few double syllable words | Few single syllable words | No language |
| Social | Poor social interaction, hyperactive | Poor social interaction, hyperactive | Poor social interaction, hyperactive | Poor social interaction, hyperactive | Poor social interaction, hyperactive | Poor social interaction |
| Motor stereotypies | Hand flapping | Hand flapping | Hand flapping, clapping, head nodding versus negative myoclonus | Hand flapping, clapping, head nodding | Hand flapping | Hand flapping |
| Regression of acquired milestones | − | − | Worsening gait ataxia and neurocognitive abilities | Worsening gait ataxia and neurocognitive abilities | − | |
| Neurological findings | ||||||
| Extrapyramidal symptoms | Hand tremors | − | Hand tremors | Hand tremors | Hand tremors | − |
| Brainstem findings | − | − | Dysphagia | − | − | − |
| Nystagmus | + (horizontal) | − | − | − | + (horizontal) | + (horizontal) |
| Motor weakness (upper and/or lower extremities) | + | + | + | + | + | + |
| Muscle tone, bulk | Hypotonia, normal bulk | Hypotonia, normal bulk | Hypotonia, normal bulk | Hypotonia, normal bulk | Hypotonia, normal bulk | Hypotonia, normal bulk |
| Deep tendon reflexes | Brisk in all extremities | Brisk in all extremities | Brisk in all extremities | Brisk in all extremities | Brisk in all extremities | Brisk in all extremities |
| Gait | Wide based, unsteady | Unsteady supported | Wide based, progressive worsening of unsteady gait | Wide based, progressive worsening of unsteady gait | Wide based, unsteady | Cannot walk |
| Seizures | ||||||
| Onset | 3 years | 2 years | 2 years | 2 years | 11 months | − |
| Type | GTC | GTC | GTC | GTC | GTC | GTC |
| Electroencephalogram | Multifocal | N/A | Multifocal | Multifocal | Multifocal | N/A |
| Other systemic findings | Strabismus | − | − | Strabismus | Strabismus, recurrent bone fractures | Strabismus |
(+) present, (−) absent. HC, head circumference; GTC, generalised tonic-clonic seizure; SD, standard deviation; N/A, not available.
Family 341 presented with an affected boy of 8 years (341–3–4) and a girl of 3 years (341–3–5), family 350 presented with a girl aged 12 years (350–3–5) and a boy aged 10 years (350–3–6) and family 1712 presented with two male siblings aged 10 years (1712–3–1) and 2 years (1712–3–3), respectively (figure 1B). All patients manifested microcephaly from birth, global developmental delay, intellectual disability, autistic features such as poor social interaction, language impairment and repetitive automatism behaviours and generalised tonic-clonic seizures (table 1). By the time of evaluation, all patients had reduced head circumference (more than −3 SD) and short stature (−2 to −3 SD), although weight was average for age and gender. No obvious facial dysmorphism was noted besides prominent ears owing to small head size (figure 1C). Neurological examination was remarkable for global hypotonia, generalised weakness with normal muscle bulk, brisk deep tendon reflexes in all extremities and ataxic gait in all six patients. Patient 1712–3–3 was still not yet walking at the last evaluation at 2 years of age. Three patients had mild horizontal nystagmus, four manifested resting and postural hand tremors and one patient showed jaw and perioral dystonia (350–3–6, see online Supplementary video). Two patients (350–3–5 and 350–3–6) manifested worsening of ataxic gait, lost the capability to eat by themselves and became overall more dependent at 23 and 21 years of age, respectively, suggesting a slowly progressive course. Brain MRI was available from families 350 and 1712 (figure 1C), which showed cortical atrophy (350–3–5, 350–3–6, 1712–3–3), thin corpus callosum (all patients) and cerebellar and brainstem atrophy (350–3–5, 350–3–6). In patient 350–3–5, these structural findings worsened over time as compared with imaging at younger age (see online Supplementary figure 1), consistent with a potential neurodegenerative course of the disease. Interictal EEG was available in four patients, and all showed intermittent multifocal epileptiform discharges (table 1), despite the clinical appearance of generalised seizures.
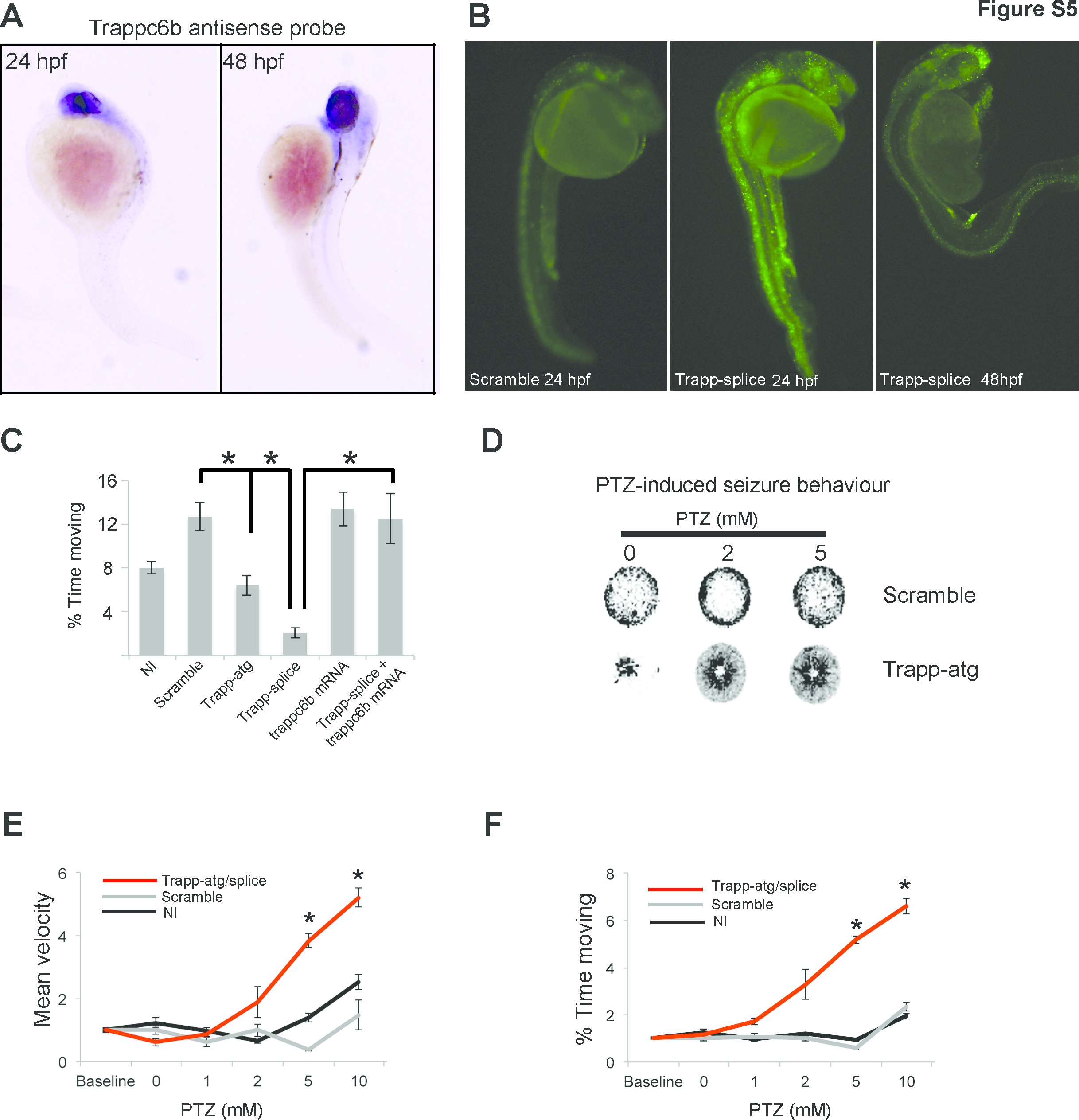
WES analysis of each affected member from family 341 and 350 detected a homozygous mutation in a putative, well-conserved splice site in TRAPPC6B (hg19:14:g.39628756T>C:TRAPPC-6B:NM_177452) (figure 1D, table 1), falling in a linkage peak with LOD >2 identified in both families (see online Supplementary figure 2). The same mutation was subsequently identified in the affected members of family 1712 (see online Supplementary figure 2). Haplotype analysis for families 341, 350 and 1712 identified a maximal potential sharing of about 32 kb surrounding the TRAPPC6B splice site variant (see online Supplementary figure 2), suggesting a common founder mutation. The minor allele frequency according to gnomAD was 0.00004991, and the allele frequency of our internal database of 2608 Egyptians was 0.0023. However, since most of the sequenced individuals in our database were selected based on developmental disorders, this value might overestimate the allele frequency of the resident Egyptian population. The identified TRAPPC6B variant in our patients was predicted to interfere with mRNA splicing. Thus, we obtained dermal fibroblasts from affected and unaffected members of families 350 and 1712 and tested TRAPPC6B mRNA stability. As predicted in silico, dermal fibroblasts from affected individuals showed an abnormal shorter transcript (figure 1E). DNA from PCR bands was extracted for sequencing, which confirmed that the shorter transcript skipped exon 2, leading to a reading frame shift and protein truncation (p.E28Vfs*11) (figure 1F). Fibroblasts from patients did not show obvious differences, neither in the ER or Golgi morphology, nor defects in trafficking using time lapse analysis of transfected cDNA encoding vesicle stomatitis virus ts045 G protein fused to green fluorescent protein (VSVG-GFP) (see online Supplementary figures 4 and 5).
To further investigate whether TRAPPC6B is required for neurodevelopment in vertebrates, we designed a zebrafish model. In humans, TRAPPC6B is expressed in many tissues, including the fetal brain and different regions of the adult brain and spinal cord (figure 1G). In zebrafish, trappc6b transcript was detected in the central nervous system as early as 24 hours postfertilisation (see online Supplementary figure 5). To mimic the mutation carried by our patients in zebrafish, we knocked down trappc6b using two different morpholinos (MOs), a translation blocking (trappc6b-atg, GAAAAAGAGCCTCATCTGCCATCTC) and a splice blocking MO (trappc6b-splice, ATTCCAATACTGTGCAAACCTGGT). The trappc6b-splice MO was designed to mask the splice-donor site of exon 2, the same exon which is abnormally spliced in patients carrying the TRAPPC6B mutation. Trappc6b-atg morphants manifested a decreased survival rate, and both the trappc6b-atg and the trappc6b-splice morphants had reduced head size but normal body length (figure 2A–B). In addition, trappc6b morphants showed an increased number of apoptotic cells when stained with acridine orange (see online Supplementary figure 5), indicating that the reduction in head size might result from cell death during early development.
Figure 2.
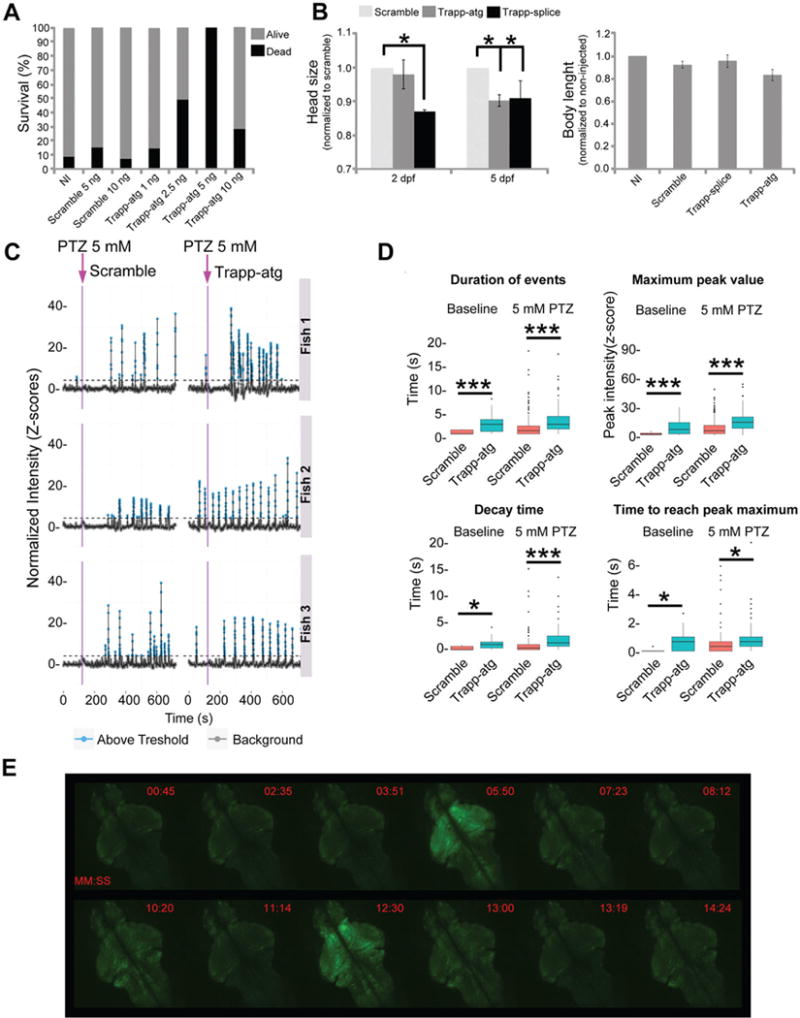
Trappc6b zebrafish model shows neurodevelopmental abnormalities and brain hyperexcitability. (A) The survival rate of trappc6b-atg morphants was lower than scramble morphants and non-injected (NI) fish, mostly with higher concentrations of morpholino. (B) Trappc6b translation blocking (Trapp-atg) and splice blocking (Trapp-splice) morpholinos caused reduced head size relative to scramble morpholino at 2 and 5 dpf. Head size was normalised to scramble at the same developmental stage. In contrast, no differences in body length were observed between groups. (C) Neuronal activity was measured by quantification of calcium transients in the telencephalon, tectum and hindbrain of 5 dpf Tg(elavl3:GCaMP5G) fish injected with scramble or trappc6b-atg morpholinos. Baseline neuronal activity was calculated for a period of 3 min. At the end of the baseline recording, pentylenetetrazol (PTZ) was added at the final concentration of 5 mM and activity was recorded for additional 15 min. Representative traces for three scramble and three trappc6b-atg morphants are shown. Purple arrows indicate PTZ addition. Trappc6b-atg morphants showed a significant increase in spontaneous neuronal activity (baseline) as well as a significant increase in transients when exposed to 5 mM PTZ. (D) Quantification of average duration of each transient event, maximum intensity, decay time and time to reach the maximum intensity showed values significantly different for the trappc6b-atg morphants when compared with controls. *p<0.05, ***p<0.005 (t-test). n>2 experiments for a total of n=9 animal per genotype. Error bars represent SD. (E) Representative whole brain Ca2+ imaging of 5 dpf trappc6b-atg morphants treated with 5 mm PTZ (MM:SS). Pictures were captured at specific time points in order to register the burst of fluorescence.
Since patients manifested seizures, we used the zebrafish model to test the effect of trappc6b knockdown on neuronal excitability and assess seizure threshold using pentylenetetrazol (PTZ). In order to monitor seizure-like phenotypes, we employed an automatic tracking system and compared fish behaviour in the presence of increasing PTZ concentrations. Notably, trappc6b morphants showed abnormal behaviour in all tested conditions (see online Supplementary figure 5), indicating a lower PTZ-induced seizure threshold and underscoring reduced baseline motility. Furthermore, we were interested in studying how the observed behaviour could be explained by abnormal neuronal activity. Thus, we monitored basal neuronal activity employing functional calcium imaging in GCaMP5-expressing zebrafish.6 Trappc6b morphants manifested a significant increase in spontaneous neuronal firing, as well as an increased activity in response to low dose of PTZ (figure 2C–E). Calcium transients in trappc6b morphants were found to be significantly more frequent, longer and of higher intensity. Increase in calcium transient length correlated with a slower calcium transient ramp-up and decay time. Altogether, trappc6b disruption in zebrafish impairs brain growth, leads to neuronal hyperexcitability and lowers seizure threshold.
DISCUSSION
This work expands the range of representative diseases associated with TRAPP subunits. In contrast to most TRAPP-associated conditions, our patients manifested a neurodevelopmental disorder of childhood onset characterised by impaired brain growth, severe psychomotor delay, epilepsy and autistic features. Other relevant findings include extrapyramidal symptoms such as hand tremors and dystonia, motor stereotypies and unstable gait (table 1). The two siblings from family 350 manifested signs of disease progression, such as worsening of ataxic gait and loss of some psychomotor skills in their early 20s, which indicates that this condition may have a neurodegenerative course. The clinical worsening of these two patients correlates well with the severe cerebellar atrophy and the moderate cortical and brainstem atrophy, as compared with previous brain imaging at younger age (see online Supplementary figure 1). Recently, a splice site mutation in TRAPPC6B of likely dominant inheritance was reported in one family with restless leg syndrome (RLS),7 an extrapyramidal condition characterised by the uncontrollable urge to move the legs due to dysfunction of the dopaminergic system. In our patient cohort, a formal diagnosis of RLS could not be established, but the presence of the aforementioned extra-pyramidal symptoms and the fact that most patients manifested hyperactive behaviour may indicate that mutations in this gene could compromise the dopaminergic system, in addition to other neural structures.
Thus far, TrappC9 and TrappC11 are the other subunits that have been associated with disorders of brain development.3,8–13 Besides its role in membrane trafficking as part of complex TRAPP II, TrappC9 is also involved in the nuclear factor-kappa B signalling pathway.1,14 This protein has been implicated in neural differentiation,15 and inactivation in PC12 cells prevents nerve growth factor-induced neurite outgrowth.14 Its expression is highest in the human cerebellum, suggesting that TrappC9 may have a brain-specific signalling function outside of its general role in membrane trafficking.16 Several children with intellectual disability and microcephaly have been found to carry mutations in TRAPPC9.8–11,17 The clinical phenotype is similar to that of our patients, consisting of moderate-to-severe intellectual disability, microcephaly and focal and generalised seizures. In contrast to our cohort, none of the TRAPPC9 patients show autistic features or extrapyramidal symptoms, and the disease does not worsen over time. Brain MRI reveals similar structural abnormalities to TRAPPC6B patients, including thin corpus callosum and global brain atrophy (mostly of cortex and cerebellum), and also shows brainstem hypoplasia. On the contrary, the clinical phenotype of patients with mutations in TRAPPC11 is widespread, involving primarily the skeletal muscle, bone and brain. Patients manifest childhood-onset limb girdle muscular dystrophy, hip dysplasia and scoliosis, intellectual disability, choreiform movements and truncal ataxia.3,12,13 Brain imaging is normal in some patients, and in others it shows brain atrophy.3,12 The molecular mechanism underlying TRAPPC11-associated pathology involves disruption of the anterograde transport of secretory proteins.3 Patients’ fibroblasts have disrupted Golgi architecture and delayed exit of vesicles from the Golgi to the plasma membrane. The involvement of several organs in this condition indicates that TrappC11 may have a widespread membrane trafficking regulatory function as part of the TRAPP III complex, in contrast to TrappC6B in which the clinical phenotype involves primarily the nervous system and in which patient fibroblasts do not show obvious structural or trafficking defects. Our results in zebrafish corroborate the specific role of TRAPPC6B in brain growth and excitability as seen in patients. Further studies are needed to uncover its function at the cellular level in the brain.
The other TRAPP subunit associated with brain disease is TrappC6A, a paralog of TrappC6B and also part of complex TRAPP I. A haplotype at the TRAPPC6A locus has been implicated in non-verbal reasoning in individuals lacking the APOE ε4 allele, suggesting that it may play a role in Alzheimer’s disease (AD).18 It has been shown that the N-terminal internal deletion isoform, TrappC6AΔ, forms aggregates in the brain extracellular matrix, particularly in the cortex and hippocampus of patients with AD.19 This process is prevented by tumour suppressor WW domain-containing oxidoreductase (WWOX), such that its downregulation, as observed in the hippocampus of patients with AD and in the brain of WWOX knockout mice,19,20 favours TrappC6AΔ aggregation, plaque formation and neuronal death. This mechanism may explain the potential TRAPPC6A-associated neurodegeneration and its different clinical course compared with patients with mutations in TRAPPC6B, despite the fact that both subunits are part of the same complex. The challenge for the future will be to further elucidate the role of each individual subunit within and outside of the TRAPP complex in different cell types. As more patients carrying mutations in these subunits are reported, we will better understand their tissue-specific functions and their role in such divergent clinical conditions.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the patients and their families for participation in this study. The authors also thank Ali Crawford, Larry Goldstein, Allyson Muotri and Bruce Hamilton for discussions, and Jennifer Santini at the UCSD Microscope Core (P30 NS047101 and DK80506) for imaging support.
Funding IM–V was sponsored by Pilot Grant awarded by the Center for Basic and Translational Research on Disorders of the Digestive System at The Rockefeller University through the generosity of the Leona M and Harry B. Helmsley Charitable Trust. The authors thank the Broad Institute and Yale Mendelian Sequencing Effort. They thank the Broad Institute (U54HG003067 to E. Lander and UM1HG008900 to D. MacArthur) and the Yale Center for Mendelian Disorders (U54HG006504 to R. Lifton and M. Gunel). This work was supported by NIH grants P01HD070494, R01NS048453, R01NS052455, P01HD070494, the Simons Foundation Autism Research Initiative and Howard Hughes Medical Institute (to JGG and GN).
Footnotes
Contributors IMV, MZ and JGG designed the study. IMV, GN, AJ, BR, JWG and IF performed experiments and analysis. JWG and IF participated in data interpretation and analysis of zebrafish data. IMV, MI, GB, VS, OR, MZ and JGG participated in recruiting patients, gathering clinical information and described the clinical phenotype. IMV, GN, AJ, GB, MZ and JGG wrote the manuscript. MZ and JGG supervised the project.
Competing interests None declared.
Patient consent Consent obtained from guardian.
Ethics approval The study was consistent with Institutional Review Board guidelines and approved by the ethical committees of UC San Diego.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Brunet S, Sacher M. In sickness and in health: the role of TRAPP and associated proteins in disease. Traffic. 2014;15:803–18. doi: 10.1111/tra.12183. [DOI] [PubMed] [Google Scholar]
- 2.Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gécz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet. 1999;22:400–4. doi: 10.1038/11976. [DOI] [PubMed] [Google Scholar]
- 3.Bögershausen N, Shahrzad N, Chong JX, von Kleist-Retzow JC, Stanga D, Li Y, Bernier FP, Loucks CM, Wirth R, Puffenberger EG, Hegele RA, Schreml J, Lapointe G, Keupp K, Brett CL, Anderson R, Hahn A, Innes AM, Suchowersky O, Mets MB, Nürnberg G, McLeod DR, Thiele H, Waggoner D, Altmüller J, Boycott KM, Schoser B, Nürnberg P, Ober C, Heller R, Parboosingh JS, Wollnik B, Sacher M, Lamont RE. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet. 2013;93:181–90. doi: 10.1016/j.ajhg.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khattak NA, Mir A. Computational analysis of TRAPPC9: candidate gene for autosomal recessive non-syndromic mental retardation. CNS Neurol Disord Drug Targets. 2014;13:699–711. doi: 10.2174/18715273113129990105. [DOI] [PubMed] [Google Scholar]
- 5.Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, Olvera J, Bafna V, Zaki MS, Abdel-Salam GH, Mansour LA, Selim L, Abdel-Hadi S, Marzouki N, Ben-Omran T, Al-Saana NA, Sonmez FM, Celep F, Azam M, Hill KJ, Collazo A, Fenstermaker AG, Novarino G, Akizu N, Garimella KV, Sougnez C, Russ C, Gabriel SB, Gleeson JG. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78. doi: 10.1126/scitranslmed.3003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. Optimization of a GCaMP calcium Indicator for neural activity imaging. J Neurosci. 2012;32:13819–40. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aridon P, De Fusco M, Winkelmann JW, Zucconi M, Arnao V, Ferini-Strambi L, Casari GA. TRAPPC6B splicing variant associates to restless legs syndrome. Parkinsonism Relat Disord. 2016;31:135–8. doi: 10.1016/j.parkreldis.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Abou Jamra R, Wohlfart S, Zweier M, Uebe S, Priebe L, Ekici A, Giesebrecht S, Abboud A, Al Khateeb MA, Fakher M, Hamdan S, Ismael A, Muhammad S, Nöthen MM, Schumacher J, Reis A. Homozygosity mapping in 64 syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet. 2011;19:1161–6. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, Kahrizi K, Rafiq MA, Weksberg R, Nasr T, Naeem F, Tzschach A, Kuss AW, Ishak GE, Doherty D, Ropers HH, Barkovich AJ, Najmabadi H, Ayub M, Vincent JB. Identification of mutations in TRAPPC9, which encodes the IK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am J Hum Genet. 2009;85:909–15. doi: 10.1016/j.ajhg.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mochida GH, Mahajnah M, Hill AD, Basel-Vanagaite L, Gleason D, Hill RS, Bodell A, Crosier M, Straussberg R, Walsh CA. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am J Hum Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Philippe O, Rio M, Carioux A, Plaza JM, Guigue P, Molinari F, Boddaert N, Bole-Feysot C, Nitschke P, Smahi A, Munnich A, Colleaux L. Combination of linkage mapping and microarray-expression analysis identifies NF-kappaB signaling defect as a cause of autosomal-recessive mental retardation. Am J Hum Genet. 2009;85:903–8. doi: 10.1016/j.ajhg.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koehler K, Milev MP, Prematilake K, Reschke F, Kutzner S, Jühlen R, Landgraf D, Utine E, Hazan F, Diniz G, Schuelke M, Huebner A, Sacher M. A novel TRAPPC11 mutation in two turkish families associated with cerebral atrophy, global retardation, scoliosis, achalasia and alacrima. J Med Genet. 2017;54 doi: 10.1136/jmedgenet-2016-104108. [DOI] [PubMed] [Google Scholar]
- 13.Liang WC, Zhu W, Mitsuhashi S, Noguchi S, Sacher M, Ogawa M, Shih HH, Jong YJ, Nishino I. Congenital muscular dystrophy with fatty liver and infantile-onset cataract caused by TRAPPC11 mutations: broadening of the phenotype. Skelet Muscle. 2015;5:29. doi: 10.1186/s13395-015-0056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pendergast Hu WH, Mo JS, Brambilla XM, Bracchi-Ricard R, Li V, Walters F, Blits WM, He B, Schaal L, Bethea SM., JR IBP, a novel IK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J Biol Chem. 2005;280:29233–41. doi: 10.1074/jbc.M501670200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Bitner D, Pontes Filho AA, Li F, Liu S, Wang H, Yang F, Adhikari S, Gordon J, Srinivasan S, Hu W. Expression and function of IK- and IKK2-binding protein I in mouse enteric nervous system. Neurogastroenterol Motil. 2014;26:77–97. doi: 10.1111/nmo.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Court F, Camprubi C, Garcia CV, Guillaumet-Adkins A, Sparago A, Seruggia D, Sandoval J, Esteller M, Martin-Trujillo A, Riccio A, Montoliu L, Monk D. The EG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus. Epigenetics Chromatin. 2014;7:5. doi: 10.1186/1756-8935-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marangi G, Leuzzi V, Manti F, Lattante S, Orteschi D, Pecile V, Neri G, Zollino M. TRAPPC9-related autosomal recessive intellectual disability: report of a new mutation and clinical phenotype. Eur J Hum Genet. 2013;21:229–32. doi: 10.1038/ejhg.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamilton G, Harris SE, Davies G, Liewald DC, Tenesa A, Starr JM, Porteous D, Deary IJ. Alzheimer’s disease genes are associated with measures of cognitive ageing in the lothian birth cohorts of 1921 and 1936. Int J Alzheimers Dis. 2011;2011:505984. doi: 10.4061/2011/505984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang JY, Lee MH, Lin SR, Yang LY, Sun HS, Sze CI, Hong Q, Lin YS, Chou YT, Hsu LJ, Jan MS, Gong CX, Chang NS. Trafficking protein particle complex 6A Delta (TRAPPC6AΔ) is an extracellular plaque-forming protein in the brain. Oncotarget. 2015;6:3578–89. doi: 10.18632/oncotarget.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS. Downregulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J Biol Chem. 2004;279:30498–506. doi: 10.1074/jbc.M401399200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.